

Abstract
Objective:
To assess safety and efficacy of an oral, single, low dose of octanoic acid (OA) in subjects with alcohol-responsive essential tremor (ET).
Methods:
We conducted a double-blind, placebo-controlled, crossover, phase I/II clinical trial evaluating the effect of 4 mg/kg OA in 19 subjects with ET. The primary outcome was accelerometric postural tremor power of the dominant hand 80 minutes after administration. Secondary outcomes included digital spiral analysis, pharmacokinetic sampling, as well as safety measures.
Results:
OA was safe and well tolerated. Nonserious adverse events were mild (Common Terminology Criteria for Adverse Events grade 1) and equally present after OA and placebo. At the primary outcome, OA effects were not different from placebo. Secondary outcome analyses of digital spiral analysis, comparison across the entire time course in weighted and nonweighted accelerometry, as well as nondominant hand tremor power did not show a benefit of OA over placebo. The analysis of individual time points showed that OA improved tremor at 300 minutes (dominant hand, F1,16 = 5.49, p = 0.032 vs placebo), with a maximum benefit at 180 minutes after OA (both hands, F1,16 = 6.1, p = 0.025).
Conclusions:
Although the effects of OA and placebo at the primary outcome were not different, secondary outcome measures suggest superiority of OA in reducing tremor at later time points, warranting further trials at higher dose levels.
Classification of evidence:
This study provides Class I evidence that a single 4-mg/kg dose of OA is not effective in reducing postural tremor in patients with ET at a primary outcome of 80 minutes, but is effective for a secondary outcome after 180 minutes.
Up to 74% of subjects with essential tremor (ET) reported a significant reduction in tremor intensity after ingesting small amounts of ethanol.1–3 Recently, it was shown that tremor improved up to 50% in patients with ethanol-responsive ET after an ethanol challenge.4
The long-chain alcohol 1-octanol has been demonstrated to effectively alleviate tremor symptoms in ET without causing intoxication or other clinically relevant adverse effects.5,6 Pharmacokinetic findings suggested that the effect of 1-octanol might be mediated through its metabolite octanoic acid (OA).7 In the harmaline-induced animal-model of ET, OA reduced tremor in a dose-dependent manner.8 OA was approved by the US Food and Drug Administration as a food additive, received the status GRAS (generally recognized as safe), is used as a component in high-caloric formulas, and has been studied as a component of ketogenic diet for the management of pediatric epilepsy.9
Current pharmacotherapy of ET is often limited by insufficient efficacy, unavoidable side effects, or drug interactions.10 One-third of patients eventually discontinue their treatment.11 Novel pharmacologic treatment approaches are therefore needed for ET, which causes significant impairment in activities of daily living in 3 of 4 patients.12
The aim of this phase I/II, double-blind, placebo-controlled, crossover study was to investigate the safety and efficacy of a low dose of oral OA (4 mg/kg) in patients with ET. The primary outcome was to determine the efficacy of OA in reducing postural tremor power of the dominant hand, 80 minutes after administration, compared with placebo.
METHODS
Patients.
Patients aged 21 years or older were eligible to participate in the study. Inclusion criteria were the presence of ethanol-responsive ET, which was assessed using an objective, standardized ethanol challenge, as previously described, with postural tremor measured by accelerometry as target symptom.7 ET was diagnosed according to consensus criteria for “Classical ET.”13 Detailed study inclusion and exclusion criteria are provided in the supplementary materials (appendix e-1 on the Neurology® Web site at www.neurology.org). A neurologic examination including clinical rating using The Essential Tremor Rating Scale (TETRAS© v.3.1)14 and routine screening laboratory tests were conducted.
Standard protocol approvals, registrations, drug formulation, and patents.
The study (ClinicalTrials.gov identifier: NCT00848172) was approved by the NIH Combined Neurosciences institutional review board. Study monitoring was conducted by an external clinical research organization (KAI Research, Inc.) as well as an independent medical monitor. The consent process was conducted per NIH guidelines, and written informed consent was obtained from each patient before enrollment.
For use in this study, oral OA was awarded an Investigational New Drug status (#103,671) by the US Food and Drug Administration. Together with study cosponsor Ariston Pharmaceuticals, NIH/National Institute of Neurological Disorders and Stroke has filed a patent application for 1-octanol and OA. Oral OA formulations and matching placebo were manufactured by the NIH Pharmaceutical Development Section (Bethesda, MD); OA was dispensed in capsules containing 50 mg OA formulated in 12.5 mL soybean oil, 1.9 mg lemon oil, and microcrystalline cellulose. The dose level of 4 mg/kg was extrapolated from pharmacokinetic and pharmacodynamic data of OA that were expected to be safe and possibly effective.7 The administered dose was rounded to the nearest available 50-mg increment (see appendix e-2 for individual doses).
Study design.
We conducted a double-blind, placebo-controlled, crossover, phase I/II clinical trial assessing the safety and effect of a single oral dose of OA (4 mg/kg) in subjects with ethanol-responsive ET, involving 3 study visits (figure 1). After a screening visit, patients were scheduled for a 3-day inpatient stay, which included 2 consecutive study intervention days on which OA and matching placebo capsules were administered in a randomized, balanced sequence. Patients were allocated 1:1 to a treatment sequence of OA/placebo or placebo/OA. The randomization and allocation was performed before inclusion of the first patient by the NIH Pharmaceutical Development Section, to which the investigators were blinded. The study drugs were prepared in sequentially numbered containers and delivered to the inpatient unit on the morning of administration. Patients and investigators were blinded until the end of the study, until all outcome data had been processed, and the database was locked. If no adverse events were present, subjects were discharged at the end of the third inpatient day and invited back for a follow-up safety visit 7 to 14 days after discharge. The study drugs were administered at the same time of day (6:30 am), with patients fasting starting from midnight until the final tremor recording on that study day. Study procedures were identical on both intervention days and started 30 minutes prior until 5 hours after drug administration (figure 2). For pharmacokinetic sampling, a peripherally inserted central venous catheter was placed on admission. To ensure adequate hydration, patients received IV dextrose 5% (in normal saline) during fasting periods.
Figure 1. Study design.
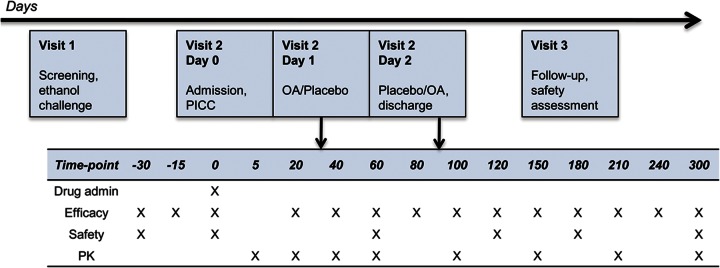
Study flow diagram including 3 visits and time plan for study interventions during the 3-day inpatient visit, during which treatment was administered. Time points in minutes. OA = octanoic acid; PICC = peripherally inserted central venous catheter; PK = pharmacokinetic sampling.
Figure 2. CONSORT 2010 flow diagram.

Enrollment, treatment allocation, follow-up, and analysis. OA = octanoic acid.
For screening and inpatient visits, patients discontinued their antitremor medication and were asked to discontinue their antitremor therapy for at least 5 plasma half-lives before the study. The study was conducted at the NIH Clinical Center in Bethesda, MD.
Efficacy.
Postural tremor was measured while patients sat in a chair, wrists and hands extended beyond armrests parallel to the ground. Tremor was recorded in the vertical z-axis using a 4g triaxial piezo-sensitive accelerometer (Kistler Instrument Corp., Amherst, NY; sensitivity 20 mV/g, measurement range ±250g) placed on the dorsum of each hand. EMG surface electrodes were placed over bilateral wrist extensors and flexors. At each time point, tremor and EMG were recorded simultaneously for 2 minutes, before and after placement of a 1-lb weight attached to each hand. Data were captured using commercial software (NeuroScan, Herndon, VA). The continuous files were broken into 8,192-millisecond epochs at a sampling rate of 1,000 Hz. Fast Fourier transformation was performed on each epoch. Total tremor power in the spectral peak was calculated and averaged across epochs using self-developed Matlab® Scripts.
The weighted condition was used for analysis of tremor power of the central tremor component. The central tremor peak was defined as the spectral accelerometric peak with corresponding EMG peak that remained unchanged in frequency compared with the nonweight condition. To account for baseline variations, baseline measures were taken at −30 minutes, −15 minutes, and at the time of drug administration, and averaged. An area under the curve ±1 Hz across the central frequency peak was calculated for primary outcome analysis; to account for outliers, smoothing of spectral time-point data was performed via moving 3-point average. Total spectral power (2–15 Hz) in the nonweighted condition representing the total tremor (central and peripheral tremor components), as well as digital spiral analysis, were used for secondary analysis.15 Efficacy outcome data recorded after drug administration (figure 2) were normalized to baseline.
Safety.
Safety was assessed using a standardized adverse-events questionnaire in accordance with the Common Terminology Criteria for Adverse Events (v.3.0), an intoxication scale as described previously,7 as well as laboratory parameters including electrolytes, glucose, liver, and kidney function parameters, complete blood count, coagulation, and lipid parameters. EKG and vital signs were obtained at baseline and multiple time points after drug administration (figure 2).
Pharmacokinetics.
Plasma samples were collected at predefined time points (5, 20, 40, 60, 100, 150, 210, and 300 minutes) and analyzed for OA content using an established high-performance liquid chromatography/tandem mass spectrometry assay.7 The lowest limit of quantitation was 20 ng/mL. For detailed methods of pharmacokinetic analysis, see appendix e-2.
Primary/secondary end points.
The primary outcome was defined as the difference in postural tremor power of the central ET component of the dominant hand between OA and placebo at 80 minutes after administration. This time point was chosen based on pharmacokinetic data on OA from previous studies of 1-octanol, expecting a peak effect 80 minutes after administration. Secondary efficacy outcomes included nondominant hand postural and spiral tremor intensities. Furthermore, all other time points were analyzed for the central tremor component and the total tremor. Pharmacokinetic analysis of OA plasma concentration across time points as well as the safety assessment were performed as secondary outcome measure.
Statistics.
A sample size of 19 subjects was determined by power analysis (power 0.8, α = 0.05), using an estimated effect size based on pharmacodynamic data of 1-octanol and OA, where a postural tremor power reduction of 50% was observed.5–7 Primary and secondary efficacy outcomes were analyzed using a linear mixed-model analysis using an unstructured covariance. First, interaction between treatment period (treatment day) and group (OA or placebo) was examined using the linear mixed model with treatment period and treatment group as main factors either at each time point separately or all together. If the interaction was significant, the difference between OA and placebo was analyzed by using either Wilcoxon rank sum test or 2-sample t test, as appropriate, separately for each treatment day with lowering the significance level to 0.025. Safety outcomes are reported descriptively and using linear mixed-model statistics. As an exploratory analysis, within-subject benefit ratio was calculated (OA effect minus placebo effect per subject) and analyzed using a Friedman test in order to examine the differences across all time points.
An overall p value of <0.05 was considered statistically significant. Pharmacokinetic data were analyzed using standard noncompartmental analysis.
Classification of evidence.
This study provides Class I evidence that a single 4-mg/kg dose of OA is not effective in reducing postural tremor in patients with ET at a primary outcome of 80 minutes, but is effective for a secondary outcome after 180 minutes.
RESULTS
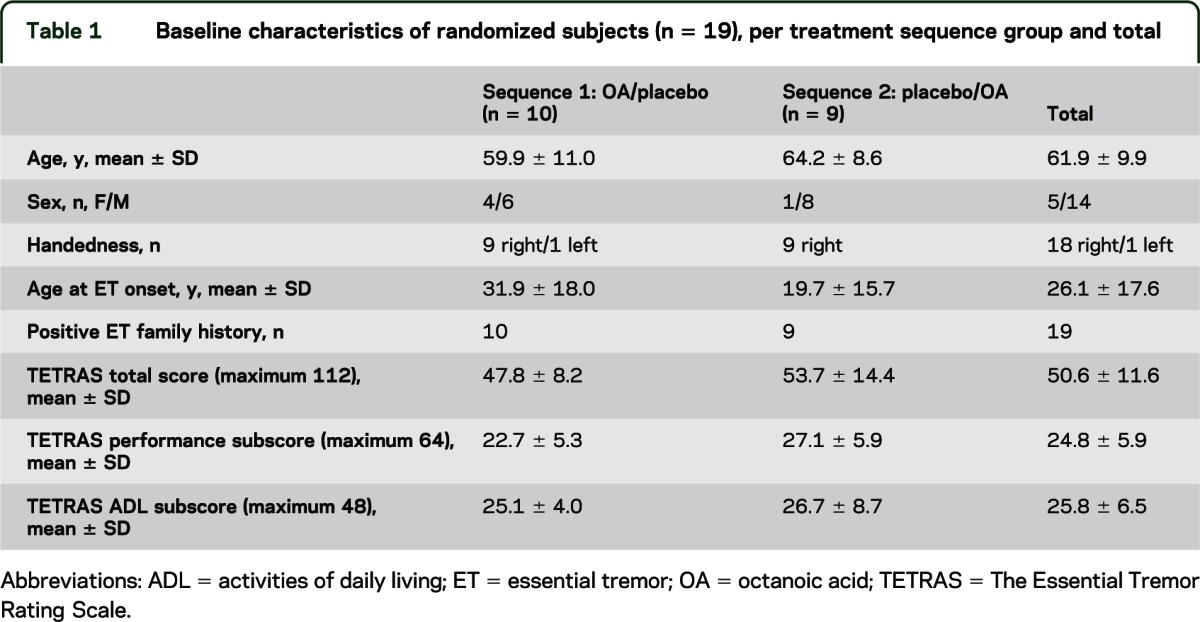
A total of 29 subjects (12 female) were screened for eligibility (figure 2). Recruitment started in June 2009, and the last patient was followed up in August 2010. Ten subjects were considered screening failures for the following reasons: failure to confirm ET according to diagnostic criteria (n = 7), other medical conditions precluding a safe participation (n = 2), or lack of objective alcohol response (n = 1). Nineteen subjects were randomized (table 1). One subject was withdrawn from the study before OA was administered because of an unrelated serious adverse event (SAE). OA was administered to 18 subjects in a mean dose of 352.8 ± 69.9 mg. All 18 subjects completed the trial. At time of offline data processing, the central postural tremor component of one subject's dominant hand recordings was not detectable, but it was present during screening. Because of a lack of the target symptom, this subject was removed from the analysis of the primary outcome, but remained in the cohort for secondary outcome analyses.
Table 1.
Baseline characteristics of randomized subjects (n = 19), per treatment sequence group and total
Primary outcome.
In the analysis of the primary study outcome (weighted condition, dominant hand at t = 80), we found no significant interactions between period (treatment day) and treatment as well as no significant difference between OA and placebo (F1,16 = 0.95, p = 0.345).
Secondary outcomes.
At the final time point 300 minutes after administration, OA significantly improved dominant hand tremor over placebo (F1,16 = 5.49, p = 0.032) with a trend to benefit over placebo starting at t = 150 (F1,16 = 3.43, p = 0.083; figure 3A). At all other time points, differences between the treatment arms were not significant. Across all time points, there was no overall difference between OA and placebo, neither in the weighted nor in the nonweighted accelerometric condition. OA reduced dominant hand postural tremor power up to 41% compared with baseline (t = 240, normalized tremor power 0.59, interquartile range 0.47–1.16, baseline = 1). Using linear mixed-model analysis, digital spiral-analysis measures were not different between OA and placebo.
Figure 3. Octanoic acid effect on tremor power up to 300 minutes after administration.

Normalized tremor power of postural hand tremor at the spectral tremor frequency peak, measured with weighted accelerometry, across time points up to 300 minutes after administration, shown (A) for the dominant hand and (B) both hands. Because individual time-point data were not normally distributed, plot shows median and interquartile range. X-axes represent normalized tremor power (baseline = 1); values >1 represent larger tremor power, <1 reduction in tremor power; *time points with significant differences between octanoic acid and placebo (p < 0.05).
Other outcomes and post hoc analyses.
When analyzing central tremor power at peak frequency of both hands together, a significant benefit of OA over placebo was seen at t = 180 (F1,16 = 6.1, p = 0.025) and t = 300 (F1,16 = 5.57, p = 0.031), with a trend starting at t = 150 (F1,16 = 4.20, p = 0.057; figure 3B). Analysis of benefit ratios of the central tremor component showed that averages of benefit ratios were significantly different favoring OA for the dominant hand (p = 0.001) as well as for both hands together (p < 0.0001). Analysis of benefit ratios in kinetic tremor showed a significant difference on averages of the benefit ratios favoring OA for the dominant hand (p < 0.001).
Safety.
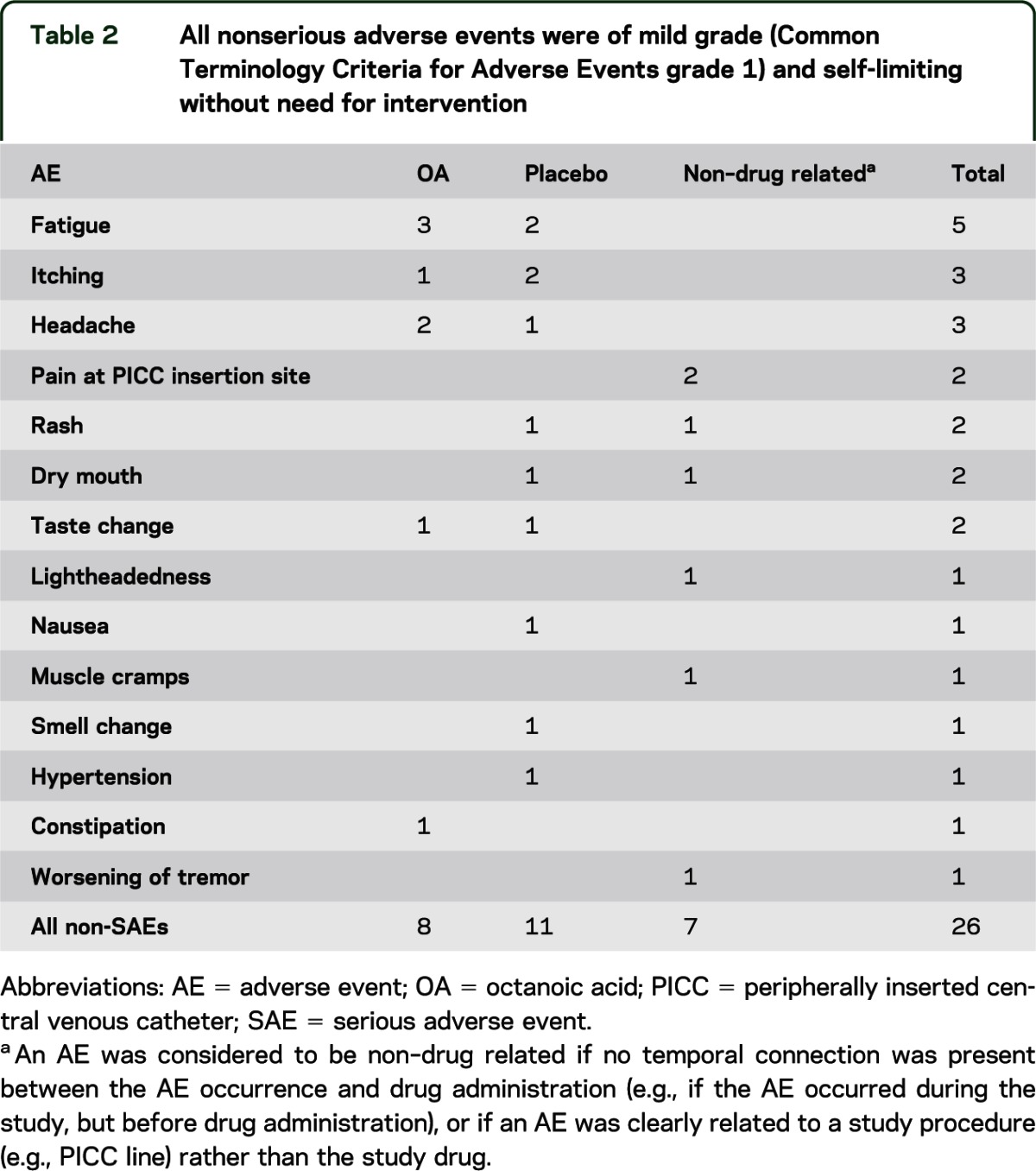
OA was well tolerated. No signs of intoxication were observed. Non-SAEs were mild (Common Terminology Criteria for Adverse Events grade 1), self-limited, and resolved without the need for intervention. They were equally present after the administration of OA and placebo (table 2). Two SAEs were not related to the study drug (1 food-borne illness with consecutive troponin I elevation before OA administration and 1 peripherally inserted central venous line–related SAE). There were no significant changes in vital signs, EKG, or laboratory parameters.
Table 2.
All nonserious adverse events were of mild grade (Common Terminology Criteria for Adverse Events grade 1) and self-limiting without need for intervention
Pharmacokinetics.
Pharmacokinetic parameters obtained using standard noncompartmental analyses (Phoenix™ WinNonlin® 6.0) for all 18 subjects receiving a dose of OA with the corresponding averages and an average concentration-time profile are summarized in appendix e-2. OA was absorbed very quickly: at 5 minutes after administration, OA was already detectable in all subjects, and its average plasma level was 301.1 ng/mL. Maximum concentrations were reached approximately 70 minutes after administration (tmax = 72.8 ± 34.3 minutes). The apparent volume of distribution was relatively large (Vd/F = 389 L), and the average apparent clearance was also relatively high (CL/F = 186.8 L/h). The average elimination half-life was t1/2 = 83.5 minutes, corresponding to an elimination constant of λz = 0.0098 minute−1.
DISCUSSION
In this study, we evaluated OA as a novel therapeutic agent in subjects with alcohol-responsive ET. Objective tremor accelerometry was chosen purposefully as primary efficacy outcome measure to be able to capture possible subclinical effects. Because of interindividual variability in tremor severity, a crossover design was selected so that each subject could be their own control.
The study failed to meet its primary efficacy outcome at 80 minutes after administration, at which time a maximum effect was anticipated. However, an effect of OA was measurable later than expected, and was more pronounced when tremor power of both hands were analyzed together, suggesting a systemic drug effect. The observation of a trend to improvement of tremor at 80 minutes after both OA and placebo as seen in figure 3 might be explained by a placebo effect in both groups, with a larger magnitude than any potential OA effect at that time point, and any true effects of OA separating from placebo at later time points. Because a single time point was chosen as primary outcome to describe the peak effect of this low dose, future trials examining the therapeutic benefit in an outpatient setting should assess longer-term effects of OA.
The crossover design allowed the analysis of effects within each subject. In this exploratory analysis, differences of OA compared with placebo within subjects showed a significant benefit of OA across time.
The average plasma concentration/time profile corresponded well to typical absorption/elimination profiles characteristic for oral administrations. The average elimination half-life and the corresponding elimination constant obtained here are in excellent agreement with our previous pharmacokinetic study of OA after the administration of 1-octanol, which indicated a relatively fast elimination.7 Elimination was not entirely first-order, as the presence of a slower, second phase was noticeable in the elimination time profile (see figure in appendix e-2). The dissociation between the time points of highest plasma level (t = 70 minute) and first measurable effect on tremor vs placebo (t = 180 minute) could be explained by a possible second compartment (e.g., the CNS), which is responsible for the effect after redistribution takes place. Animal studies showed a high permeability of OA across the blood-brain barrier.16 Therefore, a rapid resorption and transport across the blood-brain barrier with prolonged effect in the CNS serves as a possible explanation.
The overall duration of effect was longer than expected, which raises a concern of an adequate washout period between treatment days in a crossover design. Our analysis of the treatment sequence effects, however, did not suggest a carryover effect.
The focus of this study on objective outcome parameters instead of clinical scores might be considered a study limitation. However, because a low dose of OA was administered in patients with ET for this trial, we aimed to capture even possible subclinical effects of reduction in postural hand tremor power that might not yet translate into a reasonable clinical effect in a motor task affected by kinetic tremor. This was eventually the case, because at specific time points, differences were apparent using high-sensitive accelerometry measuring postural tremor, but not in our task of kinetic tremor (spiral drawing). However, our exploratory analysis of within-subject OA effects showing a significant benefit over time in both postural and kinetic tremor conditions can be considered a promising observation. The fact that via accelerometry an effect was measurable at the central tremor peak, but not the overall tremor power between 2 and 15 Hz in the nonweighted condition could be a further argument for a specific CNS effect.
One limitation of the study might be that attention span or reaction time as potential confounder was not assessed formally, although we do not expect a major effect on our primary efficacy outcome measure accelerometry of postural limb tremor in an isometric position. The lack of difference between OA and placebo in the spiral task might also be explained by a training effect. To our knowledge, it is not known whether repeated spiral drawing is leading to amplitude reductions due to a learning or adaptation effect. Future studies on training effects in motor tasks in ET are needed to quantify this potential bias.
Although the exact mechanism of OA in ET remains speculative, this study was built on the effects of ethanol and 1-octanol, where a similar pathophysiologic mechanism within the olivocerebellar circuits could be expected with long-chain alcohols or their acids.17 Because of these considerations, only ethanol responders were recruited for this study. Therefore, our results are only applicable to the subpopulation of ethanol responders.
Although no adverse events clearly associated with OA were observed, because of the small number of patients, our study might be too imprecise to rule out a significant difference in adverse effects frequencies between OA and placebo. Furthermore, because this was a single-dose administration study, it is not known whether any concerns would arise when administered chronically, such as whether any accumulation might occur. However, previous studies on OA as a component of ketogenic diets, taken up to several years, did not mention significant tolerability concerns at the dose levels intended to be effective in ET.9,18
Although the primary outcome parameter was not met, this study showed efficacy in secondary outcome data of OA in a double-blind, placebo-controlled design using objective outcomes. These results warrant future studies to investigate the safety and efficacy of OA at higher doses.
Supplementary Material
GLOSSARY
- ET
essential tremor
- OA
octanoic acid
- SAE
serious adverse event
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dietrich Haubenberger, MD, was responsible for developing the study concept and design, acquisition of data, study coordination, obtaining funding, analysis and interpretation of data, as well as drafting of the manuscript. Gayle McCrossin, RN, was responsible for acquisition of data and study coordination. Codrin Lungu, MD, was responsible for data acquisition. Elaine Considine, RN, was responsible for study coordination. Camilo Toro, MD, was responsible for contribution of vital tools for the study (digital spiral-analysis software), analysis and interpretation of data, as well as critical review of the manuscript. Fatta Nahab, MD, was responsible for contributing to the study concept and design, interpretation of data, as well as critical review of the manuscript. Sungyoung Auh, PhD, was responsible for statistical analysis. Peter Buchwald, PhD, was responsible for the analysis of pharmacokinetic data. George J. Grimes, RPh, was responsible for the formulation of the study drug and conducting the stability analysis. Judith Starling, RPh, was responsible for randomization and drug dispensing. Gopal Potti, PhD, was responsible for the formulation of the study drug and conducting the stability analysis. Linda Scheider, BS, Daniel Kalowitz, BS, Daniel Bowen, BS, and Andrea Carnie, BS, were responsible for contribution to analysis of data. Mark Hallett, MD, was responsible for developing the study concept and design, interpretation of data, obtaining funding, study supervision, and critical review of the manuscript.
STUDY FUNDING
This study was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program, as well as a Cooperative Research and Development Agreement (CRADA, National Institute of Neurological Disorders and Stroke Ref. No. 02036) with Ariston Pharmaceuticals, Inc. Dr. Haubenberger was supported by the Austrian Science Fund (Erwin Schroedinger Fellowship J2783-B09).
DISCLOSURE
D. Haubenberger received research support through the National Institute of Neurological Disorders and Stroke Intramural Research Program and the Austrian Science Fund FWF (Erwin Schroedinger Fellowship, project number J2783-B09). Dr. Haubenberger serves as member of the Medical Advisory Board of the International Essential Tremor Foundation. Dr. Haubenberger received honoraria and conference support from Ipsen and UCB. G. McCrossin reports no disclosures. C. Lungu, E. Considine, and C. Toro report no disclosures. F. Nahab has received research support from the International Essential Tremor Foundation, National Institute of Neurological Disorders and Stroke/NIH, and is an inventor for patent applications of 1-octanol and octanoic acid filed by the National Institute of Neurological Disorders and Stroke/NIH and Ariston. S. Auh, P. Buchwald, G. Grimes, J. Starling, G. Potti, L. Scheider, D. Kalowitz, and A. Carnie report no disclosures. M. Hallett serves as Chair of the Medical Advisory Board for and receives funding for travel from the Neurotoxin Institute; serves as Chair of the Medical Advisory Board of the Benign Essential Blepharospasm Foundation and Chair of the Medical Advisory Board of the International Essential Tremor Foundation; may accrue revenue on US Patent #6,780,413 B2 (issued August 24, 2004): immunotoxin (MAB-Ricin) for the treatment of focal movement disorders; US Patent #7,407,478 (issued August 5, 2008): coil for magnetic stimulation and methods for using the same; receives research support from Ariston Pharmaceuticals, NIH/National Institute of Neurological Disorders and Stroke (Intramural Program), and the US Department of Defense (Army); has received license fee payments from the NIH (from Brainsway) for licensing the patent for the H-coil. Dr. Hallett's research at the NIH is largely supported by the NIH Intramural Program. Supplemental research funds are provided by the US Army via the Henry Jackson Foundation, Ariston Pharmaceutical Company via a Cooperative Research and Development Agreement (CRADA) with the NIH, and the Kinetics Foundation via a Clinical Trials Agreement with the NIH. Dr. Hallett is an inventor for patent applications of 1-octanol and octanoic acid filed by National Institute of Neurological Disorders and Stroke/NIH and Ariston. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Deuschl G, Wenzelburger R, Löffler K, Raethjen J, Stolze H. Essential tremor and cerebellar dysfunction: clinical and kinematic analysis of intention tremor. Brain 2000;123(pt 8):1568–1580 [DOI] [PubMed] [Google Scholar]
- 2.Koller WC, Busenbark K, Miner K. The relationship of essential tremor to other movement disorders: report on 678 patients. Essential Tremor Study Group. Ann Neurol 1994;35:717–723 [DOI] [PubMed] [Google Scholar]
- 3.Lou JS, Jankovic J. Essential tremor: clinical correlates in 350 patients. Neurology 1991;41:234–238 [DOI] [PubMed] [Google Scholar]
- 4.Hague S, Rogaeva E, Hernandez D, et al. Early-onset Parkinson's disease caused by a compound heterozygous DJ-1 mutation. Ann Neurol 2003;54:271–274 [DOI] [PubMed] [Google Scholar]
- 5.Bushara KO, Goldstein SR, Grimes GJ, Burstein AH, Hallett M. Pilot trial of 1-octanol in essential tremor. Neurology 2004;62:122–124 [DOI] [PubMed] [Google Scholar]
- 6.Shill HA, Bushara KO, Mari Z, Reich M, Hallett M. Open-label dose-escalation study of oral 1-octanol in patients with essential tremor. Neurology 2004;62:2320–2322 [DOI] [PubMed] [Google Scholar]
- 7.Nahab FB, Wittevrongel L, Ippolito D, et al. An open-label, single-dose, crossover study of the pharmacokinetics and metabolism of two oral formulations of 1-octanol in patients with essential tremor. Neurotherapeutics 2011;8:753–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nahab FB, Handforth A, Brown T, et al. Octanoic acid suppresses harmaline-induced tremor in mouse model of essential tremor. Neurotherapeutics 2012;9:635–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sills MA, Forsythe WI, Haidukewych D, MacDonald A, Robinson M. The medium chain triglyceride diet and intractable epilepsy. Arch Dis Child 1986;61:1168–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deuschl G, Raethjen J, Hellriegel H, Elble R. Treatment of patients with essential tremor. Lancet Neurol 2011;10:148–161 [DOI] [PubMed] [Google Scholar]
- 11.Louis ED, Rios E, Henchcliffe C. How are we doing with the treatment of essential tremor (ET)? Persistence of patients with ET on medication: data from 528 patients in three settings. Eur J Neurol 2010;17:882–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Louis ED, Barnes L, Albert SM, et al. Correlates of functional disability in essential tremor. Mov Disord 2001;16:914–920 [DOI] [PubMed] [Google Scholar]
- 13.Deuschl G, Bain P, Brin M. Consensus statement of the movement disorder society on tremor. Ad Hoc Scientific Committee. Mov Disord 1998;313 suppl:2–23 [DOI] [PubMed] [Google Scholar]
- 14.Elble R, Lewitt PA, Lyons K, et al. Reliability of the essential tremor rating assessment scale (TETRAS). Mov Disord 2012;27(suppl 1):S409 [Google Scholar]
- 15.Haubenberger D, Kalowitz D, Nahab FB, et al. Validation of digital spiral analysis as outcome parameter for clinical trials in essential tremor. Mov Disord 2011;26:2073–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oldendorf WH. Carrier-mediated blood-brain barrier transport of short-chain monocarboxylic organic acids. Am J Physiol 1973;224:1450–1453 [DOI] [PubMed] [Google Scholar]
- 17.Mostile G, Jankovic J. Alcohol in essential tremor and other movement disorders. Mov Disord 2010;25:2274–2284 [DOI] [PubMed] [Google Scholar]
- 18.Sills MA, Forsythe WI, Haidukewych D. Role of octanoic and decanoic acids in the control of seizures. Arch Dis Child 1986;61:1173–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.