

Abstract
The Rubiaceae species, Ophiorrhiza pumila, accumulates camptothecin, an anti-cancer alkaloid with a potent DNA topoisomerase I inhibitory activity, as well as anthraquinones that are derived from the combination of the isochorismate and hemiterpenoid pathways. The biosynthesis of these secondary products is active in O. pumila hairy roots yet very low in cell suspension culture. Deep transcriptome analysis was conducted in O. pumila hairy roots and cell suspension cultures using the Illumina platform, yielding a total of 2 Gb of sequence for each sample. We generated a hybrid transcriptome assembly of O. pumila using the Illumina-derived short read sequences and conventional Sanger-derived expressed sequence tag clones derived from a full-length cDNA library constructed using RNA from hairy roots. Among 35,608 non-redundant unigenes, 3,649 were preferentially expressed in hairy roots compared with cell suspension culture. Candidate genes involved in the biosynthetic pathway for the monoterpenoid indole alkaloid camptothecin were identified; specifically, genes involved in post-strictosamide biosynthetic events and genes involved in the biosynthesis of anthraquinones and chlorogenic acid. Untargeted metabolomic analysis by Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) indicated that most of the proposed intermediates in the camptothecin biosynthetic pathway accumulated in hairy roots in a preferential manner compared with cell suspension culture. In addition, a number of anthraquinones and chlorogenic acid preferentially accumulated in hairy roots compared with cell suspension culture. These results suggest that deep transcriptome and metabolome data sets can facilitate the identification of genes and intermediates involved in the biosynthesis of secondary products including camptothecin in O. pumila.
Keywords: Anthraquinone, Camptothecin, Hairy root, Metabolome, Ophiorrhiza pumila, Transcriptome
The nucleotide sequences reported in this paper have been submitted to DDBJ under the accession numbers, HX703880–HX739052, for ESTs; and DDBJ Sequence Read Archive under the accession number, DRA000930, for the Illumina reads.
Introduction
In the post-genomic era, integrated analysis of comprehensive gene expression (transcriptome) and metabolic profiling (metabolomics) has been successful in plant functional genomics (Saito et al. 2008, Saito and Matsuda 2010). In particular, a number of examples in Arabidopsis thaliana have reported the identification and/or the prediction of gene function employing a coupled approach using transcriptome and metabolome data sets (Hirai et al. 2005, Tohge et al. 2005, Hirai et al. 2007, Yonekura-Sakakibara et al. 2007, Yonekura-Sakakibara et al. 2008, Okazaki et al. 2009, Matsuda et al. 2010, Yonekura-Sakakibara et al. 2012). In A. thaliana, these successes are due in large part to the availability of genome sequence data, expression profiles and genetic resources such as tagged mutants and natural variants that enable functional genomics research. The application of integrated ‘omics’ analyses to medicinal plants, however, is not necessarily straightforward, as typically few, if any, sequence or expression profiling data are available (Yonekura-Sakakibara and Saito 2009). Nevertheless, the recent technological advances in DNA sequencing, the so-called ‘next-generation sequencing’, allow holistic profiling of RNA expression (Wang et al. 2009, Ozsolak and Milos 2011) in non-model plant species in which limited molecular genetics studies have been performed. Deep transcriptome technology, also known as RNA sequencing (RNA-seq), provides whole-transcriptome expression profiles of selected plant tissues or cells, thereby permitting the integrated analysis of transcriptomics and metabolomics in any plant species.
The anti-cancer alkaloid, camptothecin (compound 7 in Fig. 1), was identified first in the Chinese tree Camptotheca acuminata (Nyssaceae) during an extensive screening of natural anti-cancer compounds (Wall et al. 1966). This alkaloid inhibits DNA topoisomerase I and has potent anti-tumor activity (Hsiang et al. 1985). In the clinical field, semi-synthetic water-soluble camptothecin derivatives are used throughout the world as chemotherapy agents for cancers of the lung, cervix, ovary, colon and other organs (Sirikantaramas et al. 2007). Though the skeleton structure is a pentacyclic quinoline-type alkaloid, camptothecin is synthesized via the monoterpenopid indole alkaloid pathway (Hutchinson et al. 1979, Yamazaki et al. 2003a, Yamazaki et al. 2004) (Fig. 1).
Fig. 1.

Possible biosynthetic pathway of camptothecin and related alkaloids.
The genus Ophiorrhiza (Rubiaceae) encompasses approximately 150 species distributed throughout tropical and subtropical Asia (Darwin 1976), with some species producing camptothecin and related indole alkaloids (Aimi et al. 1989, Arbain et al. 1993, Kitajima et al. 2005, Viraporn et al. 2011). Ophiorrhiza pumila specimens in Japan accumulate camptothecin and its potential biosynthetic intermediates (Aimi et al. 1989, Aimi et al. 1990). Ophiorrhiza pumila also produces anthraquinones, flavonoids and chlorogenic acid, in addition to indole alkaloids (Kitajima et al. 1998, Yamazaki et al. 2003b). Several genes involved in the biosynthesis of camptothecin have been characterized (Yamazaki et al. 2003a), and possible intermediates in camptothecin biosynthesis have been proposed (Asano et al. 2012). Nevertheless, a large part of the information on the genes and their encoded enzymes for camptothecin biosynthesis is as yet unknown.
We established an O. pumila hairy root culture (Saito et al. 2001) which produces camptothecin and anthraquinones, and an O. pumila cell suspension culture derived from hairy roots (Asano et al. 2012) which does not accumulate these secondary metabolites. These two cultured cells provide contrasting sources for integrated and differential analysis of transcriptome and metabolome data to facilitate the prediction of candidate genes involved in the biosynthesis of these secondary metabolites. In the present study, we utilized deep transcriptomics combined with untargeted metabolic profiling by in-fusion Fourier transform-ion cyclotron resonance-mass spectrometry (FT-ICR-MS) using a hairy root culture and a cell suspension culture of O. pumila to identify candidate genes associated with secondary metabolite production. Through our combined analyses, we identified differential transcripts and metabolites in the two tissues that are presumed to be associated with the biosynthesis of secondary products including camptothecin. These data sets are useful resources for further studies involving intermediary metabolites.
Results
Assembly of deep transcriptome data from O. pumila
Deep transcriptome analysis was performed using RNA from hairy roots and cell suspension culture, exhibiting differential accumulation of secondary metabolites, using the Illumina HiSeq 2000 platform. Over 20 million paired-end reads for each sample representing >2 Gb were generated, and a transcriptome was generated with these two data sets using the Oases transcriptome assembler (Schulz et al. 2012). A full-length cDNA library was constructed from O. pumila hairy roots, and 35,208 expressed sequence tags (ESTs) from 19,400 clones were sequenced using Sanger dideoxy sequencing. The Sanger-derived EST data were used in a hybrid assembly with the Oases-derived transcript assemblies, yielding 35,608 non-redundant unigenes that represent the O. pumila transcriptome. To estimate expression abundances in the two tissues, reads from the hairy root and the cell suspension culture RNA-seq data sets were mapped to the O. pumila unigene set using Bowtie (Langmead et al. 2009); in total, 87–91% of reads could be mapped to the assembled non-redundant unigenes (Supplementary Fig. S1).
Overview of differentially expressed genes between hairy roots and cell suspension culture
Expression levels in the hairy roots or cell suspension culture libraries were evaluated using Cufflinks (Trapnell et al. 2010) (Supplementary Fig. S1). Differential expression was determined using a false discovery rate (FDR) threshold of corrected P-value <0.05 (Fig. 2A), and a heatmap diagram sorted by the magnitude of fold change (log2) is shown in Fig. 2B. Using differential expression analyses, a total of 3,649 unigenes were up-regulated in hairy roots compared with cell suspension culture, whereas 1,777 unigenes were down-regulated in hairy roots compared with cell suspension culture (Supplementary Table S1).
Fig. 2.
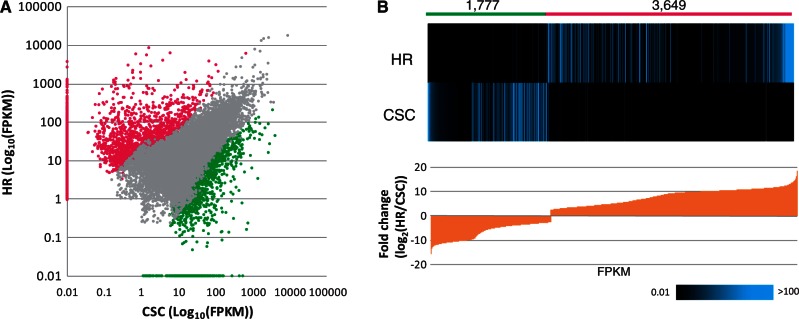
Deep transcriptome analysis of hairy roots and cell suspension cultures of O. pumila. (A) Expression profile of all unigenes plotted by log10 FPKM in hairy roots (HR) (y-axis) and cell suspension culture (CSC) (x-axis). The unigenes differentially expressed with a threshold of an FDR-corrected P-value <0.05 are shown in color: red, up-regulated inHR; green, up-regulated in CSC. (B) The differentially expressed unigenes are shown in a heatmap diagram sorted by the magnitude of fold change (log2). The 3,649 and 1,777 unigenes from HR and CSC are preferentially expressed in the respective samples.
Gene ontology (GO) enrichment analysis was performed with the 3,649 unigenes up-regulated in hairy roots compared with cell suspension cells (Supplementary Fig. S2, Supplementary Table S2). The GO terms ‘oxidation–reduction process’ (GO: 0055114) and ‘organic substance transport’ (GO: 0071702) were the most significantly enriched, followed by ‘developmental maturation’ (GO: 0021700) and ‘transition metal ion transport’ (GO: 0000041). Other significantly enriched GO terms in the set of 3,649 up-regulated unigenes included various ‘transport’ ontologies, ‘response to oxidative stress’, ‘phenylpropanoid metabolic process’ and those related to developmental processes including the terms ‘cell maturation’ and ‘root hair cell differentiation’. The GO analysis suggested that the genes related to secondary metabolism and hairy root development by Agrobacterium rhizogenes infection were particularly enriched.
Specific genes involved in the biosynthesis of secondary metabolites in hairy roots
Camptothecin biosynthetic genes
For the biosynthesis of camptothecin in O. pumila, several genes encoding enzymes involved in the pathway, as well as DNA topoisomerase I which is the target site of camptothecin toxicity, have been cloned (Yamazaki et al. 2003a, Sirikantaramas et al. 2008). The preferential expression of these known camptothecin biosynthetic pathway genes was examined in the transcriptome data set (Table 1). Three unigenes encoding tryptophan decarboxylase and one unigene for strictosidine synthase, both involved in the early steps of the biosynthesis of monoterpenoid indole alkaloids, were highly differentially expressed in hairy roots compared with cell suspension cells. However, unigenes of two enzymes which are not specifically involved in the pathway were equally expressed in hairy roots and cell suspension culture. These are NADPH-cytochrome P450 reductase, which transfers an electron from NADPH to Cyt P450 non-specifically, and DNA topoisomerase I, which is involved in DNA replication and acts as the target site of camptothecin toxicity.
Table 1.
Expression of previously characterized genes related to camptothecin biosynthesis and self-resistance of O. pumila
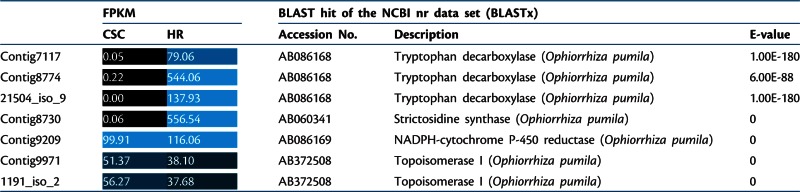 |
Expression was measured as fragments per kilobase of transcript per million fragments mapped (FPKM). Functional annotation was assigned from identical BLAST hit’s description of the NCBI nr data set. The expression levels of unigenes are represented with a black–blue color code and FPKM values.
CSC, cell suspension culture; HR, hairy roots.
Anthraquinone biosynthetic genes
Anthraquinones are a major group of secondary products found in the plants of the Rubiaceae family including O. pumila (Kitajima et al. 1998, Chan et al. 2005). Anthraquinones in Rubiaceae species have been shown to be synthesized by a combination of the isochorismate and plastidic hemiterpenoid 2-C-methyl-d-erythritol 4-phosphate (MEP) pathways (Han et al. 2001, Han et al. 2002). The anthraquinone skeleton is formed by coupling of a 1,4-dihydroxy-2-naphthoyl derivative with dimethylallyl diphosphate (Fig. 3). Thus, we examined the expression of unigenes encoding enzymes for these two pathways based on fragments per kilobase of transcript per million fragments mapped (FPKM). All the unigenes encoding the enzymes involved in the formation of 1,4-dihydroxy-2-naphthoyl-CoA from chorismate were significantly highly expressed in the hairy roots compared with cell suspension culture. On the other hand, regarding the plastidic hemiterpenoid MEP pathway, only the unigenes encoding four enzymes were highly differentially expressed in the hairy roots compared with cell suspension culture (Fig. 3). The unigenes that encode 1-deoxy-d-xylulose-5-phosphate synthase (DXPS; EC 2.2.1.7) (Contig11115, Contig2771 and Contig8042), 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase (CDPMEK; EC 2.7.1.148), (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (HDS; EC 1.17.7.1) and 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR; EC 1.17.1.2) were highly differentially expressed in the hairy roots, suggesting that these steps may be rate limiting in the formation of dimethylallyl diphosphate leading to anthraquinones.
Fig. 3.

Plausible biosynthetic pathway and expression of unigenes involved in the biosynthesis of anthraquinones in O. pumila. The expression levels of unigenes encoding the enzymes of each step are shown. Separated proteins of MenD, MenH and MenC in bacteria are known to be fused to a single protein in Arabidopsis (Gross et al. 2006). HR, hairy roots; CSC, cell suspension culture; MenF, menaquinone-specific isochorismate synthase; MenD, 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase; MenH, 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate synthase; MenC, o-succinylbenzoate synthase; MenE, o-succinylbenzoic acid-CoA ligase; MenB, naphthoate synthase; DXPS, 1-deoxy-d-xylulose-5-phosphate synthase; CLA, cloroplastos alterados; DXR, 1-deoxy-d-xylulose 5-phosphate reductoisomerase; ISPD, 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase; CDPMEK, 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase; ISPF, 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase; HDS, (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase; HDR, 4-hydroxy-3-methylbut-2-enyl diphosphate reductase. The expression levels of unigenes are represented with a black–blue color code and FPKM values.
Chlorogenic acid biosynthetic genes
In addition to camptothecin and anthraquinones, chlorogenic acid (3-O-caffeoyl quinic acid) (21) preferentially accumulates in O. pumila hairy roots (see below) (Yamazaki et al. 2003b). Chlorogenic acid is synthesized by a two-step reaction from p-coumaroyl-CoA; namely p-coumaroyl transfer to quinic acid by shikimate O-hydroxycinnamoyltransferase (HCT; EC 2.3.1.133) and subsequent hydroxylation of the 3′ position of coumaroylquinate by coumaroylquinate(coumaroylshikimate) 3′-monooxygenase (CYP98A3, C3′H; EC 1.14.13.36), or alternatively by hydroxylation of the 3′ postion of p- coumaroyl-CoA first and transfer to quinic acid (Koshiro et al. 2007) (http://www.kegg.jp/kegg-bin/show_pathway?map00941, http://pmn.plantcyc.org/PLANT/NEW-IMAGE?type = PATHWAY&object = PWY-6039) (Kanehisa et al. 2010, Chae et al. 2012). Unigenes with sequence similarity to Arabidopsis HCT and C3′H, were highly differentially expressed in hairy roots compared with cell suspension culture (Table 2). These unigenes presumably encode HCT and the C3′H involved in the formation of chlorogenic acid in O. pumila.
Table 2.
Expression abundances of unigenes potentially involved in the biosynthesis of chlorogenic acid
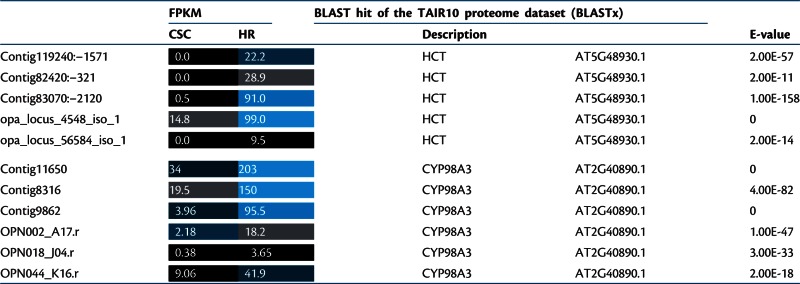 |
Expression was measured as fragments per kilobase pair of transcript per million fragments mapped (FPKM). Functional annotation was deduced from BLAST best homolog’s description of the TAIR10 proteome. The expression levels of unigenes are represented with a black–blue color code and FPKM values.
HR, hairy roots; CSC, cell suspension culture.
Collectively, these results indicate that differential expression analysis can assist in the delineation of genes involved in the biosynthesis of secondary products, including camptothecin, anthraquinones and chlorogenic acid in O. pumila hairy roots.
Untargeted metabolome analysis by in-fusion FT-ICR-MS and differential accumulation of secondary metabolites
We previously investigated the metabolic profiles of hairy roots and cell suspension culture using liquid chromatography–mass spectrometry (LC-MS) analysis (Yamazaki et al. 2003b). In the present study, we adopted an untargeted metabolomic analysis using in-fusion FT-ICR-MS (Table 3; Supplementary Table S3) (Hirai et al. 2005, Tohge et al. 2005). Annotation of detected peaks in the analyses was based on comparison of accurate m/z values observed with the theoretical values deduced from the elemental composition within a certain tolerance range. The determined elemental composition was used for putative identification of compounds by retrieving data within plant metabolite databases, KNApSAcK Core (Afendi et al. 2012) and A Dictionary of Natural Products (Hocking 1997). A total of 599 peaks were differentially accumulated, ranging from 0.022- to 303-fold (hairy root/cell suspension) (P-value <0.01, t-test) (Supplementary Table S3). Among them, 327 peaks were preferentially accumulated in hairy roots, and 272 peaks were preferentially accumulated in the cell suspension culture. Most of the proposed intermediates in the pathway of camptothecin biosynthesis accumulated in a hairy root-specific manner. In addition to camptothecin pathway intermediates, a number of anthraquinone-related compounds were highly differentially accumulated in hairy roots compared with the cell suspension culture. Chlorogenic acid and some other unidentified metabolites, which have been previously detected in hairy roots by LC-MS (Yamazaki et al. 2003b), were preferentially synthesized in hairy roots. However, no metabolites which preferentially accumulated in the cell suspension culture have been successfully annotated. Overall, the accumulation pattern of secondary products and their potential intermediates were mostly correlated with the expression of their biosynthetic genes in the transcriptome data.
Table 3.
The accumulation of annotated metabolites involved in secondary metabolite synthesis in O. pumila
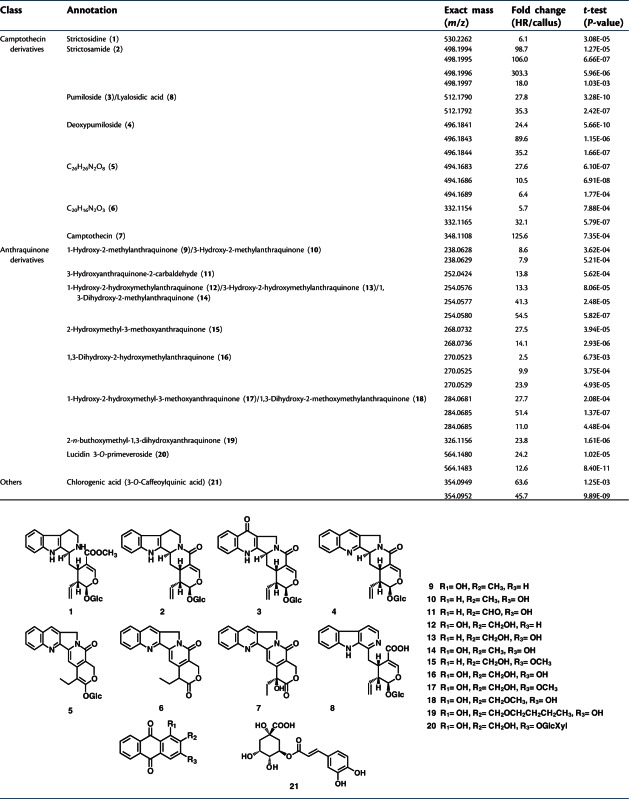 |
The annotation of metabolites was performed on the basis of observed m/z values from in-fusion FT-ICR-MS analyses in comparison with theoretical values.
HR, hairy roots.
Discussion
Deep transcriptome analysis for medicinal plants provides an opportunity to discover genes involved in the pathways that lead to the synthesis of plant specialized products. If biological resources, e.g. mutants, cell lines or developmental tissues, are available they can facilitate pathway discovery through the differential accumulation of a compound of interest. In particular, if one can combine transcriptome data with metabolic profiling data, one can generate correlations of genes and metabolites in given biological systems on a whole-genome and metabolome scale. Such an integrated ‘omics’ approach can greatly facilitate prediction of new and novel genes committed in the pathways of interest (Saito et al. 2008, Fukushima et al. 2009, Saito and Matsuda 2010). In this study, data sets for predicting the genes and metabolites involved in the camptothecin and anthraquinone biosynthetic pathways in O. pumila hairy roots were generated. These data sets will be useful for further discovery of the genes and metabolites committed in these secondary metabolic pathways as discussed in other articles of this special issue (see Muranaka and Saito; Ramilowski et al.). In particular, generation of a hypothesis for the candidate genes involved in the particular biosynthetic pathways can be achieved by a comparison of gene expression and metabolite accumulation, as demonstrated recently by a similar approach (Geu-Flores et al. 2012; Yonekura-Sakakibara et al. 2013; Higashi and Saito 2013).
Regarding camptothecin biosynthesis, the steps after strictosamide to camptothecin are not well characterized in terms of which chemical reactions, intermediary metabolites and enzymes are involved. Some candidates have been proposed by recent gene knockdown experiments (Asano et al. 2012). The present study on untargeted metabolic profiling by FT-ICR-MS supports the presence of possible intermediates (Fig. 1). The post-strictosamide events of camptothecin biosynthesis may include a number of steps such as oxidative ring expansion, reduction, dehydration, isomerization (double bond migration), deglycosylation and hydroxylation. In addition to these structural conversion reactions, the mechanisms of transport across organelles and even cells may be required. The unigenes which encode these proteins potentially involved in these steps could be preferentially expressed in the hairy roots.
Cyt P450s are presumably involved in some of the steps post-strictosamide, and 229 unigenes exhibited similarity to Cyt P450s (Supplementary Table S4). Of these, 130 unigenes were classified into 33 P450 subfamilies and were up-regulated in hairy roots compared with cell suspension culture. The CYP71B subfamily was the most abundant subfamily (21 unigenes), followed by the CYP6C subfamily (13 unigenes), the CYP716A subfamily (12 unigenes) and the CYP71A subfamily (11 unigenes). As P450s belonging to these subfamilies are known to catalyze certain reactions of plant secondary metabolism (Mizutani and Ohta 2010, Bak et al. 2011), these contigs are candidates to encode the Cyt P450s that catalyze the reactions involved in the post-strictosamide events of camptothecin biosynthesis. Similarly, unigenes encoding glycosidase-like proteins were examined. A total of 1,245 unigenes exhibited similarity with glycosidase-like proteins, and 93 unigenes were up-regulated in hairy roots compared with cell suspension culture. These unigenes belong to 21 subfamilies classified by the CAZy database (http://www.cazy.org/Glycoside-Hydrolases.html) (Supplementary Table S4). The GH1 family was the most abundant family (38 unigenes), followed by GH3 (15 unigenes). There is a gene (Contig8678) with sequence similarity to the reported strictosidine and raucaffricine acting as substrates for β-glucosidases from Rauvolfia serpentina (Xia et al. 2011). This gene might be involved in the deglycosylation reaction in the biosynthesis of camptothecin or degradation of strictosidine.
With respect to the biosynthesis of anthraquinones in O. pumila, little is known regarding the genes in this pathway, though the biosynthetic pathway has been well defined by tracer experiments in cell culture (Leistner 1985, Han et al. 2001). Our present study has the potential to narrow down the genes involved in this pathway by a combination of deep transcriptome and metabolome analysis. Functional characterization of these delineated genes, together with the detailed investigation of anthraquinone biosynthesis, will be undertaken in a future study.
Several national or international genomics and functional genomics projects focused on medicinal plants have emerged recently, including National Institutes of Health (http://medicinalplantgenomics.msu.edu/), Canada Genome (http://www.phytometasyn.com/) and 1KP Plant (http://www.onekp.com/project.html) initiatives. Accessing these data sets will be useful for further fine delineation of candidate genes, such as comparison of gene families in C. acuminata (Sun et al. 2011), Cathranthus roseus and R. serpentina, all of which produce monoterpenoid indole alkaloids.
Materials and Methods
Plant materials
Hairy roots of O. pumila were induced from aseptic plants as described previously (Saito et al. 2001). The callus and cell suspension cultures of O. pumila were induced from the hairy roots by addition of phytohormones as described elsewhere (Asano et al. 2012). The cell suspension cultures and the hairy roots were maintained in Gamborg B5 liquid medium (Gamborg et al. 1968) containing 2% sucrose with 0.5 μM naphthaleneacetic acid and 5 μM N6-benzyladenine for cell suspension cultures or without these phytohormones for hairy roots at 25°C on a rotary shaker (125 r.p.m. for cell suspension cultures or 80 r.p.m. for hairy roots) under dark conditions.
Construction of a full-length cDNA library and sequence data trimming
A full-length cDNA library was constructed from the poly(A)+ RNA obtained from the hairy roots of O. pumila (3 weeks old) as described previously (Carninci et al. 2001) and sequenced on ABI3730xl sequencers (Life Technologies) (Taji et al. 2008). Bases and quality values were determined using the Phred program (Ewing and Green 1998, Ewing et al. 1998), and low quality regions (Phred quality score <20, and >20 bases repeated) were discarded. Vector sequences were identified using the cross_match program (Ewing and Green 1998, Ewing et al. 1998) with the parameters ‘-minmatch 10 -minscore 20 parameters’. Sequences shorter than 100 bases were removed. ESTs are available in the DNA Data Bank of Japan (DDBJ) (Kodama et al. 2012) (accession Nos. HX703880 to HX739052).
Deep transcriptome analysis with the Illumina platform
Poly(A)+ RNA was isolated from 3-week-old hairy roots and cell suspension cultures of O. pumila. cDNA libraries were constructed using the Illumina TruSeq™ Sample Preparation Kits and sequenced using the paired-end method with an Illumina HiSeq 2000/cBOT platform. Each fragment was sequenced to a read length of 90 nucleotides from each end; 29,682,050 hairy root-derived and 24,617,708 cell suspension culture-derived reads were generated. The Illumina reads have been deposited in the DDBJ Sequence Read Archive as the accession No. DRA000930.
Assembly of deep transcriptome data and generation of a hybrid transcriptome unigene set
A schematic workflow of the computational analysis to assemble the O. pumila transcriptome data is shown in Supplementary Fig. S1. The FASTX tool kit was used to pre-process paired-end reads of 90 bp length. Based on the quality score distributions, reads were trimmed to 75 bp and then homopolymers were removed and only sequences with a quality score ≥20 and a minimum length of 70 bp were retained (Blankenberg et al. 2010); Version 0.0.13, http://hannonlab.cshl.edu/fastx_toolkit). A de novo transcriptome assembly of cleaned paired-end reads was performed using the Oases assembler with a k-mer length of 31 (Schulz et al. 2012). Oases-derived contigs and the ESTs from the full-length cDNAs derived from Sanger sequencing were assembled into a non-redundant transcript data set (unigene) using the CAP3 program (Huang and Madan 1999) with a parameter of sequence identity >90% (–p 90). Transcripts in the hybrid assembly will be available to download via our site, http://ngs-data-archive.psc.riken.jp/pub/ophiorrhiza_pumila/download/.
Differential expression analysis of RNA seq data
The Illumina reads were mapped to the unigene set using the Bowtie program with a default setting in the single end mode. Transcripts were quantified and differential expression assessed using Cufflinks v.0.9.0 (Trapnell et al. 2010). The cuffmerge command was used to calculate FPKM values of each gene in the tissues of cell suspension culture or hairy roots. The cuffdiff command was used to test and identify differentially expressed unigenes with an FDR-corrected P-value of <0.05. Additionally, unigenes specifically expressed in either tissue with significant expression level were annotated as differentially expressed genes.
Functional annotations
To predict the function of the unigenes, we performed a similarity search of the unique gene set as query data using the BLASTx program (E-value <10–5) (Altschul et al. 1997) against protein databases of the NCBI nr and the A. thaliana proteome (TAIR10) (Lamesch et al. 2012). The BLAST result with NCBI nr (Benson et al. 2012) proteins was applied to identify putative Cyt P450-encoding genes in O. pumila based on sequence similarity. The BLAST result with A. thaliana proteins was applied to identify enriched GO terms using the GOrilla web server (Eden et al. 2009) as well as to identify putative glycosidase-encoding genes by reference to A. thaliana glycosidase genes classified in the CAZy database (Cantarel et al. 2009).
Untargeted metabolomics analysis
Untargeted metabolomic analyses were carried out by in-fusion FT-ICR-MS method. Briefly, high, middle and non-polar extracts of plant tissues (hairy roots and cell suspension cultures; 3 weeks old) were subjected to triplicate analyses with FT-ICR-MS (APEX III, Bruker Daltonics) as described previously (Aharoni et al. 2002, Hirai et al. 2004, Tohge et al. 2005). Data were processed using DISCOVArray (Phenomenome Discoveries). Metabolite peaks were annotated based on the elemental composition calculated from accurate m/z values followed by a metabolite search with databases (Hocking 1997, Afendi et al. 2012). The fold changes of the peaks were calculated based on the average signal intensities obtained from hairy roots and cell suspension cultures, respectively.
Supplementary data
Supplementary data are available at PCP online.
Funding
The Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan [Gants-in-Aid for Scientific Research (22108008, 24570041)]; the Japan Science and Technology Agency (JST) [CREST]; the National Institute of General Medical Sciences, USA [a grant (1RC2GM092521) to C.R.B. for work on medicinal plant transcriptomes].
Supplementary Material
Acknowledgments
We thank all of the technical staff of the Sequence Technology Team at RIKEN GSC for their assistance.
Glossary
Abbreviations
- C3′H, coumaroylquinate (coumaroylshikimate) 3′-monooxygenase
EST
- expressed sequence tag
FDR
- false discovery rate
FPKM
- fragments per kilobase of transcript per million fragments mapped
FT-ICR-MS
- Fourier transform-ion cyclotron resonance-mass spectrometry
GO
- gene ontology
HCT
- shikimate O-hydroxycinnamoyltransferase
LC-MS
- liquid chromatography–mass spectrometry
MEP
- 2-C-methyl-D-erythritol 4-phosphate
References
- Afendi FM, Okada T, Yamazaki M, Hirai-Morita A, Nakamura Y, Nakamura K, et al. KNApSAcK family databases: integrated metabolite–plant species databases for multifaceted plant research. Plant Cell Physiol. 2012;53:e1. doi: 10.1093/pcp/pcr165. [DOI] [PubMed] [Google Scholar]
- Aharoni A, Ric de Vos CH, Verhoeven HA, Maliepaard CA, Kruppa G, Bino R, et al. Nontargeted metabolome analysis by use of Fourier transform ion cyclotron mass spectrometry. OMICS. 2002;6:217–234. doi: 10.1089/15362310260256882. [DOI] [PubMed] [Google Scholar]
- Aimi N, Hoshino H, Nishimura M, Sakai S-i, Haginiwa J. Chaboside, first natural glycocamptothecin found from Ophiorrhiza pumila. Tetrahedron Lett. 1990;31:5169–5172. [Google Scholar]
- Aimi N, Nishimura M, Miwa A, Hoshino H, Sakai S-i, Haginiwa J. Pumiloside and deoxypumiloside; plausible intermediates of camptothecin biosynthesis. Tetrahedron Lett. 1989;30:4991–4994. [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbain D, Putra D, Sargent M. The alkaloids of Ophiorrhiza filistipula. Aust. J. Chem. 1993;46:977–985. [Google Scholar]
- Asano T, Kobayashi K, Kashihara E, Sudo H, Sasaki R, Iijima Y, et al. Suppression of camptothecin biosynthetic genes results in metabolic modification of secondary products in hairy roots of Ophiorrhiza pumila. Phytochemistry. 2012 doi: 10.1016/j.phytochem.2012.04.019. (in press) doi.org/10.1016/j.phytochem.2012.04.019. [DOI] [PubMed] [Google Scholar]
- Bak S, Beisson F, Bishop G, Hamberger B, Höfer R, Paquette S, et al. Cytochromes P450. The Arabidopsis Book. 2011:e0144. doi: 10.1199/tab.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2012;40:D48–D53. doi: 10.1093/nar/gkr1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A. Manipulation of FASTQ data with Galaxy. Bioinformatics. 2010;26:1783–1785. doi: 10.1093/bioinformatics/btq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Shibata Y, Hayatsu N, Itoh M, Shiraki T, Hirozane T, et al. Balanced-size and long-size cloning of full-length, cap-trapped cDNAs into vectors of the novel lambda-FLC family allows enhanced gene discovery rate and functional analysis. Genomics. 2001;77:79–90. doi: 10.1006/geno.2001.6601. [DOI] [PubMed] [Google Scholar]
- Chae L, Lee I, Shin J, Rhee SY. Towards understanding how molecular networks evolve in plants. Curr. Opin. Plant Biol. 2012;15:177–184. doi: 10.1016/j.pbi.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Chan H-H, Li C-Y, Damu AG, Wu T-S. Anthraquinones from Ophiorrhiza hayatana OHWI. Chem. Pharm. Bulll. 2005;53:1232–1235. doi: 10.1248/cpb.53.1232. [DOI] [PubMed] [Google Scholar]
- Darwin SP. The Pacific species of Ophiorrhiza L. (Rubiaceae) Lyonia. 1976;1:47–102. [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Fukushima A, Kusano M, Redestig H, Arita M, Saito K. Integrated omics approaches in plant systems biology. Curr. Opin. Chem. Biol. 2009;13:532–538. doi: 10.1016/j.cbpa.2009.09.022. [DOI] [PubMed] [Google Scholar]
- Gamborg OL, Miller RA, Ojima K. Nutrient requirements of suspension cultures of soybean root cells. Exp. Cell Res. 1968;50:151–158. doi: 10.1016/0014-4827(68)90403-5. [DOI] [PubMed] [Google Scholar]
- Geu-Flores F, Sherden NH, Courdavault V, Burlat V, Glenn WS, Wu C, et al. An alternative route to cyclic terpenes by reductive cyclization in iridoid biosynthesis. Nature. 2012;492:138–142. doi: 10.1038/nature11692. [DOI] [PubMed] [Google Scholar]
- Gross J, Cho WK, Lezhneva L, Falk J, Krupinska K, Shinozaki K, et al. A plant locus essential for phylloquinone (vitamin K1) biosynthesis originated from a fusion of four eubacterial genes. J. Biol. Chem. 2006;281:17189–17196. doi: 10.1074/jbc.M601754200. [DOI] [PubMed] [Google Scholar]
- Han Y-S, Heijden Rvd, Lefeber AWM, Erkelens C, Verpoorte R. Biosynthesis of anthraquinones in cell cultures of Cinchona ‘Robusta’ proceeds via the methylerythritol 4-phosphate pathway. Phytochemistry. 2002;59:45–55. doi: 10.1016/s0031-9422(01)00296-5. [DOI] [PubMed] [Google Scholar]
- Han Y-S, Van der Heijden R, Verpoorte R. Biosynthesis of anthraquinones in cell cultures of the Rubiaceae. Plant Cell Tissue Org. Cult. 2001;67:201–220. [Google Scholar]
- Higashi Y, Saito K. Network analysis for gene discovery in plant-specialized metabolism. Plant, Cell Environ. 2013 doi: 10.1111/pce.12069. in press, doi: 10.1111/pce.12069. [DOI] [PubMed] [Google Scholar]
- Hirai MY, Klein M, Fujikawa Y, Yano M, Goodenowe DB, Yamazaki Y, et al. Elucidation of gene-to-gene and metabolite-to-gene networks in arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 2005;280:25590–25595. doi: 10.1074/jbc.M502332200. [DOI] [PubMed] [Google Scholar]
- Hirai MY, Sugiyama K, Sawada Y, Tohge T, Obayashi T, Suzuki A, et al. Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl Acad. Sci. USA. 2007;104:6478–6483. doi: 10.1073/pnas.0611629104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai MY, Yano M, Goodenowe DB, Kanaya S, Kimura T, Awazuhara M, et al. Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA. 2004;101:10205–10210. doi: 10.1073/pnas.0403218101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking GM. A Dictionary of Natural Products: Terms in the Field of Pharmacognosy Relating to Natural Medicinal and Pharmaceutical Materials and the Plants, Animals, and Minerals from Which They are Derived. Plexus Publishing: Medford, NJ; 1997. [Google Scholar]
- Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson CR, Heckendorf AH, Straughn JL, Daddona PE, Cane DE. Biosynthesis of camptothecin. 3. Definition of strictosamide as the penultimate biosynthetic precursor assisted by carbon-13 and deuterium NMR spectroscopy. J. Amer. Chem. Soc. 1979;101:3358–3369. [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima M, Fischer U, Nakamura M, Ohsawa M, Ueno M, Takayama H, et al. Anthraquinones from Ophiorrhiza pumila tissue and cell cultures. Phytochemistry. 1998;48:107–111. [Google Scholar]
- Kitajima M, Fujii N, Yoshino F, Sudo H, Saito K, Aimi N, et al. Camptothecins and two new monoterpene glucosides from Ophiorrhiza liukiuensis. Chem. Pharm. Bull. 2005;53:1355–1358. doi: 10.1248/cpb.53.1355. [DOI] [PubMed] [Google Scholar]
- Kodama Y, Mashima J, Kaminuma E, Gojobori T, Ogasawara O, Takagi T, et al. The DNA Data Bank of Japan launches a new resource, the DDBJ Omics Archive of functional genomics experiments. Nucleic Acids Res. 2012;40:D38–D42. doi: 10.1093/nar/gkr994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiro Y, Jackson MC, Katahira R, Wang ML, Nagai C, Ashihara H. Biosynthesis of chlorogenic acids in growing and ripening fruits of Coffea arabica and Coffea canephora plants. Z. Naturforsch. C. 2007;62:731–742. doi: 10.1515/znc-2007-9-1017. [DOI] [PubMed] [Google Scholar]
- Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;40:D1202–D1210. doi: 10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leistner E. Biosynthesis of chorismate-derived quinones in plant cell cultures. In: Neumann KH, Barz W, Reinhardt E, editors. Primary and Secondary Metabolism of Plant Cell Cultures. Berlin: Springer; 1985. pp. 215–224. [Google Scholar]
- Matsuda F, Hirai MY, Sasaki E, Akiyama K, Yonekura-Sakakibara K, Provart NJ, et al. AtMetExpress development: a phytochemical atlas of Arabidopsis development. Plant Physiol. 2010;152:566–578. doi: 10.1104/pp.109.148031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani M, Ohta D. Diversification of P450 genes during land plant evolution. Annu. Rev. Plant Biol. 2010;61:291–315. doi: 10.1146/annurev-arplant-042809-112305. [DOI] [PubMed] [Google Scholar]
- Okazaki Y, Shimojima M, Sawada Y, Toyooka K, Narisawa T, Mochida K, et al. A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell. 2009;21:892–909. doi: 10.1105/tpc.108.063925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Hirai MY, Yonekura-Sakakibara K. Decoding genes with coexpression networks and metabolomics - Majority report by precogs. Trends Plant Sci. 2008;13:36–43. doi: 10.1016/j.tplants.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Saito K, Matsuda F. Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 2010;61:463–489. doi: 10.1146/annurev.arplant.043008.092035. [DOI] [PubMed] [Google Scholar]
- Saito KS, Sudo HS, Yamazaki MY, Koseki-Nakamura MK-N, Kitajima MK, Takayama HT, et al. Feasible production of camptothecin by hairy root culture of Ophiorrhiza pumila. Plant Cell Rep. 2001;20:267–271. [Google Scholar]
- Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirikantaramas S, Asano T, Sudo H, Yamazaki M, Saito K. Camptothecin: therapeutic potential and biotechnology. Curr. Pharm. Biotechnol. 2007;8:196–202. doi: 10.2174/138920107781387447. [DOI] [PubMed] [Google Scholar]
- Sirikantaramas S, Yamazaki M, Saito K. Mutations in topoisomerase I as a self-resistance mechanism coevolved with the production of the anticancer alkaloid camptothecin in plants. Proc. Natl Acad. Sci. USA. 2008;105:6782–6786. doi: 10.1073/pnas.0801038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Luo H, Li Y, Sun C, Song J, Niu Y, et al. Pyrosequencing of the Camptotheca acuminata transcriptome reveals putative genes involved in camptothecin biosynthesis and transport. BMC Genomics. 2011;12:533. doi: 10.1186/1471-2164-12-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taji T, Sakurai T, Mochida K, Ishiwata A, Kurotani A, Totoki Y, et al. Large-scale collection and annotation of full-length enriched cDNAs from a model halophyte, Thellungiella halophila. BMC Plant Biol. 2008;8:115. doi: 10.1186/1471-2229-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohge T, Nishiyama Y, Hirai MY, Yano M, Nakajima J, Awazuhara M, et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005;42:218–235. doi: 10.1111/j.1365-313X.2005.02371.x. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viraporn V, Yamazaki M, Saito K, Denduangboripant J, Chayamarit K, Chuanasa T, et al. Correlation of camptothecin-producing ability and phylogenetic relationship in the genus Ophiorrhiza. Planta Med. 2011;77:759–764. doi: 10.1055/s-0030-1250568. [DOI] [PubMed] [Google Scholar]
- Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J. Amer. Chem. Soc. 1966;88:3888–3890. [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Ruppert M, Wang M, Panjikar S, Lin H, Rajendran C, et al. Structures of alkaloid biosynthetic glucosidases decode substrate specificity. ACS Chem. Biol. 2011;7:226–234. doi: 10.1021/cb200267w. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Kitajima M, Arita M, Takayama H, Sudo H, Yamazaki M, et al. Biosynthesis of camptothecin. In silico and in vivo tracer study from [1-13C]glucose. Plant Physiol. 2004;134:161–170. doi: 10.1104/pp.103.029389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Sudo H, Yamazaki M, Aimi N, Saito K. Camptothecin biosynthetic genes in hairy roots of Ophiorrhiza pumila: cloning, characterization and differential expression in tissues and by stress compounds. Plant Cell Physiol. 2003a;44:395–403. doi: 10.1093/pcp/pcg051. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Urano A, Sudo H, Kitajima M, Takayama H, Yamazaki M, et al. Metabolite profiling of alkaloids and strictosidine synthase activity in camptothecin producing plants. Phytochemistry. 2003b;62:461–470. doi: 10.1016/s0031-9422(02)00543-5. [DOI] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Fukushima A, Nakabayashi R, Hanada K, Matsuda F, Sugawara S, et al. Two glycosyltransferases involved in anthocyanin modification delineated by transcriptome independent component analysis in Arabidopsis thaliana. Plant J. 2012;69:154–167. doi: 10.1111/j.1365-313X.2011.04779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Fukushima A, Saito K. Transcriptome data modeling for targeted plant metabolic engineering. Curr. Opin. Biotech. 2013;24:285–290. doi: 10.1016/j.copbio.2012.10.018. [DOI] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Saito K. Functional genomics for plant natural product biosynthesis. Nat. Prod. Rep. 2009;26:1466–1487. doi: 10.1039/b817077k. [DOI] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Tohge T, Matsuda F, Nakabayashi R, Takayama H, Niida R, et al. Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell. 2008;20:2160–2176. doi: 10.1105/tpc.108.058040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Tohge T, Niida R, Saito K. Identification of a flavonol 7-O-rhamnosyltransferase gene determining flavonoid pattern in Arabidopsis by transcriptome coexpression analysis and reverse genetics. J. Biol. Chem. 2007;282:14932–14941. doi: 10.1074/jbc.M611498200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.