

Abstract
Objective:
Pigmented orthochromatic leukodystrophy (POLD) and hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) are rare neurodegenerative disorders characterized by cerebral white matter abnormalities, myelin loss, and axonal swellings. The striking overlap of clinical and pathologic features of these disorders suggested a common pathogenesis; however, no genetic or mechanistic link between POLD and HDLS has been established. Recently, we reported that mutations in the colony-stimulating factor 1 receptor (CSF1R) gene cause HDLS. In this study, we determined whether CSF1R mutations are also a cause of POLD.
Methods:
We performed sequencing of CSF1R in 2 pathologically confirmed POLD families. For the largest family (FTD368), a detailed case report was provided and brain samples from 2 affected family members previously diagnosed with POLD were re-evaluated to determine whether they had HDLS features. In vitro functional characterization of wild-type and mutant CSF1R was also performed.
Results:
We identified CSF1R mutations in both POLD families: in family 5901, we found c.2297T>C (p.M766T), previously reported by us in HDLS family CA1, and in family FTD368, we identified c.2345G>A (p.R782H), recently reported in a biopsy-proven HDLS case. Immunohistochemical examination in family FTD368 showed the typical neuronal and glial findings of HDLS. Functional analyses of CSF1R mutant p.R782H (identified in this study) and p.M875T (previously observed in HDLS), showed a similar loss of CSF1R autophosphorylation of selected tyrosine residues in the kinase domain for both mutations when compared with wild-type CSF1R.
Conclusions:
We provide the first genetic and mechanistic evidence that POLD and HDLS are a single clinicopathologic entity.
Familial pigmented orthochromatic leukodystrophy (POLD) and hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) are white matter disorders. Thus far considered distinct diseases, clinical and pathologic features suggest that POLD and HDLS are of the same disease spectrum. The pigmented macrophages identified in HDLS families are similar to those in POLD,1–5 whereas axonal dilations found in POLD families coincide with axonal spheroids described in HDLS.3,5–7 Clinically, most patients with familial POLD and HDLS present with psychiatric symptoms, which progress into dementia, often of a frontotemporal phenotype.3,5,8–11 MRIs are consistent with this clinical presentation, because both POLD and HDLS patients portray frontal-predominant atrophy.12
Although clinical and pathologic data are strikingly similar between POLD and HDLS, a common genetic cause would further solidify these diseases as a single entity.
Recently, we reported mutations in the colony-stimulating factor 1 receptor (CSF1R) gene as the first known cause of HDLS.13 We identified a unique pathogenic mutation in the CSF1R kinase domain in each autopsy- or biopsy-confirmed HDLS family included in the study, and in 1 clinically diagnosed patient with corticobasal syndrome. The antemortem diagnoses among patients in these HDLS families varied significantly, including frontotemporal dementia (FTD), corticobasal syndrome, Alzheimer disease, multiple sclerosis, atypical cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and Parkinson disease,13 suggesting that HDLS may masquerade as other neurologic conditions leading to underdiagnosis. The objective of this study was to determine whether CSF1R mutations also cause familial POLD.
METHODS
Standard protocol approvals, registrations, and patient consents.
We studied 2 neuropathologically confirmed POLD families (families FTD368 and 5901) and 1,089 neurologically normal, control individuals ascertained at Mayo Clinic Jacksonville. This study was approved by the Mayo Clinic Institutional Review Board and all subjects and/or their proxies gave informed consent to take part in this study.
Neuropathology.
Paraffin-embedded fixed tissue sections (5–7 μm thick, mounted on glass slides) were available from cerebral cortex and basal ganglia on patients FTD368 II-1 and II-2. Tissue sections were stained with hematoxylin & eosin (H&E), and with Luxol fast blue/periodic Schiff. Unstained sections were processed for autofluorescence. Sections were also immunostained with a DAKO Autostainer (DAKO, Carpinteria, CA) using Envision reagents and 3,3′-diaminobenzidine as the chromogen, with the following antibodies: amyloid precursor protein (mouse monoclonal [22C11], 1:10,000; Chemicon, Temecula, CA); phosphorylated neurofilament (mouse monoclonal [SMI-31], 1:40,000; Sternberger Monoclonals, Lutherville, MD); ubiquitin (mouse monoclonal [MAB1510], 1:60,000; Chemicon, Burlingame, CA); αB-crystallin (rabbit polyclonal, 1:10,000; gift from Dr. Jack Liang, Harvard University); human leukocyte antigen-DR for microglia (mouse monoclonal [LN-3], 1:100; eBioscience, San Diego, CA); and glial fibrillary acidic protein (rabbit polyclonal, 1:5,000; Biogenex, Fremont, CA).
DNA extraction, sequencing, and haplotype analyses.
DNA was extracted from blood (FTD368 III-1; 5901 III-2), frontal cortex (FTD368 II-2), and formalin-fixed, paraffin-embedded tissue (FTD368 II-1) using Gentra Puregene kit and QIAamp FFPE methods (Qiagen, Valencia, CA). In the probands (FTD368 III-1 and 5901 III-2), CSF1R exons 12–22 were PCR-amplified using M13-tailed primers,13 and segregation analyses of the c.2345G>A (p.R782H) mutation in patients FTD368 II-1 and II-2 were performed using custom primers (5′-CACGACGTTGTAAAACGACTGTTCACTCCAGCAGGGAC-3′ and 5′-GGATAACAATTTCACACAGGCCAATCTTGGCCACATGAC-3′). PCR products were purified using the AMPure system (Beckman Coulter, Brea, CA) and sequenced in both directions using M13 primers and Big Dye Cycle Sequencing chemistry (Applied Biosystems, Foster City, CA). Sequence reactions were purified with CleanSEQ (Beckman), sequenced on an ABI3730xl Genetic Analyzer, and analyzed using Sequencher (Gene Codes, Ann Arbor, MI). A custom TaqMan single nucleotide polymorphism genotyping assay was designed for c.2345G>A (p.R782H), and controls were genotyped on the 7900HT Fast Real Time PCR system. Genotype calls were made using SDS version 2.2 software (Applied Biosystems). For haplotype sharing studies, 10 STR markers flanking CSF1R were PCR-amplified with 1 fluorescently labeled primer, analyzed on an ABI3730xl analyzer, and scored using GENOTYPER software.
CSF1R cDNA clone mutagenesis.
A cDNA construct encoding the human CSF1R gene was purchased from OriGene (Rockville, MD; RC205288). Mutations p.M875T and p.R782H were introduced independently using the QuickChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA) and mutagenesis primers (M875T: 5′-GAAGGATGGATACCAAACGGCCCAGCCTGCATTTG-3′ and 5′-CAAATGCAGGCTGGGCCGTTTGGTATCCATCCTTC-3′; R782H: 5′-GACGTGGCAGCGCATAACGTGCTGTTG-3′ and 5′-CAACAGCACGTTATGCGCTGCCACGTC-3′). Mutation-positive clones were identified by sequencing and transformed into DH5α Escherichia coli cells, after which DNA was obtained using the Nucleobond Xtra Maxi Plus EF kit (Clontech, Mountain View, CA).
Cell culture and immunoblotting.
HeLa cells were maintained in Eagle minimum essential medium with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C, 5% CO2. Cells were transfected with 2 μg of tGFP control plasmid DNA (OriGene) or CSF1R plasmid DNA (wild-type or mutant) using Lipofectamine 2000 (Life Technologies, Grand Island, NY). Two days posttransfection, the media was changed to serum-free media for 16 hours before treatment with 50 ng/mL human recombinant CSF-1 protein (R&D Systems, Minneapolis, MN). CSF-1 treatment continued for 5, 15, or 30 minutes before harvesting. HeLa cells without CSF-1 treatment were collected as controls. Sample protein was denatured in 2× Novex Tris-glycine sodium dodecyl sulfate sample buffer (Life Technologies), and equal volumes were run on 10% sodium dodecyl sulfate–polyacrylamide gels (Life Technologies), transferred to Immobilon membranes (Millipore, Billerica, MA), and immunoblotted. Total CSF1R levels were detected using the rabbit polyclonal c-Fms/CSF-1R (C-20) antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA). CSF1R autophosphorylation was detected using rabbit polyclonal CSF1R phospho-tyrosine primary antibodies against p-Y549 (1:5,000), p-Y699 (1:2,000), p-Y708 (1:5,000), p-Y723 (1:6,000), and p-Y809 (1:500) purchased from Cell Signaling Technology (Danvers, MA). A GAPDH primary antibody (1:500,000) (Meridian Life Science, Memphis, TN) was used to ensure equal protein loading. Blots were then incubated with anti-mouse or anti-rabbit secondary horseradish peroxidase–conjugated antibodies (Promega, Madison, WI) and bands were detected by enhanced chemiluminescence using Western Lightning Plus-ECL reagents (Perkin Elmer, Waltham, MA).
RESULTS
FTD368 III-1, clinical case report.
The proband of family FTD368 (FTD368 III-1; figure 1) was a white, non-Hispanic female who at the age of 51 years began to have difficulties at work. She missed a meeting, showed up for another meeting several days early, and failed a work-related examination. She experienced forgetfulness and repeated stories, but remembered conversations. She began to struggle with arithmetic. Her husband noted a large discrepancy in her checking account, potentially from impulsive buying. She became more irritable toward her husband and retired after the forgetfulness at work became apparent. After retiring, she initially functioned well on her own. She had no other medical, neurologic, or psychiatric history. On the Short Test of Mental Status (STMS), she lost 2 points on recall, 2 points on drawing, and scored 34 of 38. The remainder of her neurologic examination, including balance, coordination, alternating motion, muscle tone, and gait, was normal. An MRI scan showed prominent frontal brain atrophy, and extensive white matter hyperintensity changes throughout the anterior portion of the frontal lobes, as well as surrounding the ventricular system in the temporal, parietal, and occipital lobes.
Figure 1. Families with POLD and CSF1R mutations.

Shown are abbreviated pedigrees of 2 families, family FTD368 (A) and family 5901 (B) affected by pigmented orthochromatic leukodystrophy (POLD) included in this study. Gray-filled symbols represent clinically affected individuals, whereas black-filled symbols represent individuals with pathologically confirmed POLD. The proband of each family is indicated with an arrowhead. Cases for which DNA was available for genetic studies are noted with an asterisk. DNA sequence traces observed in a sample from the proband from each family are shown below each pedigree. The single base substitution in each trace is indicated with an arrow.
One year later, she was having greater difficulty multitasking. Her memory had worsened, she repeated herself frequently, and had some evidence of disinhibition and a tendency to say socially inappropriate things. She was still doing some cooking and driving locally, but had become slow to react to changes in situations and events. Her STMS score decreased 8 points to 26 of 38, but her motor neurologic examination was normal.
Two years after first being seen, she had worsened substantially. She spent her days sitting, had ceased all household chores, and needed assistance with dressing and bathing. She tended to put layers of clothes on, but often put them on backward. Her weight was stable. Her STMS score had decreased to 17 of 38. Her walking was slower, but balance was satisfactory. The MRI findings were not dramatically different compared with 2 years prior. At the time of the initial visit, the clinical diagnosis was behavioral variant FTD (bvFTD). PET using 18-fluorodeoxyglucose tracer showed hypometabolism when first seen in 2006 (figure 2A). The CT brain scan at that time showed enlargement of the frontal horns of the lateral ventricles. A year later, the MRI scan showed frontal lobe atrophy, frontal lobe white matter hyperintensities, and similar changes in the parietooccipital white matter, with enlargement of the frontal horns of the lateral ventricles, and milder diffuse cerebral atrophy (figure 2B). An MRI scan performed 2 years later showed progression of white matter hyperintensities and slight increase in atrophy.
Figure 2. Neuroimaging and neuropathologic findings in pigmented orthochromatic leukodystrophy.
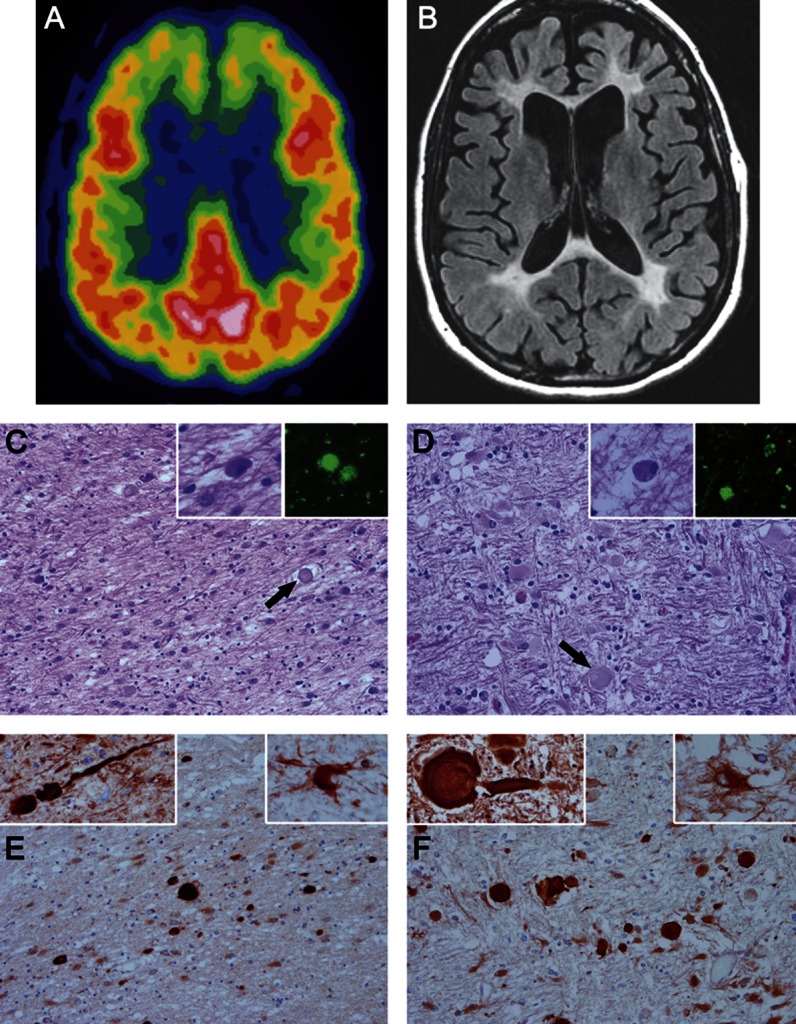
(A) 18-Fluorodeoxyglucose PET scan performed in 2006 from FTD368 II-1 shows hypometabolism in both frontal lobes. (B) MRI scan performed in 2007 shows atrophy of the frontal lobes, white matter hyperintensities in the white matter of the frontal lobes and similar changes in the parietooccipital white matter, enlargement of the frontal horns of the lateral ventricles, and milder diffuse cerebral atrophy. (C–F) Microscopic images of white matter from FTD368 II-2 (C and E) compared with typical hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) (D and F). On hematoxylin & eosin stains, the white matter has pallor because of myelinated fiber loss and gliosis, with axonal spheroids (arrows in C and D). Within the affected white matter are macrophages with basophilic to brown fine granules (left insets in C and D) that have autofluorescent pigment (right insets in C and D) when unstained sections are viewed with a fluorescent microscope. Immunohistochemistry for amyloid precursor protein (panels E and F) shows many axonal spheroids in affected white matter, which are confirmed to be axonal in origin by immunoreactivity with phosphorylated neurofilament (left insets in E and F). A characteristic feature of HDLS is the presence of bizarre hypertrophic astrocytes demonstrated with immunohistochemistry for αB-crystallin (right insets in E and F). C–F: original magnification ×200; all insets: original magnification ×400.
The proband's mother and 2 maternal aunts (FTD368 II-1, II-2, and II-3; figure 1) showed similar symptoms and were clinically diagnosed with dementia between ages 40 and 70. Her grandparents did not have dementia at the time of death. The grandfather died in his 40s of cancer. The grandmother died at age 65 years of a ruptured aortic aneurysm. Neuropathologic examination of the proband's aunt (FTD368 II-1) and mother (FTD368 II-2) led to the rare diagnosis of POLD in both patients.14
Neuropathology re-evaluation.
Archival tissue from FTD368 II-1 and II-2, reported previously,14 was re-evaluated with histologic and immunohistochemical methods as in previous reports.2,5,8,15 The findings in II-1 and II-2 were essentially the same. For comparison, FTD368 II-2 (figure 2, C and E) was compared with an HDLS case with a documented CSF1R mutation (figure 2, D and F). Cerebral white matter in both POLD and HDLS had staining pallor on H&E because of myelinated fiber loss. Axonal spheroid number varied (arrows in figure 2, C and D) but was most numerous in areas with early white matter pathology and less abundant in burnt-out white matter areas that had few axons and merely dense astrocytic gliosis. Immunohistochemistry for amyloid precursor protein most readily detected axonal spheroids (figure 2, E and F), as well as phosphorylated neurofilament (insets, figure 2, E and F). There was variable ubiquitin immunoreactivity in the spheroids (not shown). In both conditions, there were sparse to moderate pigment-laden macrophages visible on H&E (insets, figure 2, C and D) and the pigment was autofluorescent, as originally reported14 (insets, figure 2, C and D). The small heat shock protein, αB-crystallin, showed bizarre hypertrophic astrocytes in affected white matter (insets, figure 2, E and F), and scattered ballooned neurons were observed in the overlying cortex, as noted previously in HDLS.2,8
Genetic screening.
CSF1R exons 12–22 sequencing analyses in FTD368 III-1 showed a mutation in exon 18, c.2345G>A, introducing a p.R782H missense mutation in the CSF1R protein. By direct sequencing, the mutation was confirmed in the proband's mother (FTD368 II-2) and maternal aunt (FTD368 II-1) using primers designed to generate a short amplicon. The mutation was excluded in 1,089 white, non-Hispanic control subjects using a TaqMan single nucleotide polymorphism genotyping assay.
p.R782H CSF1R autophosphorylation is abrogated.
In the brain, CSF1R is a tyrosine kinase receptor highly expressed in microglia and, to a lesser extent, in neurons. Upon binding to the CSF-1 ligand, CSF1R forms homodimers before autophosphorylation of its kinase domain. This autophosphorylation precedes CSF1R-dependent phosphorylation of downstream targets, rendering this process critical for CSF-1 signaling. In our previous study, we performed functional analyses of HDLS-causing CSF1R mutations p.M875T, p.M766T, and p.E633K and found that CSF1R autophosphorylation is abrogated in mutant CSF1R-transfected cells.13 To determine whether the p.R782H mutation identified in POLD family FTD368 in this study similarly affects CSF-1–induced CSF1R autophosphorylation, we transfected wild-type CSF1R (CSF1RWT), a kinase-deficient CSF1R (CSF1RM875T),13 or CSF1RR782H in HeLa cells. Immunoblotting revealed that wild-type and mutant CSF1R overexpression levels were similar. Upon stimulation with CSF-1, autophosphorylation on multiple CSF1R tyrosine residues was observed for CSF1RWT after 5 minutes of CSF-1 exposure that gradually decreased over 30 minutes. Conversely, neither the CSF1RM875T nor the CSF1RR782H mutants showed detectable levels of autophosphorylation at any time post–CSF-1 treatment (figure 3).
Figure 3. Functional effects of CSFR1 mutations in cell culture.
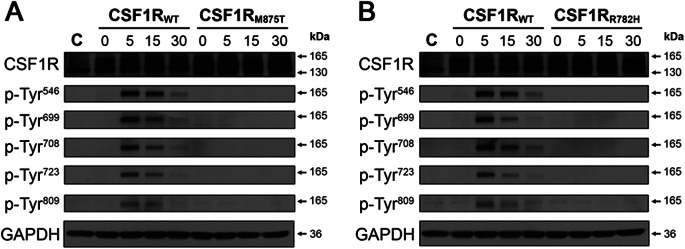
CSF-1 treatment leads to autophosphorylation of wild-type but not mutant CSF1R. HeLa cells were transfected with either a tGFP control plasmid, or a plasmid containing wild-type (CSF1RWT), the previously characterized mutant (CSF1RM875T) (A), or uncharacterized mutant (CSF1RR782H) (B) CSF1R. Cells were treated with CSF-1 for 5, 15, or 30 minutes. Each blot was probed with antibodies against total CSF1R and with antibodies that detect CSF1R autophosphorylation at several tyrosine residues (p-Tyr). Note that CSF-1 treatment only causes CSF1R autophosphorylation in cells expressing CSF1RWT. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as a loading control.
POLD family 5901.
In a recently identified unpublished family with a history of pathologically confirmed POLD, family 5901, we also analyzed the clinically affected proband's DNA (5901 III-2) for mutations in CSF1R exons 12–22. Sequencing analyses revealed the previously published and functionally characterized c.2297T>C mutation in exon 17, predicted to cause the amino acid change p.M766T.13 This mutation was previously excluded in 1,449 white, non-Hispanic controls. Genetic marker analyses did not suggest a common founder for POLD family 5901 and HDLS family CA1 (figure e-1 on the Neurology® Web site at www.neurology.org).
DISCUSSION
To date, there have been several reports with compelling evidence that the rare adult-onset neurodegenerative white matter diseases, POLD and HDLS, are 1 clinicopathologic entity. Despite the striking overlap in clinical and neuropathologic features of these diseases, a genetic or mechanistic link has not been established. One seemingly discrepant feature between these disorders had been that the HDLS inheritance pattern was usually autosomal dominant, whereas some individual POLD cases arose in families in which no parent displayed neurologic dysfunction, suggesting autosomal recessive inheritance or incomplete penetrance,7,16 and questioning the involvement of a single disease gene in both disorders.
Using a combination of genome-wide linkage analyses and exome sequencing, we recently identified mutations affecting the kinase domain of CSF1R as the genetic cause in 14 autopsy- or biopsy-proven HDLS families.13 A different CSF1R mutation was identified in each HDLS family, and the de novo occurrence of at least 1 mutation was confirmed. In the present study, we obtained DNA from one of the largest reported POLD families (FTD368) and 1 smaller unpublished pathologically confirmed POLD family (5901) to test the hypothesis that CSF1R mutations could also cause POLD. Similar to the HDLS families, sequencing analyses identified mutations in the CSF1R kinase domain in both POLD families. In family 5901, we identified the known p.M766T mutation, previously found in HDLS family CA1, whereas the mutation identified in family FTD368, p.R782H, had not been identified in our previous study. While this work was underway, the p.R782H mutation was reported by an independent group as a causative mutation in a biopsy-proven HDLS family,17 further strengthening the evidence of homogeneity between these diseases.
CSF1R is a tyrosine kinase receptor expressed on the surface of microglia, and to a lesser extent in neurons.18,19 This protein consists of 3 domains critical to its function: an extracellular ligand-binding domain, a single-pass transmembrane domain, and an intracellular kinase domain for downstream signaling.20 In a nondiseased state, CSF1R stimulation by its CSF-1 ligand induces receptor homodimerization, after which CSF1R autophosphorylates various kinase domain tyrosine residues to allow subsequent phosphorylation of its cellular targets. The CSF1R mutations recently found in HDLS families are all located within the CSF1R kinase domain and were shown to abrogate autophosphorylation on several critical tyrosine residues.13 Importantly, both CSF1R mutations identified in POLD families in this study were also located within the kinase domain. Functional analyses of these mutations (p.R782H as part of this study and p.M766T as part of our previous work) showed similar deficits in autophosphorylation in cultured cells, suggesting that familial POLD and HDLS share a common disease mechanism.
Patients with POLD and HDLS often present with varying neuropsychiatric symptoms, including depression, behavioral changes, and dementia leading to variable clinical diagnoses including FTD, Alzheimer disease, Parkinson disease, and multiple sclerosis.7,8,13–17,21–25 The clinical presentation in POLD family FTD368 included symptoms of depression, abnormal behavior, and an inability to function, progressing to dementia in the 3 siblings from the second generation. The proband (FTD368 II-1), first described in this report, was seen in the Mayo Clinic Alzheimer's Disease Research Center and presented with behavioral changes apparent at work and memory deficits that worsened over time, leading to a clinical diagnosis of bvFTD. Both the frontal and temporal lobes were affected on MRI scans, although extensive white matter hyperintensity changes were also observed, not typical of bvFTD.22,26,27 Family FTD368 illustrates the difficulties associated with clinically diagnosing POLD and HDLS, and explains why in the absence of pathologic examination, misdiagnosis is common. Based on our current findings, we argue that genetic screening for CSF1R mutations should, therefore, be considered in patients presenting with a range of neurodegenerative syndromes, including bvFTD, to provide a more accurate diagnosis.
In both kindreds described in this report, the POLD diagnosis was based on neuropathologic evaluations of autopsy or biopsy tissue.14 The brains of familial POLD cases generally show extensive myelin and axonal damage, as well as the distinctive feature of pigmented macrophages and glia accompanied by cytoplasmic autofluorescent lipofuscin granules.4,7,16,24,28 The white matter atrophy and myelin loss is similar in patients with pathologically confirmed HDLS; however, axonal spheroids are prominent in affected white matter in addition to myelinated fiber loss, gliosis, and pigment-laden glial cells.3,8,9,23,25,29 However, they can be sparse in areas of white matter with burnt-out pathology and few residual axons, and must be sought for in areas where the white matter pathology is ongoing. In a study of the originally described POLD family in 1936, it was reported that axonal dilations consistent with axonal spheroids were present in the affected brain tissue.16 Additional case reports have since described such a phenomenon in other POLD families.5
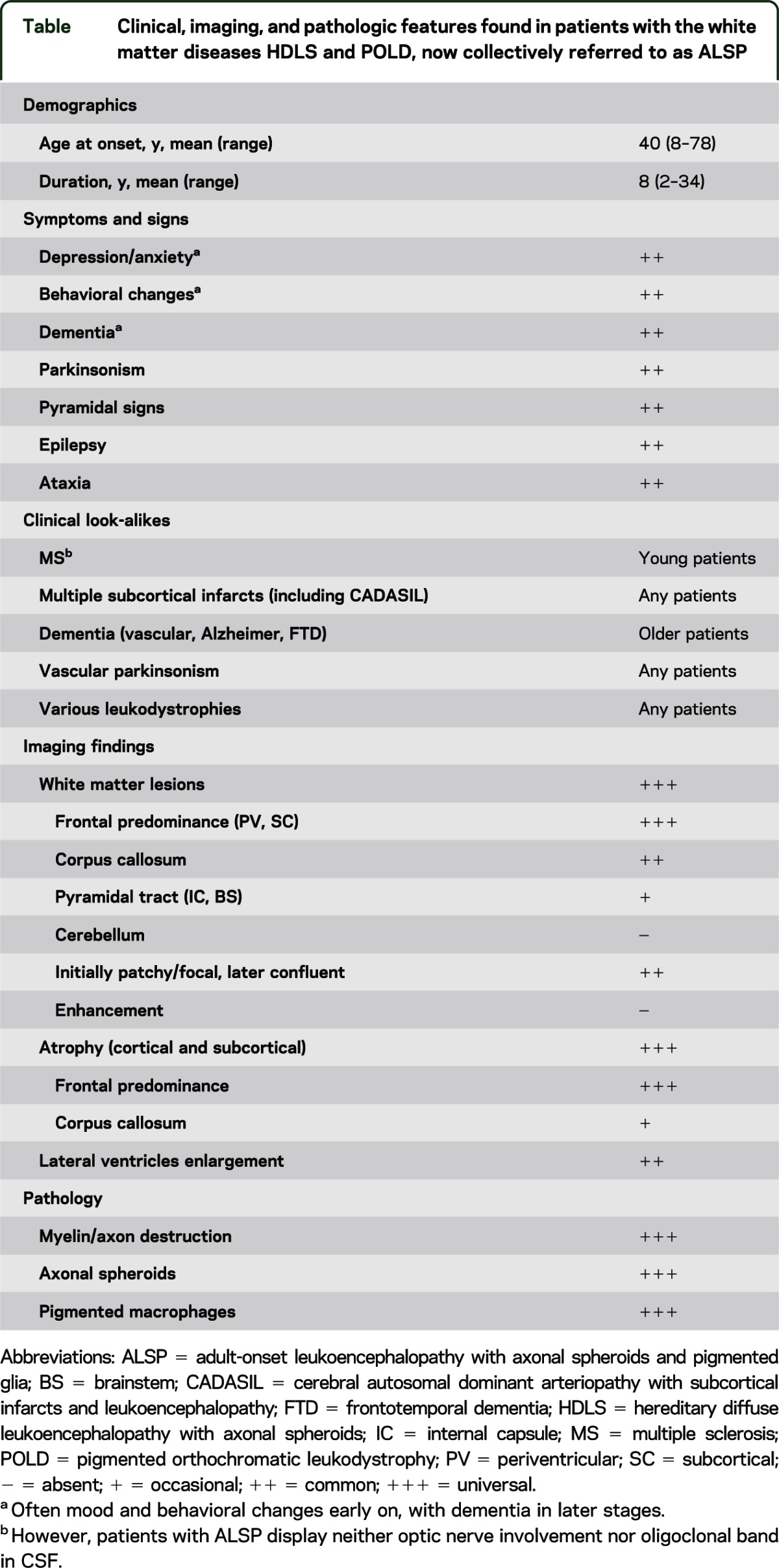
In the current HDLS and POLD classification, neither acronym adequately reflects the complete set of molecular genetic, clinical, and pathologic features observed in these 2 closely overlapping disorders. In particular, use of the term leukodystrophy in POLD implies a defect in production or catabolism of myelin, which is not the case. The discovery of defects in CSF1R, a trophic factor for microglia, suggests that the axonal spheroids in HDLS are a secondary disease process and that HDLS and POLD might be considered primary microgliopathies that lead to secondary axonal and myelin pathology. While the presence of morphologically abnormal (i.e., pigment-laden) microglia in POLD is more closely linked to the genetic defect, the specificity of autofluorescent pigment in microglia is not. Recently, we proposed the term adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) to encompass HDLS and POLD. Given that CSF1R mutations are now known to cause both disorders, we propose the continued use of ALSP to encompass the microgliopathies previously described as HDLS and POLD. Furthermore, we have included a table (table) with the clinical, imaging, and pathologic features of this CSF1R-related disease to assist in the future diagnosis of patients.
Table.
Clinical, imaging, and pathologic features found in patients with the white matter diseases HDLS and POLD, now collectively referred to as ALSP
Taken together, we identified mutations in CSF1R, the gene known to cause familial HDLS, in 2 families with autosomal dominant inheritance of pathologically confirmed POLD. This finding confirms the hypothesis that POLD and HDLS are a single disease entity.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to family members who participated in this study. The authors thank Ms. Sharleen Traynor for her assistance in collecting data on POLD family 5901.
GLOSSARY
- ALSP
adult-onset leukoencephalopathy with axonal spheroids and pigmented glia
- bvFTD
behavioral variant frontotemporal dementia
- CSF1R
colony-stimulating factor 1 receptor
- CSF1RWT
wild-type colony-stimulating factor 1 receptor
- FTD
frontotemporal dementia
- H&E
hematoxylin & eosin
- HDLS
hereditary diffuse leukoencephalopathy with axonal spheroids
- POLD
pigmented orthochromatic leukodystrophy
- STMS
Short Test of Mental Status
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Nicholson: drafting/revising the manuscript for content; analysis or interpretation of data; acquisition of data. Matt C. Baker, NiCole A. Finch, Nicola J. Rutherford: analysis or interpretation of data; acquisition of data. Drs. Wider, Graff-Radford, Nelson, Clark, Wszolek, Dickson, and Knopman: drafting/revising the manuscript for content, including medical writing for content; contribution of vital reagents/tools/patents; obtaining funding. Dr. Rademakers: drafting/revising the manuscript for content, including medical writing for content; study concept and design; analysis or interpretation of data; study supervision; obtaining funding.
STUDY FUNDING
This work is funded by a Mayo Benefactor and the Mayo Foundation and is further supported by NIH grants R01 NS065782, P50 AG016574, P50 NS072187, P30 AG028383, and P50 NS072187. A.M.N. is funded by the Association for Frontotemporal Degeneration Postdoctoral Fellowship.
DISCLOSURE
A. Nicholson, M. Baker, N. Finch, and N. Rutherford report no disclosures. C. Wider is supported by the Leenaards Foundation and the Swiss Parkinson Foundation. P. Nelson is supported by NIH grants (P30AG028383, R21 AG036875, R01 NS061933) and report no other disclosures. B. Clark is supported by NIH grants (1 PO1NS058901, R01 NS070815, and R37-NS022920). N. Graff-Radford serves as board member of The Neurologist, serves as a consultant for Codman, and receives research support from Medivation, Janssen, Allon, Forest, Pfizer, and NIA. Z. Wszolek is supported by the NIH/NINDS NS057567, 1RC2NS070276, P50 NS072187, Mayo Clinic Florida Research Committee CR program, and the Dystonia Medical Research Foundation. D. Dickson is supported by NIH grants (P50 AG16574, P50 NS72187, P01 AG03949), the Mangurian Foundation, CurePSP, and the Robert E. Jacoby Professorship for Alzheimer's Research. D. Knopman serves as Deputy Editor for Neurology®; serves on a Data Safety Monitoring Board for Lilly Pharmaceuticals; is an investigator in clinical trials sponsored by Janssen Pharmaceuticals, and receives research support from the NIH. R. Rademakers receives research support from the NIH (R01 NS065782, R01AG02651, and P50 AG16574), the ALS Therapy Alliance, and the Consortium for Frontotemporal Degeneration Research. Dr. Rademakers further received honoraria for lectures or educational activities not funded by industry and serves on the medical advisory board of the Association for Frontotemporal Degeneration. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Freeman SH, Hyman BT, Sims KB, et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol 2009;19:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Gerpen JA, Wider C, Broderick DF, Dickson DW, Brown LA, Wszolek ZK. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology 2008;71:925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marotti JD, Tobias S, Fratkin JD, Powers JM, Rhodes CH. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol 2004;107:481–488 [DOI] [PubMed] [Google Scholar]
- 4.Hanaya R, Arita K, Itoh Y, Kiura Y, Iida K, Kurisu K. Zygomatic osteotomy for resection of medial temporal cavernous angioma in dominant hemisphere after subdural grid electroencephalographic study. Hiroshima J Med Sci 2006;55:39–43 [PubMed] [Google Scholar]
- 5.Wider C, Van Gerpen JA, DeArmond S, Shuster EA, Dickson DW, Wszolek ZK. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology 2009;72:1953–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belec L, Gray F, Louarn F, et al. Pigmentary orthochromatic leukodystrophy: van Bogaert and Nyssen disease [in French]. Rev Neurol 1988;144:347–357 [PubMed] [Google Scholar]
- 7.Okeda R, Matsuo T, Kawahara Y, et al. Adult pigment type (Peiffer) of sudanophilic leukodystrophy: pathological and morphometrical studies on two autopsy cases of siblings. Acta Neuropathol 1989;78:533–542 [DOI] [PubMed] [Google Scholar]
- 8.Hughes TR, Hiley SL, Saltzman AL, Babak T, Blencowe BJ. Microarray analysis of RNA processing and modification. Methods Enzymol 2006;410:300–316 [DOI] [PubMed] [Google Scholar]
- 9.Hancock N, Poon M, Taylor B, McLean C. Hereditary diffuse leucoencephalopathy with spheroids. J Neurol Neurosurg Psychiatry 2003;74:1345–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Constantinidis J, Wisniewski TM. The dominant form of the pigmentary orthochromatic leukodystrophy. Acta Neuropathol 1991;82:483–487 [DOI] [PubMed] [Google Scholar]
- 11.Oepen H. Clinical, anatomopathologic and genealogic study of a late adult leukodystrophy [in German]. Arch Psychiatr Nervenkr 1964;206:115–130 [PubMed] [Google Scholar]
- 12.Sundal C, Van Gerpen JA, Nicholson A, et al. MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology 2012;79:566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2011;44:200–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knopman D, Sung JH, Davis D. Progressive familial leukodystrophy of late onset. Neurology 1996;46:429–434 [DOI] [PubMed] [Google Scholar]
- 15.Sundal C, Lash J, Aasly J, et al. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): a misdiagnosed disease entity. J Neurol Sci 2012;314:130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Bogaert L, Nyssen R. Le type tardif de la leukodystrophie progressive familiale. Rev Neurol 1936;65:21–45 [Google Scholar]
- 17.Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T, Ikeda SI. Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci 2012;318:115–118 [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Berezovska O, Fedoroff S. Expression of colony stimulating factor-1 receptor (CSF-1R) by CNS neurons in mice. J Neurosci Res 1999;57:616–632 [PubMed] [Google Scholar]
- 19.Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, McGeer PL. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer's disease and amyotrophic lateral sclerosis. Brain Res 1994;639:171–174 [DOI] [PubMed] [Google Scholar]
- 20.Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol 2004;14:628–638 [DOI] [PubMed] [Google Scholar]
- 21.Ali ZS, Van Der Voorn JP, Powers JM. A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: a role for oxidative damage. J Neuropathol Exp Neurol 2007;66:660–672 [DOI] [PubMed] [Google Scholar]
- 22.Wong JC, Chow TW, Hazrati LN. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia can present as frontotemporal dementia syndrome. Dement Geriatr Cogn Disord 2011;32:150–158 [DOI] [PubMed] [Google Scholar]
- 23.Axelsson R, Roytta M, Sourander P, Akesson HO, Andersen O. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand Suppl 1984;314:1–65 [PubMed] [Google Scholar]
- 24.Gray F, Destee A, Bourre JM, et al. Pigmentary type of orthochromatic leukodystrophy (OLD): a new case with ultrastructural and biochemical study. J Neuropathol Exp Neurol 1987;46:585–596 [DOI] [PubMed] [Google Scholar]
- 25.Terada S, Ishizu H, Yokota O, et al. An autopsy case of hereditary diffuse leukoencephalopathy with spheroids, clinically suspected of Alzheimer's disease. Acta Neuropathol 2004;108:538–545 [DOI] [PubMed] [Google Scholar]
- 26.Mascalchi M, Gavazzi C, Morbin M, et al. CT and MR imaging of neuroaxonal leukodystrophy presenting as early-onset frontal dementia. AJNR Am J Neuroradiol 2006;27:1037–1039 [PMC free article] [PubMed] [Google Scholar]
- 27.Savoiardo M, Grisoli M. Imaging dementias. Eur Radiol 2001;11:484–492 [DOI] [PubMed] [Google Scholar]
- 28.Tunon T, Ferrer I, Gallego J, Delgado G, Villanueva JA, Martinez-Penuela JM. Leucodystrophy with pigmented glial and scavenger cells (pigmentary type of orthochromatic leucodystrophy). Neuropathol Appl Neurobiol 1988;14:337–344 [PubMed] [Google Scholar]
- 29.van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, Kamphorst W, Weinstein HC. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology 2000;54:463–468 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.