

Abstract
Objective:
Recently, vacuolar protein sorting 35 (VPS35) and eukaryotic translation initiation factor 4 gamma 1 (EIF4G1) have been identified as 2 causal Parkinson disease (PD) genes. We used whole exome sequencing for rapid, parallel analysis of variations in these 2 genes.
Methods:
We performed whole exome sequencing in 213 patients with PD and 272 control individuals. Those rare variants (RVs) with <5% frequency in the exome variant server database and our own control data were considered for analysis. We performed joint gene-based tests for association using RVASSOC and SKAT (Sequence Kernel Association Test) as well as single-variant test statistics.
Results:
We identified 3 novel VPS35 variations that changed the coded amino acid (nonsynonymous) in 3 cases. Two variations were in multiplex families and neither segregated with PD. In EIF4G1, we identified 11 (9 nonsynonymous and 2 small indels) RVs including the reported pathogenic mutation p.R1205H, which segregated in all affected members of a large family, but also in 1 unaffected 86-year-old family member. Two additional RVs were found in isolated patients only. Whereas initial association studies suggested an association (p = 0.04) with all RVs in EIF4G1, subsequent testing in a second dataset for the driving variant (p.F1461) suggested no association between RVs in the gene and PD.
Conclusions:
We confirm that the specific EIF4G1 variation p.R1205H seems to be a strong PD risk factor, but is nonpenetrant in at least one 86-year-old. A few other select RVs in both genes could not be ruled out as causal. However, there was no evidence for an overall contribution of genetic variability in VPS35 or EIF4G1 to PD development in our dataset.
Parkinson disease (PD) is the second most common neurodegenerative brain disorder and affects approximately 2% of the population older than 65 years.1 A few families have a mendelian form of PD (<10%). Research of these families has led to the subsequent discovery of at least 5 causal genes (SNCA, PARK2, PINK1, PARK7, and LRRK2).2–6 These genes have been consistently shown to segregate with disease in the families studied. Additional genes have been identified, but reports on their pathogenicity remain inconsistent: HTRA2,7 UCHL1,8 and ATP13A2.9 Recently, 2 new genes have been implicated as causal genes for PD: vacuolar protein sorting 35 (VPS35)10 gene and eukaryotic translation initiation factor 4 gamma 1 (EIF4G1) gene.11
Next-generation sequencing technology—including whole exome sequencing (WES)—provides the opportunity to investigate all these genes and possible new genes in parallel.
We set out to utilize the advantages of WES to investigate the genetic involvement of the recently identified genes EIF4G1 and VPS35 in PD development, both on a single variant and gene level.
METHODS
Subject ascertainment.
Subjects used in this study were collected by the University of Miami, Morris K. Udall Parkinson Disease Research Center of Excellence (J.M. Vance, principal investigator), and 13 centers of the Parkinson Disease Genetics Collaboration.12 All the patients and controls were examined by a neurologist, the majority movement disorder specialists. A standard neurologic examination including the Unified Parkinson's Disease Rating Scale was performed and has been described previously.12 Unaffected individuals demonstrated no signs of the disease at age of examination. Two hundred thirteen cases of PD were included in an initial WES discovery dataset. All individuals are of white, non-Hispanic/Latino descent. Among the 213 PD cases, 188 are unrelated. Two groups were used as controls. Eighty-five white, non-Hispanic/Latino control individuals from the Udall control dataset were sequenced here by WES. All had normal neurologic examinations and normal Modified Mini-Mental State Examination. In addition, the Hussman Institute for Human Genomics has whole exome data for 188 self-reported white, non-Hispanic/Latino subjects; these include unrelated samples from families with diagnoses of autism, hereditary spastic paraplegia, multiple sclerosis, and thrombotic storm. These “internal control database” (ICD) samples are analyzed using the same sequencing protocol as our initial PD dataset. Although no individuals had clearly symptomatic PD, they were not necessarily examined by a neurologist. However, because the frequency of PD in the general population is low (0.3%), misclassification in this “control” group would be very low and negligible to the analysis. We used these samples as additional control data to minimize costs.
Standard protocol approvals, registrations, and patient consents.
All subjects were collected with IRB approval at the University of Miami and provided written informed consent.
Whole exome sequencing.
Sequence capture.
Genomic DNA was extracted from blood and processed according to the Illumina Paired-End Sample Preparation Guide with modifications listed in Agilent's protocol (Agilent Technologies Inc., Santa Clara, CA; v2.0.1, May 2010). Fragmented DNA was captured using the SureSelect Human All Exon Kit, designed to cover 38 or 50 Mb of human genomic sequences. We used the 38-Mb capture kit in the initial 21 individuals (all patients), and updated to the 50-Mb kit for the remaining samples (n = 277).
Next-generation sequencing.
The libraries were loaded onto an Illumina cBot for cluster generation (HiSeq Paired End Cluster Generation Kit v1.5 and later version TruSeq PE Cluster Kit v2.5-cBot-HS; Illumina, Inc., San Diego, CA). One lane of each flow cell was reserved for a PhiX control. The primer-hybridized flow cells were then transferred to HiSeq2000 sequencers and paired-end sequencing was done with TruSeq SBS Kit-HS (200 cycle) (Illumina) in a 2 × 101b mode.
Alignment and base calling.
The base calling was done by Illumina CASAVA 1.6 pipeline, and aligned to hg19, using Genome Analysis Tool Kit v1.1. The Unified Genotyper from the Genome Analysis Tool Kit calls both variants and indels and performs VQS (variant quality score) recalibration and genotype refinement to make accurate variant calls.13 Additionally, the Unified Genotyper generates normalized Phred-scaled likelihood scores without priors, for each alternate genotype. Variants with VQSLOD <−3 and alternate Phred-scaled likelihood scores <99 are excluded from the remainder of the analysis presented here. All of the remaining variants were annotated using ANNOVAR.14 Variations were screened against the Exome Variant Server (EVS) version ESP5400 from the NHLBI Exome Sequencing Project (Seattle, WA) (URL: http://evs.gs.washington.edu/EVS/) for previously observed variants.
In silico analysis of rare variants.
Conservation scores GERP (Genomic Evolutionary Rate Profiling) and phastCons (Phylogenetic Analysis with Space/Time Models) are both obtained through ANNOVAR. PolyPhen2 was used to predict effects of amino acid changes on protein functional status.15 Programs SpliceView, NNsplice, and ESEfinder were used to predict the effect of variants on splicing.16–18
Statistical analysis.
We performed joint gene-based tests for association between PD and VPS35 or EIF4G1 using 2 methods: the CA sum test19 implemented in version 1.11 of the RVASSOC program19a and the Sequence Kernel Association Test (SKAT).20 Both combine squared single-variant score statistics, which makes them robust to the inclusion of neutral and protective variants and more powerful than pooling tests. The CA sum test considers the sum of single-variant Cochran-Armitage trend χ2 statistics and calculates a permutation p value with 1M permutations. SKAT is based on a general linear model framework. Version 0.75 of the SKAT R package was run with default settings and without covariates. With complete genotype data and without covariates, the tests are expected to yield similar inferences. However, the tests differ in the handling of missing data and covariates.
Sanger sequencing.
To determine whether variant p.R1205H in the family lies on the same background haplotype as previously published, we performed Sanger sequencing for those variants not covered in WES (primer sequences available upon request).
Genotyping assay.
Samples for genotyping were analyzed using a TaqMan® allelic discrimination Assay-By-Design method (ABI; Applied Biosystems, Foster City, CA) (primer and probe sequences available upon request).
RESULTS
Whole exome sequencing.
Identified variants are shown in tables 1 and 2. The samples had an average depth of 57.3x/51.8x and an average coverage of 87.1% (80%–94%) (38-Mb capture kit)/84.6% (69%–95%) (50-Mb capture kit) at minimum coverage of 8x. Specifically, at least 99% and 91% of the coding sequence of VPS35 and EIF4G1, respectively, were covered at a depth of at least 8x in the PD samples and controls.
Table 1.
Coding variants identified in VPS35
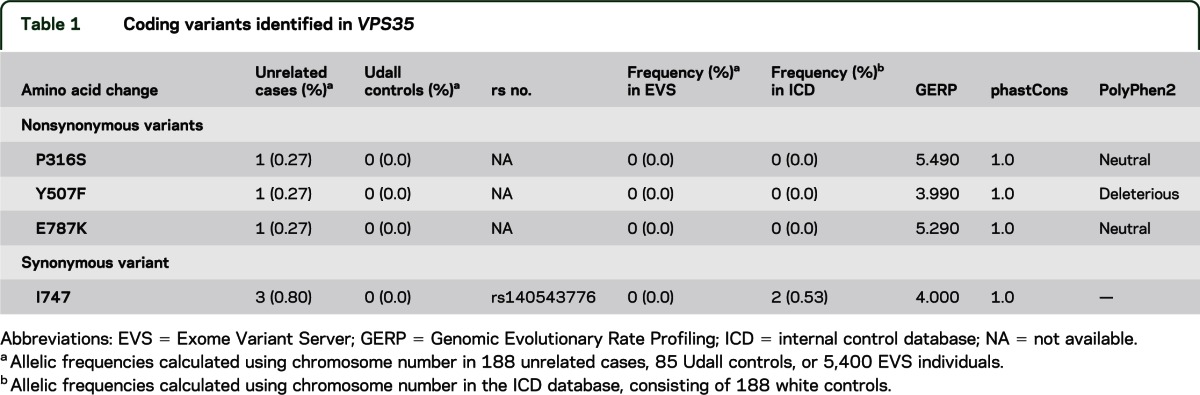
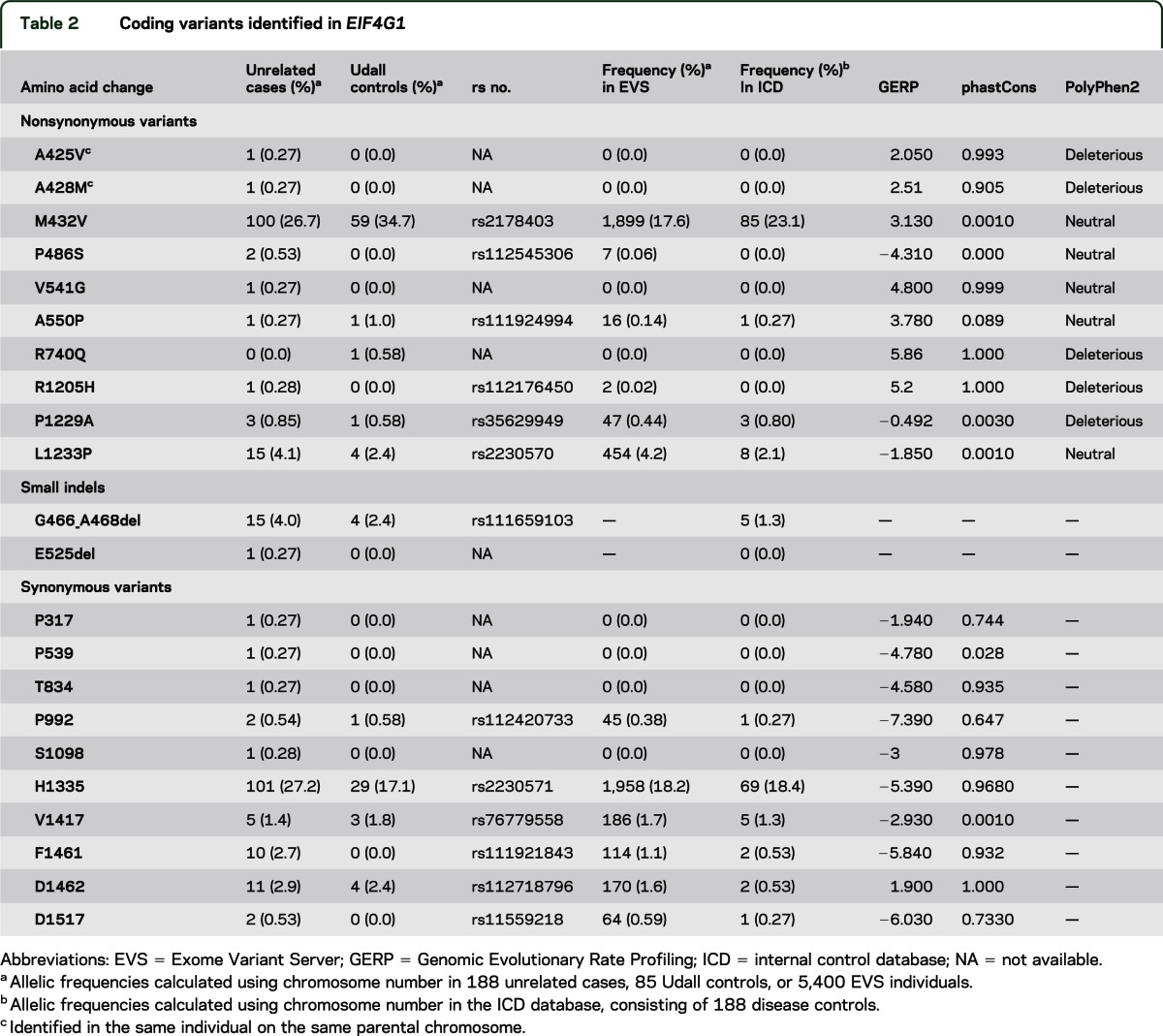
Table 2.
Coding variants identified in EIF4G1
VPS35.
WES of 213 patients with PD revealed 4 variants in the regions coding for protein (exome) in VPS35 meeting our QC criteria: 3 variants that changed the amino acid coded in the protein (nonsynonymous rare variants [RVs] or nsRVs) variants (p.P316S; p.Y507F; p.E787K) and 1 variant that did not change an amino acid (synonymous, sRV) variant with minor allele frequency ≤5% (table 1). None of the 3 nonsynonymous variants was observed in the 85 Udall controls of this study, the ICD, or the EVS. We observed the p.E787K variant in a patient with an age at onset (AAO) of 60 years but not in his uncle, who was diagnosed with PD at the age of 81 years. The first 2 nsRVs (p.P316S; p.Y507F) were each identified in 1 patient with early AAO (26 years and 29 years, respectively). However, the mother of the patient with the p.Y507F variant also carried this variation and was unaffected at the age of 73 years. We identified the p.P316S variant in a sporadic patient. In silico analysis predicts a “probably damaging” effect for p.Y507F, but supports a “probable benign” effect of the changes in variants p.P316S and p.E787K.
EIF4G1.
WES of 213 patients with PD revealed 19 coding RVs in EIF4G1, of which 8 are nsRVs (including p.R1205H), 9 sRVs, and 2 small deletions (table 2). The other 2 coding variants constitute common single-nucleotide polymorphisms (SNPs). The patients with PD carrying the 8 nsRVs (24/188 = 12.8%) had an average AAO of 54 years (range 35–80 years).11 Seven of the RVs are not present in the EVS: 3 nsRVs (p.A425V; p.A428M; p.V541G) and 4 sRVs. We identified all these novel nsRVs in 1 patient each and not in any of the 85 Udall controls or ICD controls. Interestingly, both p.A425V and p.A428M were identified in the same singleton patient. Sanger sequencing in the unaffected parents of this patient showed that both variants were inherited from the same unaffected parent (age at examination 71 years). We observed nsRV p.V541G in 1 sporadic patient with no other family members available for testing. Four of the 5 remaining nsRVs (p.R1205H [discussed below]; p.P486S; p.A550P; p.P1229A) had frequencies <1% in EVS and ICD (table 2). Variant p.P486S was present in 2 patients and none of the 85 Udall controls or ICD controls. We observed the other 2 variants (p.A550P; p.P1229A) in both patients and controls. These patients are sporadic or singleton individuals, so no further segregation analysis could be performed for these. Interestingly, we identified only 1 control-specific variation in the 85 Udall controls (p.R740Q). We did not observe this variation in the ICD. In silico analyses of the 8 nsRV positions are shown in table 2.
Additionally, we observed 2 small coding deletions in EIF4G1. Indels are currently not included in EVS so no frequency in a large population could be obtained. The novel p.E525del was confirmed in 1 of 2 affected siblings.
The location of the observed variants is shown in figure 1. Variations p.V541G, p.A550P, and p.E525del are positioned between p.A502V—previously reported to perturb EIF4E and EIF4G1 interaction11—and the EIF4E interaction site, suggesting that these variants might also influence EIF4E interaction. One more variant is located within 25 base pairs of a functional domain or pathogenic mutation (p.P486S).
Figure 1. EIF4G1 protein and its interaction domains.
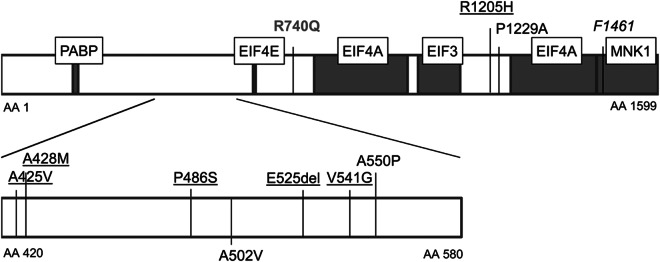
Amino acids located in the protein are denoted by AA prefix; protein accession number: NP_937884.1. Gray boxes are interaction sites; boxed texts are the interacting proteins. Missense variations above the protein box were found in this study with frequency <1%. Underlined missense variants are identified in patients only. p.R740Q (gray) was observed in controls only. Variants below the protein box were reported to be pathogenic by the original publication.11
Mutation p.R1205H.
We observed the previously reported pathogenic mutation p.R1205H in 1 individual with a positive family history. Subsequent Sanger sequencing showed segregation of the mutation with its family members with clinical PD (figure 2). These patients had AAO of 54 years, 57 years, and 73 years. However, the mutation also segregated with 2 other individuals. One uncle (II.1), examined at age 90 years, had an “unclear” PD diagnosis. He displayed dementia and postural instability early in the course of symptoms and was wheelchair-bound. The second individual (II.3) had no symptoms of PD on examination at the age of 86 years. All individuals carrying p.R1205H shared the same SNP alleles as in the founder haplotype reported for this mutation11 (figure 2). We did not observe this mutation in the 2 control groups. Both conservation measures and PolyPhen2 support a pathogenic effect of this variation. Interestingly, this variation was observed twice in EVS.
Figure 2. Sharing analysis in p.R1205H family.

Sex is masked to maintain confidentiality. Darkened individuals in the pedigree are affected. ? = unclear status; N/A = not available. Box alleles represent haplotype reported by the original publication.11
Gene-based statistical analysis.
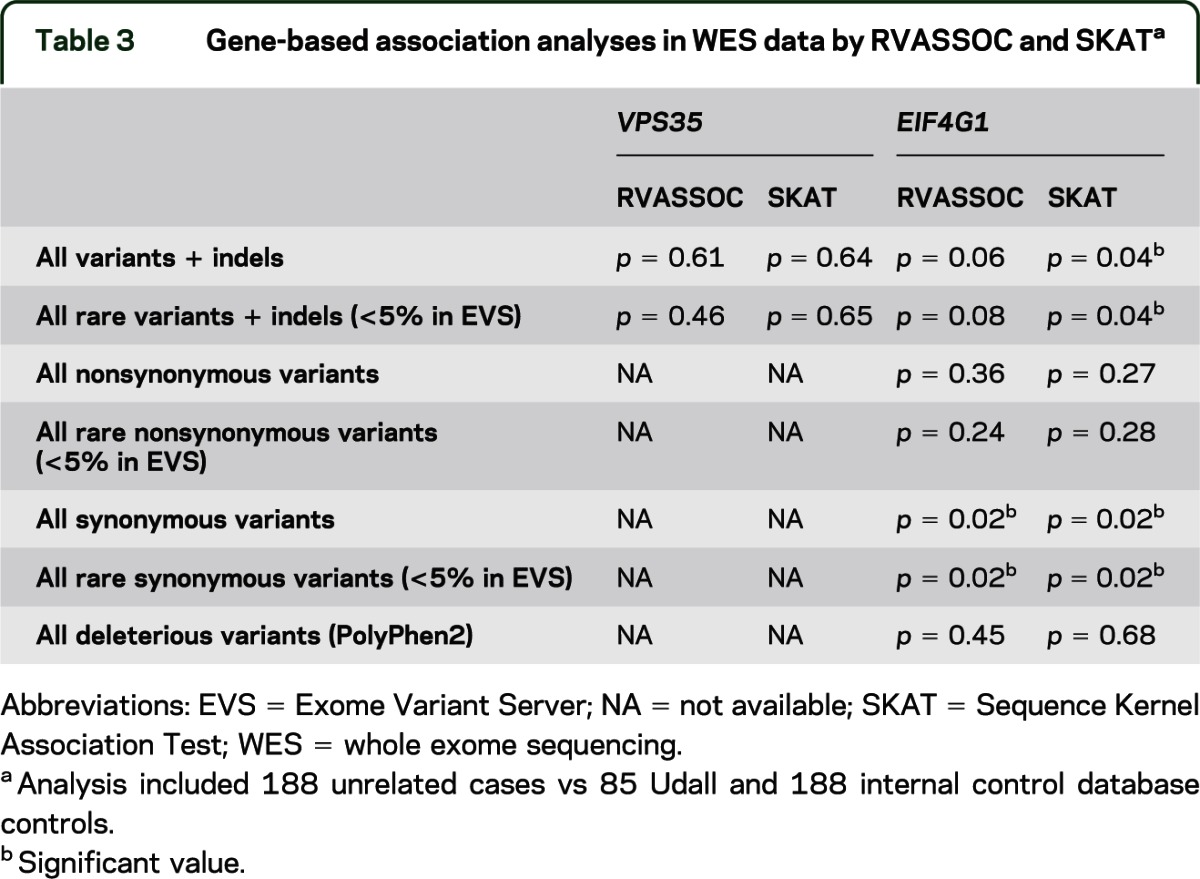
Because the evidence supporting the common disease-RV hypothesis is increasing, we wanted to test the hypothesis that overall the genes VPS35 and EIF4G1 are associated with PD, which would provide further evidence of the involvement of the RVs that we identified. We used 2 different programs for this purpose, RVASSOC and SKAT. Neither RVASSOC nor SKAT showed association between PD and VPS35 for all variants or all RVs (table 3). Because only 4 coding variants were observed with only 1 predicted to be deleterious (table 1), no additional association analyses on functional variants were performed. When analyzing the variants and indels in EIF4G1, we obtained initial evidence of association between PD and EIF4G1 when including all variants or just all RVs (SKAT p = 0.04 for both analyses) (table 3). We broke down these groups further, and unexpectedly found association for the synonymous variants (p = 0.02 in RVASSOC and SKAT), but not the nonsynonymous or functionally deleterious variants. The association with the synonymous variants was driven primarily by p.F1461 (single variant analysis p = 0.002; 10/188 patients vs 2/272 controls). We did not observe any of the other nsRVs in haplotype with p.F1461, which seemed to rule out p.F1461 as a tagging SNP. In silico analysis of this variant showed no effect on splicing (SpliceView, NNsplice) or binding of exonic splicing enhancers (ESEfinder). Thus, we performed association analysis in a larger population of 826 unrelated patients and 218 controls. This analysis showed consistent direction of the effect but did not confirm the previous strong association (p = 0.14).
Table 3.
Gene-based association analyses in WES data by RVASSOC and SKATa
DISCUSSION
In this new age of high-throughput sequencing, we will identify “private” genetic changes or specific, rare genetic changes (RVs) that are found in only a few individuals. Some of these changes may be clinically relevant and lead to a significantly increased risk for disease. However, many, if not most, of these RVs will provide a moderate, mild, or lack of susceptibility to disease. Although we can only state the likelihood of each single change being important to the disease, we can get some measure of the overall importance of the gene's contribution to PD risk. To do this, we examine whether the total number of specific RVs are significantly more abundant in the PD population than controls.
In this report, we strongly support that the p.R1205H variant is involved in PD, with the segregation of p.R1205H in 3 patients with PD in 1 large family. However, unlike in the first report, we also found this variant in an 86-year-old unaffected family member, suggesting the mutation has a reduced penetrance. Interestingly, another study21 recently reported the presence of p.R1205H in 3 general population controls, but the authors indicated that these carriers may have or develop the disease at a later age. Of the remaining novel nsRVs, p.A425 and p.A428M are unlikely to be causal based on the presence of the variant in the unaffected parent of the carrier. p.V541G has now been reported in 2 patients21 and no controls, and is well conserved in vertebrates, although the change is not predicted to be functionally significant. Therefore, it remains as a possible causal variant. p.P486S has also now been reported in 4 patients,22 but also 7 controls in the EVS, and is poorly conserved in evolution and predicted to be functionally neutral. It therefore seems unlikely to contribute to PD risk.
However, it is interesting that, similar to others, we also found that most of the variants we identified in EIF4G1 are clustered within close proximity (<50 amino acids) of the functional domains involving PABP and EIF4E (figure 1), suggesting they might affect the protein's function. A recent analysis of the EVS dataset22 suggests, however, that the high number of variants surrounding p.A502V in controls as well as patients suggests they may not be functionally related, but rather could indicate a higher tolerance for genetic variability in this region. The lack of significance in RVs between patients and controls for EIF4G1 in this study supports the latter hypothesis. However, as data on more individuals is gathered, specific locations within this region may still prove to be important in PD risk.
In 2011, the first novel causal gene identified through next-generation sequencing for PD was described: VPS35.10 This and additional studies reported the p.D620N change in 9 families as well as other missense variants in sporadic cases.23,24 In VPS35, we identified 3 nsRVs, of which p.Y507F suggests a noncausal effect based on family segregation. The p.P316S variant has been previously reported in 2 affected individuals and 1 unaffected individual in 1 family,10 but in no controls and is highly evolutionarily conserved. Therefore, it remains as a variant with a possible strong risk effect. The p.E787K variant is less clear. The carrier's affected uncle did not carry this variation. The uncle's AAO was 21 years later than the carrier, however, so this difference could suggest a possible different etiology of PD in this individual. This chromosomal position is also highly conserved, so it is possible that other factors contributed to the uncle's disease, and the variant remains a possible contributor to PD risk.
The first EIF4G1 study11 reported functional analyses that suggested both of the mutations they reported in EIF4G1 disrupted interaction of the protein with its physiologic binding partners, cap-binding protein EIF4E, and RNA helicase EIF4A, which form the EIF4F translation initiation complex. We thus also evaluated the WES data for EIF4E and EIF4A, but did not see any evidence for RVs contributing to PD in these genes. Interestingly, we previously reported association of decreased PD AAO with variants in EIF2B3 coding for the gamma subunit of another translation initiation factor complex (EIF2B), specifically regulating protein synthesis under mild cellular stress.25 Because many of the known PD genes are involved in protection of the mitochondria against cellular stress (PARK2, PINK1, PARK7, HtrA26,26–30), genes involved in handling this cellular stress could contribute to risk of developing PD. The field's knowledge of EIF4G1 interaction partners denotes that its complex is part of the larger RNA transport/translation regulation pathway (KEGG; http://www.genome.jp/kegg/). This EIF4F complex interacts with FMR1 interacting protein (KEGG) in the regulation of the actin cytoskeleton pathway. This pathway was ranked as the seventh strongest association in our recent joint pathway analysis in PD using both genome-wide associa-tion studies and gene expression analysis.31 VPS35 pathways are less clarified at present.
It seems intuitive that the uniqueness of the pathogenic strength of the p.R1205H variation in EIF4G1, relative to the other RVs in the gene, suggests that the function disrupted by this variation, and its downstream effects, is a potentially important piece of information in understanding the pathogenesis of PD.
One method to measure the cumulative importance of RVs in a gene is by using the newer gene-based statistical association techniques to find whether RV tests across a gene show differences in cases and controls, as we did here with 2 different algorithms. The original study reported a nonsignificant overall 3-fold difference in number of novel RVs in cases only vs controls only,11 suggesting a possible RV effect. However, although we saw an initial weak association with all RVs, subsequent testing of the driving variation (p.F1461) demonstrated no association in a larger dataset, suggesting that the initial p.F1461 association was likely by chance or the second dataset provided a false-negative result. However, although major effects of most variations in EIF4G1 or VPS35 are not suggested by our analysis, larger datasets could still detect an overall smaller susceptibility effect. The WES data presented here, however, agrees with several more recent reports that also do not support a major involvement of VPS35 variability, besides p.D620N, in the development of PD.24,32–34
In general, no evidence for an overall significant contribution of VPS35 or EIF4G1 variability to PD development was identified in our dataset. This study emphasizes the importance of examining RVs individually and in larger datasets before they are declared “pathogenic.” Our data confirmed that p.R1205H is a strong PD risk factor, but is nonpenetrant in one 86-year-old individual. Several additional RVs in this study remain potential strong contributors to PD risk, but more data are required before this can be confirmed.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to the families and staffs who participated in this study, and to Drs. Nahab and Singer (Department of Neurology, Miller School of Medicine, University of Miami, FL) who have contributed to this study through collection of PD samples. In addition, the authors thank Drs. Pericak-Vance and McCauley (Hussman Institute for Human Genomics) who contributed to the ICD data collection. Some of the samples used in this study were collected while the Udall PDRCE was based at Duke University.
GLOSSARY
- AAO
age at onset
- EIF4G1
eukaryotic translation initiation factor 4 gamma 1
- EVS
Exome Variant Server
- ICD
internal control database
- nsRV
nonsynonymous rare variant
- PD
Parkinson disease
- RV
rare variant
- SKAT
Sequence Kernel Association Test
- SNP
single-nucleotide polymorphism
- sRV
synonymous rare variant
- VPS35
vacuolar protein sorting 35
- WES
whole exome sequencing
Footnotes
Editorial, page 974
AUTHOR CONTRIBUTIONS
Design or conceptualization of the study: K.N., G.B., S.Z., E.R.M., W.K.S., J.M.V. Analysis or interpretation of the data: K.N., V.I., A.D., D.D.K., A.M., G.W.B., E.R.M. Drafting or revising manuscript for intellectual content: K.N., S.Z., L.W., W.K.S., J.M.V.
STUDY FUNDING
This work was supported by NIH grants NS039764, NS071674 (J.M.V.), and 5RC2HG005605-02 (E.R.M.).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson's disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54:S21–S23 [PubMed] [Google Scholar]
- 2.Bonifati V. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003;299:256–259 [DOI] [PubMed] [Google Scholar]
- 3.Kitada T. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608 [DOI] [PubMed] [Google Scholar]
- 4.Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 2004;44:595–600 [DOI] [PubMed] [Google Scholar]
- 5.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997;276:2045–2047 [DOI] [PubMed] [Google Scholar]
- 6.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004;304:1158–1160 [DOI] [PubMed] [Google Scholar]
- 7.Strauss KM. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet 2005;14:2099–2111 [DOI] [PubMed] [Google Scholar]
- 8.Lincoln S, Vaughan J, Wood N, et al. Low frequency of pathogenic mutations in the ubiquitin carboxy-terminal hydrolase gene in familial Parkinson's disease. Neuroreport 1999;10:427–429 [DOI] [PubMed] [Google Scholar]
- 9.Ramirez A, Heimbach A, Grundemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006;38:1184–1191 [DOI] [PubMed] [Google Scholar]
- 10.Vilarino-Guell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011;89:162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 2011;89:398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards TL, Scott WK, Almonte C, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 2010;74:97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogozin IB, Milanesi L. Analysis of donor splice sites in different eukaryotic organisms. J Mol Evol 1997;45:50–59 [DOI] [PubMed] [Google Scholar]
- 17.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comput Biol 1997;4:311–323 [DOI] [PubMed] [Google Scholar]
- 18.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 2003;31:3568–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinnamon DD, Hershberger RE, Martin ER. Reconsidering association testing methods using single-variant test statistics as alternatives to pooling tests for sequence data with rare variants. PLoS One 2012;7:e30238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Kinnamon DD. RVASSOC: Rare Variant ASSOCiation (version 1.11). [Source code on Internet.] Available at: http://rvassoc.sourceforge.net. Accessed October 4, 2011 [Google Scholar]
- 20.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 2011;89:82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulte EC, Mollenhauer B, Zimprich A, et al. Variants in eukaryotic translation initiation factor 4G1 in sporadic Parkinson's disease. Neurogenetics 2012;13:281–285 [DOI] [PubMed] [Google Scholar]
- 22.Tucci A, Charlesworth G, Sheerin UM, Plagnol V, Wood NW, Hardy J. Study of the genetic variability in a Parkinson's disease gene: EIF4G1. Neurosci Lett 2012;518:19–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheerin UM, Charlesworth G, Bras J, et al. Screening for VPS35 mutations in Parkinson's disease. Neurobiol Aging 2012;33:838.e1–838.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimprich A, Benet-Pages A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011;89:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliveira SA, Li YJ, Noureddine MA, et al. Identification of risk and age-at-onset genes on chromosome 1p in Parkinson disease. Am J Hum Genet 2005;77:252–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in drosophila parkin mutants. Proc Natl Acad Sci USA 2003;100:4078–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 2001;8:613–621 [DOI] [PubMed] [Google Scholar]
- 28.Canet-Aviles RM. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci USA 2004;101:9103–9108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183:795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in drosophila PINK1 mutants is complemented by parkin. Nature 2006;441:1157–1161 [DOI] [PubMed] [Google Scholar]
- 31.Edwards YJ, Beecham GW, Scott WK, et al. Identifying consensus disease pathways in Parkinson's disease using an integrative systems biology approach. PLoS One 2011;6:e16917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verstraeten A, Wauters E, Crosiers D, et al. Contribution of VPS35 genetic variability to LBD in the Flanders-Belgian population. Neurobiol Aging 2012;33:1844.e11–1844.e13 [DOI] [PubMed] [Google Scholar]
- 33.Lesage S, Condroyer C, Klebe S, et al. Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology 2012;78:1449–1450 [DOI] [PubMed] [Google Scholar]
- 34.Guo JF, Sun QY, Lv ZY, et al. VPS35 gene variants are not associated with Parkinson's disease in the mainland Chinese population. Parkinsonism Relat Disord 2012;18:983–985 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.