

Abstract
Objective:
To model the temporal trajectory of β-amyloid accumulation using serial amyloid PET imaging.
Methods:
Participants, aged 70–92 years, were enrolled in either the Mayo Clinic Study of Aging (n = 246) or the Mayo Alzheimer's Disease Research Center (n = 14). All underwent 2 or more serial amyloid PET examinations. There were 205 participants classified as cognitively normal and 55 as cognitively impaired (47 mild cognitive impairment and 8 Alzheimer dementia). We measured baseline amyloid PET-relative standardized uptake values (SUVR) and, for each participant, estimated a slope representing their annual amyloid accumulation rate. We then fit regression models to predict the rate of amyloid accumulation given baseline amyloid SUVR, and evaluated age, sex, clinical group, and APOE as covariates. Finally, we integrated the amyloid accumulation rate vs baseline amyloid PET SUVR association to an amyloid PET SUVR vs time association.
Results:
Rates of amyloid accumulation were low at low baseline SUVR. Rates increased to a maximum at baseline SUVR around 2.0, above which rates declined—reaching zero at baseline SUVR above 2.7. The rate of amyloid accumulation as a function of baseline SUVR had an inverted U shape. Integration produced a sigmoid curve relating amyloid PET SUVR to time. The average estimated time required to travel from an SUVR of 1.5–2.5 is approximately 15 years.
Conclusion:
This roughly 15-year interval where the slope of the amyloid SUVR vs time curve is greatest and roughly linear represents a large therapeutic window for secondary preventive interventions.
The most well-established biomarkers of Alzheimer disease (AD) at this time can be divided into 2 major categories: 1) measures of neuronal injury and degeneration1–4 and 2) measures of brain amyloid beta (Aβ) deposition including CSF Aβ421,2 and PET amyloid imaging.5–7 Some of us recently proposed a model describing the temporal evolution of AD biomarkers.8 Based on autopsy9 and in vivo amyloid PET studies10–12 available at the time we hypothesized that amyloid load follows a sigmoid trajectory as a function of time.8 A sigmoid magnitude vs time shape implies that the rate of amyloid accumulation in the brain initially accelerates then later decelerates. Our objective in this article was to test the hypothesis that amyloid PET load vs time can be reasonably modeled by a sigmoid function.
METHODS
Participants.
Participants were drawn from either the Mayo Clinic Study of Aging (MCSA) or the Mayo Alzheimer's Disease Research Center (ADRC). The MCSA (total current enrollment = 1,847) is a longitudinal population-based observational study of cognitive aging that was established in Olmsted County, Minnesota, in 2004. MCSA subjects were 70 years or older and did not have dementia at the time of enrollment. The ADRC (total current enrollment = 421) is a longitudinal observational sample of subjects recruited from the behavioral neurology practice at Mayo Clinic.
To be included, participants must have had 2 or more serial amyloid PET studies, and have clinical diagnoses of cognitively normal (CN), mild cognitive impairment (MCI), or AD dementia. Otherwise there were no other exclusion criteria. The amyloid PET studies were obtained over the period March 2006–July 2012. A total of 260 participants met these inclusion criteria (205 CN, 47 MCI, and 8 AD) and all were included in these analyses. Of these 260, 220 participants had 2 imaging visits, 38 had 3, and 2 had 4. Participants were categorized as impaired (defined as either MCI or AD dementia) or CN. All 205 CN participants were from the MCSA. The impaired group (n = 55) was composed of 41 MCI from the MCSA, 6 MCI from the ADRC, and 8 AD dementia from the ADRC.
Standard protocol approvals, registrations, and patient consents.
These studies were approved by the Mayo institutional review board and informed consent was obtained from all participants or their surrogates.
PET methods.
PET images were acquired using a PET/CT scanner. The 11C Pittsburgh compound B–PET scan consisting of 4 5-minute dynamic frames was acquired from 40 to 60 minutes after injection. Image analysis was done using our in-house fully automated image processing pipeline.12 A cortical amyloid PET retention ratio (SUVR) was formed by calculating the median uptake over voxels in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus regions of interest (ROIs) for each subject and dividing this by the median uptake over voxels in the cerebellar gray matter ROI of the atlas.13 The atlas and image registration steps were based on a 3D T1-weighted volume MRI scan.
The primary analyses were performed with MRI-based partial volume-corrected (PVC) amyloid PET. PVC was accomplished by creating voxel-wise tissue probability maps for each participant's MRI.12 Each PET voxel was divided by the estimated proportion of gray plus white matter in the voxel. While we did employ PVC for all reported PET analyses, we found the effect of PVC to be negligible. Lin concordance coefficient between PVC and non-PVC amyloid PET SUVR was 0.95 at baseline and 0.94 for annual change in SUVR.
Statistical methods.
We used 2-sample t tests and χ2 tests to assess differences between the CN and impaired groups.
We estimated an intraclass correlation (ICC) to evaluate the measurement error associated with longitudinal amyloid PET SUVR measurements by fitting a random-intercepts plus random-slopes linear mixed effect model among CN participants, with time as a fixed effect. This model removes from the repeated measurements within each subject the variability due to a linear rate of change in SUVR and partitions the remaining variation as follows: 1) variability due to intersubject differences in baseline SUVR, denoted by 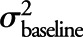 ; 2) variability due to intersubject differences in rates of accumulation, denoted by
; 2) variability due to intersubject differences in rates of accumulation, denoted by 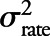 ; and 3) residual variability primarily consisting of measurement error, denoted by
; and 3) residual variability primarily consisting of measurement error, denoted by 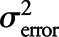 . We report the ICC as
. We report the ICC as 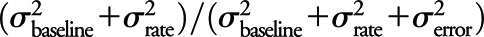 and report the residual standard error σerror by itself and scaled by the median SUVR of 1.38, using this last quantity as our metric of relative measurement error.
and report the residual standard error σerror by itself and scaled by the median SUVR of 1.38, using this last quantity as our metric of relative measurement error.
For our primary analysis, using all available time points with amyloid PET SUVR measured, we fit a slope for each subject to represent the annual amyloid accumulation rate. We then used linear regression models to predict these subject-specific rates of amyloid accumulation given the participants' baseline amyloid PET SUVR. Age, sex, APOE genotype, and baseline clinical diagnosis were evaluated as covariates. Baseline amyloid PET SUVR was modeled with a restricted cubic spline with 4 knots to allow the accumulation rate to vary nonlinearly with baseline SUVR. The knots were placed at SUVR levels of 1.3, 1.5, 2.0, and 2.5 and chosen to allow for flexibility at SUVR that were interpretable and of interest. In our parameterization of the restricted cubic splines there is a coefficient representing the linear component of the predictor and 2 coefficients representing the nonlinear components. We tested linearity with an F test by testing the null hypothesis that both nonlinear coefficients were zero. We used a modified Euler method to solve the first-order differential equation for amyloid accumulation rate vs baseline amyloid PET SUVR as described by the fitted spline function and subject to the condition that time would be set to 0 for an SUVR of 1.3. In this process, we had fixed SUVR, and solved for times between those values. The resulting integral curve was smooth and continuous, with amyloid PET SUVR on the vertical axis, and predicted time from a value of 1.3 on the horizontal axis.
RESULTS
Age, sex, or education did not differ between CN and impaired participants (table). As expected, the impaired group had a higher proportion of APOE ε4 carriers and had a lower median Mini-Mental State Examination than the CN group. Baseline amyloid PET SUVR was higher in impaired participants (p < 0.001) and in APOE ε4 carriers (p < 0.001). Age and sex were not associated with baseline amyloid load.
Table.
Characteristics overall and by clinical diagnosis
In our mixed effects model analysis, we observed a high ICC of 0.99. The residual standard error was found to be σerror = 0.038. This value, when divided by the median SUVR among controls of 1.38, is approximately 3%. An ICC of 0.99, coupled with a relative measurement error of 3%, suggests that measurement error was quite small.
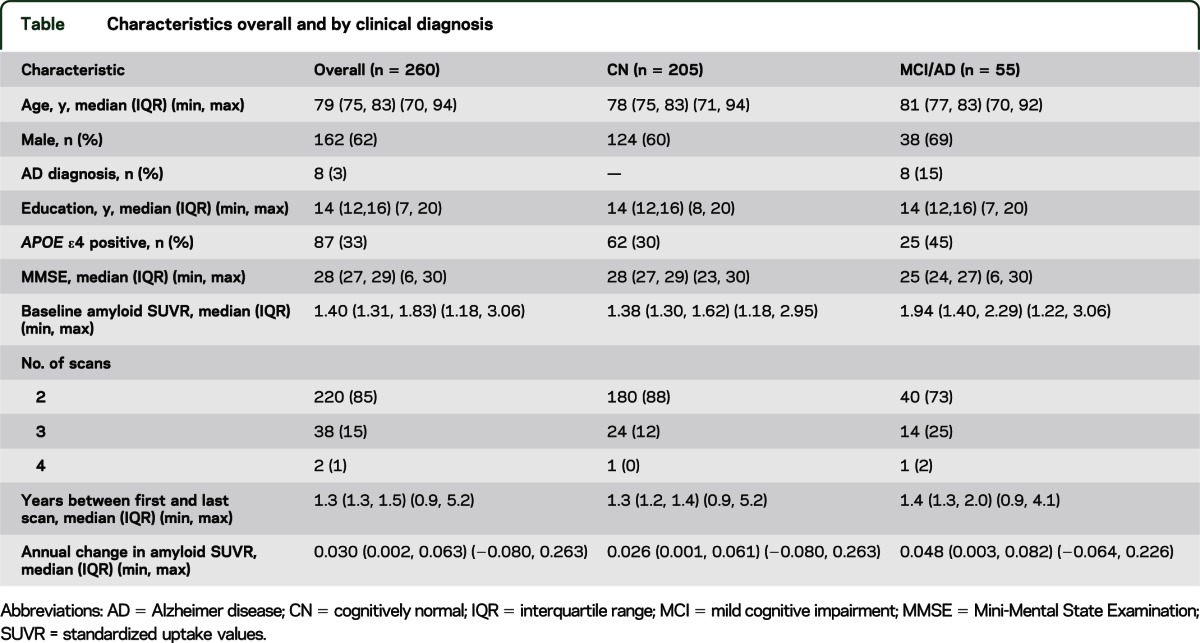
Including all participants, the rate of amyloid accumulation as a function of baseline amyloid PET SUVR had an inverted U shape (p < 0.001, test of nonlinearity) (figure 1). Rates of accumulation were low at low baseline amyloid PET SUVR. Rates increased to a maximum at baseline SUVR around 2.0 (95% confidence interval [CI] 1.9–2.1), above which rates declined—reaching amyloid accumulation rates near zero at baseline SUVR greater than 2.7.
Figure 1. Rates as a function of baseline amyloid standardized uptake values.
Scatterplot of annual rate of change of amyloid PET standardized uptake values (SUVR) vs baseline amyloid PET SUVR for all subjects (n = 260). The estimated mean slope from a linear regression model is shown with 95% confidence limits. Age, sex, diagnosis, and APOE genotype were not significant covariates in the model and are therefore not included here. Cognitively normal (CN) participants who are APOE ε4− are represented by light blue circles, CN participants who are APOE ε4+ are represented by dark blue plus signs, impaired participants who are APOE ε4− are represented by light orange circles, and impaired participants who are APOE ε4+ are represented by dark orange plus signs. AD = Alzheimer disease; MCI = mild cognitive impairment.
We evaluated age, sex, APOE genotype, and clinical diagnosis as potential covariates in the model predicting rate of accumulation as a function of baseline SUVR. Age was not associated with rate of amyloid accumulation. A trend for higher rates of amyloid accumulation among men (p = 0.06) was attenuated after including baseline amyloid PET SUVR in the model (p = 0.11). Similarly, a trend for higher rates of amyloid accumulation among impaired vs CN participants (p = 0.06) was attenuated after including baseline amyloid PET SUVR (p = 0.23) in the model. Rates of amyloid accumulation were greater in APOE ε4 carriers than noncarriers (p < 0.001) and remained nearly significant after including baseline amyloid PET SUVR in the model (p = 0.05). There were no significant interactions with age, sex, APOE genotype, or clinical diagnosis and baseline amyloid PET SUVR on rate of amyloid accumulation. Therefore, our final model did not include any of these covariates.
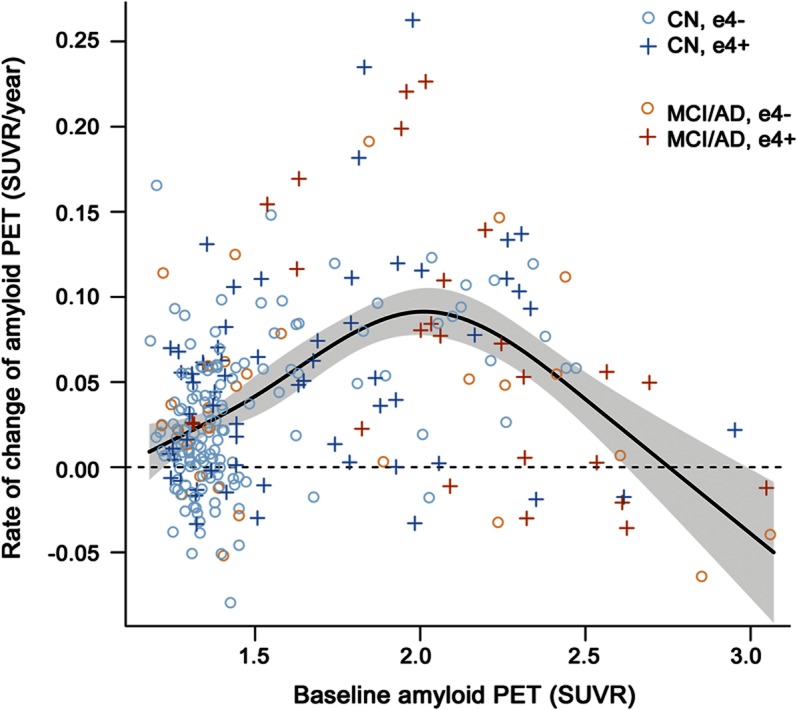
In figure 2A, we plot the rate of amyloid accumulation per year as a function of baseline amyloid load in SUVR units. In figure 2B, we plot the integral with respect to time of the data in figure 2A, which models amyloid level in SUVR units as a function of increasing time in years. These plots are provided based on fits among all participants (n = 260, in red) and also among only those participants whose baseline amyloid PET was 1.3 SUVR or greater (n = 212, in blue). However, truncating the lower limit at 1.3 had little effect on either the plots of rate of amyloid accumulation vs baseline amyloid SUVR, or of amyloid SUVR vs time (figure 2, A and B). For both the lower truncated (at 1.3 SUVR) and nontruncated analyses, the upper SUVR in the model was truncated at 2.7 due to sparseness of the data at SUVR values higher than this.
Figure 2. Relating the inverted U-shaped amyloid rates as a function of baseline standardized uptake values to sigmoid-shaped trajectory of amyloid accumulation with time.
(A) The rate of amyloid accumulation per year as a function of baseline amyloid load in standardized uptake value (SUVR) units. The red plot in A is the same as the plot in figure 1, except for the upper bound truncation (see below). (B) The integral with respect to time of the data in (A) which models amyloid level in SUVR units as a function of increasing time in years. These plots are provided for all participants (n = 260, in red) and for only those participants whose baseline amyloid PET was 1.3 SUVR or greater (n = 212, in blue). For both the lower truncated (at 1.3 SUVR) and nontruncated analyses, the upper SUVR in the model was truncated at 2.7.
The findings above imply a sigmoidal relationship between amyloid load and time. This sigmoidal relationship is not directly tied to a specific age—1 subject might start increasing on the curve at 70, while another might start increasing on the curve at 80. Consequently, the x axes in figures 2B and 3 are in units of time, not age. Our data suggest that the midpoint in the sigmoid function (i.e., where rates of accumulation reach a maximum and beyond which begin to decelerate) occurs at amyloid PET SUVR around 2.0 (95% CI 1.9–2.1) and that amyloid load approaches a horizontal asymptote (i.e., plateaus) at SUVR somewhat above 2.7 (figure 2).
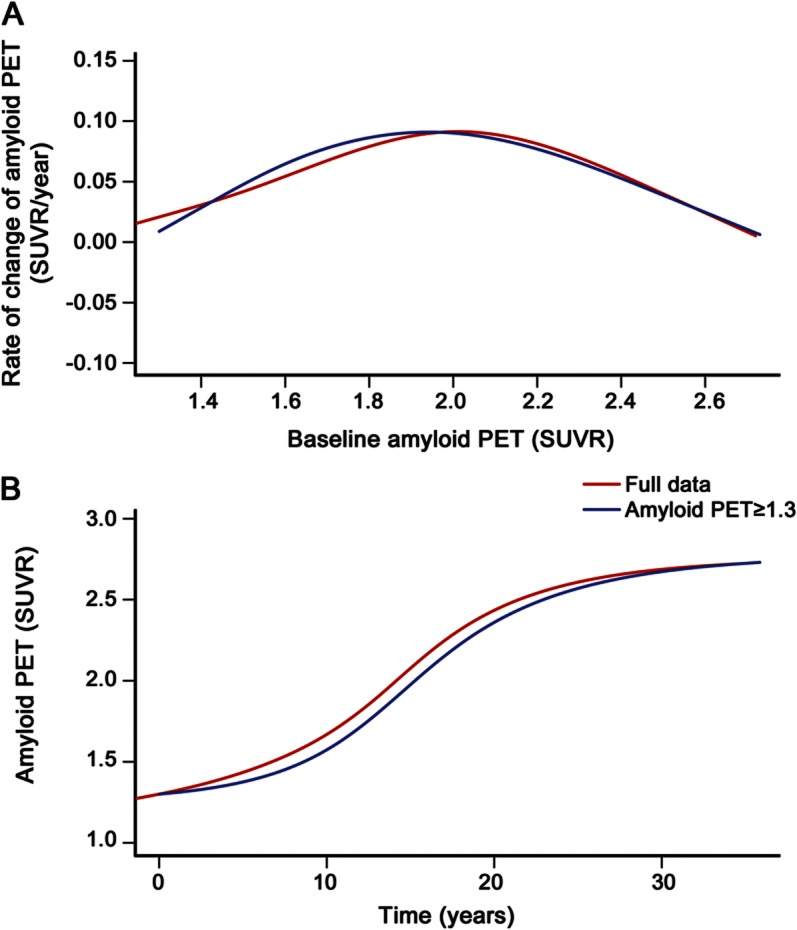
Figure 3. Estimates of time required to travel from a baseline amyloid PET load of 1.5 standardized uptake values to greater values.
This is illustrated in the plot of amyloid PET load vs time in participants whose baseline standardized uptake value (SUVR) was greater than or equal to 1.3 (n = 212, in blue from figure 2B).
Symmetric bracket points in the sigmoid curve around the midpoint of 2.0 where the rate is maximum, and over which the function is fairly linear, occur at SUVR of roughly 1.5 and 2.5. In figure 3, we provide estimates of the time (in years) required to travel from a baseline amyloid PET SUVR of 1.5 to a series of higher values among those participants whose baseline SUVR was greater than or equal to 1.3. The average estimated time required to travel from an SUVR of 1.5–2.5 is approximately 15 years.
For reference purposes, we plotted within-participant trajectories (spaghetti plots) of amyloid accumulation as a function of age among all 195 participants (figure 4).
Figure 4. Within-subject trajectories of amyloid PET standardized uptake values by age for all subjects (n = 260).
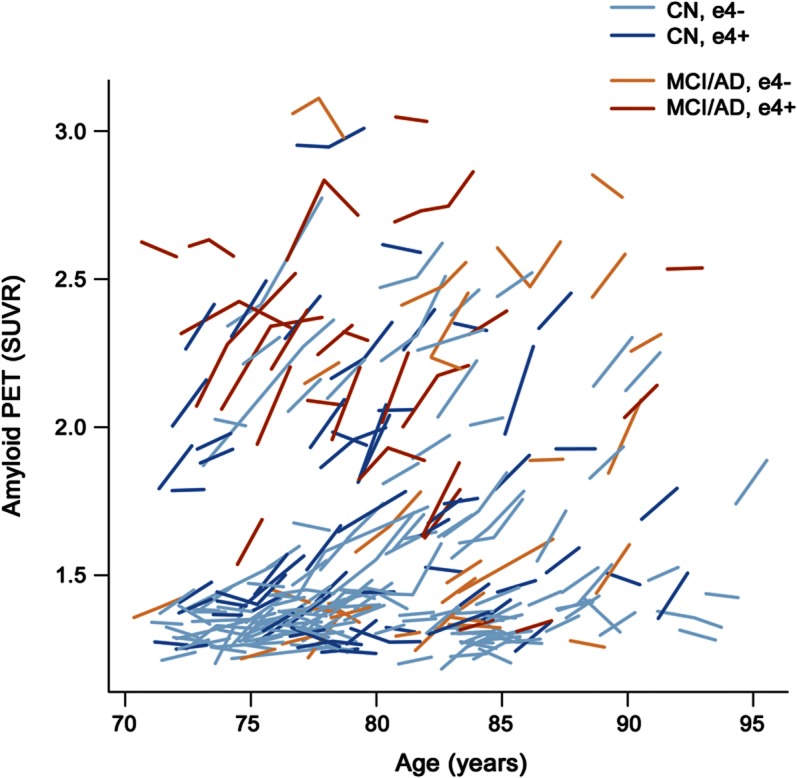
Cognitively normal (CN) participants are represented by blue lines and impaired participants by orange lines. Lighter colored lines represent APOE ε4− participants and darker colors represent APOE ε4+ participants. AD = Alzheimer disease; MCI = mild cognitive impairment.
DISCUSSION
Ideal conditions for modeling AD biomarker trajectories require collecting a complete data battery at multiple time points in many individuals over the entire course of the disease. Given that the course of the disease evaluable by currently available biomarkers may span 30 or more years it will take decades to collect such an idealized dataset. An alternative, however, is to piece together shorter time interval data in many participants to create plausible long-term disease models. This is only possible, however, if the individual short-interval series of within-subject observations can be pieced together in a way that places each subject's observations in an appropriate order along a continuous measure of pathophysiologic severity. We believe that indexing individual participants by amyloid PET SUVR provides a valid solution to this problem in the current context where we are evaluating an amyloid biomarker. While each individual subject's PET trajectory was assumed to be linear, this does not preclude making nonlinear inferences about amyloid deposition vs time over the duration of the disease. The median time from first to last scan was 1.3 years, which represents a tiny fraction of the total duration of the disease. As the sampling interval becomes shorter, all nonlinear functions can be approximated locally by a linear fit.
A different approach to AD biomarker modeling that we and others have pursued previously is to index participants by degree of cognitive impairment.14–17 This approach is flawed, however, because all cognitive tests have floor and ceiling effects. Cognitive function is also mediated by cognitive reserve, rendering the relationship between biomarker measures of underlying brain pathophysiology and cognition variable.18–20
Age likewise is not a reasonable way to measure AD pathophysiologic severity. In a given elderly cohort, some participants will have entered the AD pathophysiologic pathway and some will not. Among those who have, individual participants will have entered at different ages. Therefore combining different participants who are indexed by age along the x axis does not model amyloid load as a function of disease severity and may obscure fundamental relationships. For example, while we see evidence of acceleration then deceleration in rates of amyloid accumulation when participants are indexed along the disease severity pathway by baseline amyloid SUVR (figures 1 and 2A), this relationship is not evident when participants are indexed by age, as in figure 4.
Of the major AD biomarkers, only those that measure brain amyloid are specific for AD pathophysiology.21–24 Biomarkers of AD neurodegeneration are also abnormal in non-AD conditions including head trauma and stroke.3 This specificity of amyloid PET allowed us to assess the relationship between baseline amyloid SUVR and rate of change in participants who we were more confident were in the AD pathophysiologic path. Based on recent studies favoring lenient amyloid PET cutpoints23,25 to detect earliest evidence of brain amyloidosis, we used a cutpoint of 1.3 SUVR to select participants (n = 212) for this subanalysis; however, the results in figures 2 and 3 were not noticeably different with higher cutpoints of 1.4–1.5. In addition, as illustrated in figure 2, results were only slightly different when all participants vs only those with baseline SUVR greater than 1.3 were included.
Our data in figure 4 are similar to other reports where amyloid PET magnitude is plotted as a function of age.7,26–29 The fact that, unlike some others,6,26,29,30 we did not see an overall increase in baseline SUVR or rate of amyloid accumulation with age may be due to the absence of individuals younger than 70 years in our dataset. Recent autopsy and amyloid PET data6,28,29 indicate that the late 50s is the age when some subjects, particularly APOE ε4 carriers,6,26 first demonstrate significant amyloid pathophysiology.
Baseline amyloid PET SUVR and rates of amyloid accumulation were greater in APOE ε4 carriers and in impaired participants. These data were largely consistent with existing literature.6,23,26,27,29,31–33 However, with baseline amyloid PET SUVR in the model, the rate of amyloid accumulation was not associated with clinical group and, while close, this association did not meet significance at p < 0.05 for APOE either. This implies that the rate of amyloid accumulation is more closely related to amyloid load than to APOE ε4, cognitive impairment, or age.
Some of our subjects with higher amyloid PET SUVR were impaired while others were not. This is consistent with the concept that the relationship between a given amount of amyloid deposition and clinical impairment is indirect. Cognitive impairment is a direct consequence of neurodegeneration while amyloid deposition is an earlier, “upstream” pathophysiologic event.11,34–37 Moreover, the ultimate clinical result of a given amount of amyloid can be modified by many factors. For a given amount of amyloid, cognitive decline may occur sooner in participants with risk-enhancing exposures such as comorbid brain pathologies, low education, or risk amplification genes. Likewise, decline may be delayed in participants with risk-reducing exposures such as high education18–20 or protective genes.
In figure 2, it is evident that average rates decline monotonically with baseline SUVR for baseline SUVR greater than 2.0. The downward trend in rate above baseline SUVR of 2.0 is primarily driven by the 47 subjects with baseline SUVR between 2.0 and 2.7, not the few subjects with baseline SUVR greater than 2.7. Specific absolute SUVR will likely vary with different PET ligands and different PET image analysis approaches.22,23,29,38 We believe, however, that our conclusions about the biology of AD should not be affected by methodologic variation.
An ICC of 0.99 coupled with a relative measurement error of 3% suggests that the precision of our rate measurements seems more than adequate. Questions may also arise concerning bias in rate measures in impaired subjects. But of the 51 subjects with baseline SUVR greater than 2.0, 25 were CN and 26 were impaired. Thus, the observed decline in rates of amyloid accumulation with baseline SUVR above 2.0 can not be attributed to bias from rate measures only in impaired subjects in this range.
Rates of change were negative in all 4 participants with baseline SUVR greater than 2.7 (figure 1), which might imply that absolute amyloid load declines over time at very high baseline levels. Because the CI modeling rate as a function of baseline SUVR includes 0 at the highest SUVR (figure 1) though, our data do not support a conclusion other than that rates seem to approach zero at the upper range of observed amyloid load. An accumulation rate of 0 by definition implies a plateau in absolute amyloid load.
A sigmoid-shaped trajectory of brain amyloid burden as a function of time could be attributed to 2 different effects. One relates to sensitivity limits of any measurement technique at extremes. Floor (detection) and ceiling (saturation) measurement sensitivity effects would impart a sigmoid shape to a data distribution. A second more profound explanation is that this is a fundamental biologic phenomenon. A sigmoid-shaped function with respect to time would be consistent with reaching a state of equilibrium at high amyloid load.9
The sigmoidal nature of amyloid accumulation with time also has implications for formulating treatment strategies.39,40 The data in figure 3 imply a roughly 15-year interval from an SUVR of 1.5 to 2.5. This period where the slope of the amyloid SUVR vs time curve is greatest and roughly linear represents a large therapeutic window for secondary preventive interventions. However, our data also imply that therapeutic interventions designed to reduce the rate of new amyloid deposition (note the distinction from removing previously deposited amyloid) may be less effective in patients who have already reached plateau levels of amyloid deposition.
GLOSSARY
- AD
Alzheimer disease
- ADRC
Mayo Alzheimer's Disease Research Center
- CI
confidence interval
- CN
cognitively normal
- ICC
intraclass correlation
- MCI
mild cognitive impairment
- MCSA
Mayo Clinic Study of Aging
- PVC
partial volume-corrected
- ROI
region of interest
- SUVR
standardized uptake values
Footnotes
Editorial, page 878
AUTHOR CONTRIBUTIONS
Clifford R. Jack: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data, statistical analysis, study supervision, obtaining funding. Heather Wiste: drafting/revising the manuscript, analysis or interpretation of data, statistical analysis. Timothy G. Lesnick: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, statistical analysis. Stephen D. Weigand: drafting/revising the manuscript, analysis or interpretation of data, statistical analysis. David S. Knopman: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data. Prashanthi Vemuri: drafting/revising the manuscript, analysis or interpretation of data. Vernon Shane Pankratz: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, statistical analysis. Matthew L. Senjem: drafting/revising the manuscript, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data, statistical analysis. Jeffrey L. Gunter: drafting/revising the manuscript, analysis or interpretation of data, statistical analysis. Michelle M. Mielke: drafting/revising the manuscript, analysis or interpretation of data. Val J. Lowe: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data, study supervision, obtaining funding. Bradley F. Boeve: analysis or interpretation of data, acquisition of data. Ronald C. Petersen: drafting/revising the manuscript, obtaining funding.
STUDY FUNDING
Funding sources include the NIH, National Institute on Aging R01 AG11378, P50 AG16574, and U01 AG06786. The General Electric Corporation also provided funding for some imaging studies used in this analysis. Also supported by US NIH Construction Grant (NIH C06 RR018898), The Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation, USA, and the Robert H. and Clarice Smith Alzheimer's Disease Research Program of the Mayo Foundation.
DISCLOSURE
C.R. Jack serves as a consultant for Janssen, Bristol-Meyer-Squibb, General Electric, and Johnson and Johnson, and is involved in clinical trials sponsored by Allon and Baxter, Inc. He receives research funding from the NIH (R01-AG011378, RO1-AG037551, U01-HL096917, U01-AG032438, U01-AG024904) and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation. H. Wiste, T. Lesnick, and S. Weigand report no disclosures. D. Knopman serves as Deputy Editor for Neurology®, serves on a Data Safety Monitoring Board for Lilly Pharmaceuticals, is an investigator in clinical trials sponsored by Janssen Pharmaceuticals, and receives research support from the NIH. P. Vemuri reports no disclosures. V.S. Pankratz is funded by the NIH (R01AG040042, U01AG06786, Mayo Clinic Alzheimer's Disease Research Center/Core C P50AG16574/Core C, and R01AG32990). M. Senjem, J. Gunter, and M. Mielke report no disclosures. V. Lowe serves on scientific advisory boards for Bayer Schering Pharma and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, the NIH (NIA, NCI), the MN Partnership for Biotechnology and Medical Genomics, and the Leukemia & Lymphoma Society. B. Boeve has served as a consultant to GE Healthcare; receives publishing royalties for The Behavioral Neurology of Dementia (Cambridge University Press, 2009); and receives research support from Cephalon, Inc., Allon Therapeutics, Inc., the NIH/NIA, the Alzheimer's Association, and the Mangurian Foundation. R. Petersen reports receiving consulting fees from Elan Pharmaceuticals and GE Healthcare, receiving royalties from Oxford University Press, and serving as chair of data monitoring committees for Pfizer and Janssen Alzheimer Immunotherapy; and receives research support from the NIH/NIA. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Fagan AM, Head D, Shah AR, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol 2009;65:176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jagust WJ, Zheng L, Harvey DJ, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol 2008;63:72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vemuri P, Whitwell JL, Kantarci K, et al. Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage 2008;42:559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 2004;55:306–319 [DOI] [PubMed] [Google Scholar]
- 6.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–1283 [DOI] [PubMed] [Google Scholar]
- 7.Sojkova J, Zhou Y, An Y, et al. Longitudinal patterns of beta-amyloid deposition in nondemented older adults. Arch Neurol 2011;68:644–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004;62:925–931 [DOI] [PubMed] [Google Scholar]
- 10.Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain 2006;129:2856–2866 [DOI] [PubMed] [Google Scholar]
- 11.Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009;132:1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopresti BJ, Klunk WE, Mathis CA, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med 2005;46:1959–1972 [PubMed] [Google Scholar]
- 14.Jack CR, Jr, Vemuri P, Wiste HJ, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol 2011;68:1526–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caroli A, Frisoni GB. The dynamics of Alzheimer's disease biomarkers in the Alzheimer's disease Neuroimaging Initiative cohort. Neurobiol Aging 2010;31:1263–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo RY, Hubbard AE, Shaw LM, et al. Longitudinal change of biomarkers in cognitive decline. Arch Neurol 2011;68:1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jack CR, Jr, Vemuri P, Wiste HJ, et al. Shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch Neurol 2012;69:856–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rentz DM, Locascio JJ, Becker JA, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 2010;67:353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vemuri P, Weigand SD, Przybelski SA, et al. Cognitive reserve and Alzheimer's disease biomarkers are independent determinants of cognition. Brain 2011;134:1479–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reed BR, Mungas D, Farias ST, et al. Measuring cognitive reserve based on the decomposition of episodic memory variance. Brain 2010;133:2196–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain 2008;131:1630–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 2011;305:275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol 2011;68:1404–1411 [DOI] [PubMed] [Google Scholar]
- 24.Sojkova J, Driscoll I, Iacono D, et al. In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol 2011;68:232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mormino EC, Brandel MG, Madison CM, et al. Not quite PIB-positive, not quite PIB-negative: slight PIB elevations in elderly normal control subjects are biologically relevant. Neuroimage 2012;59:1152–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol 2011;69:181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vlassenko AG, Mintun MA, Xiong C, et al. Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C]Pittsburgh compound B data. Ann Neurol 2011;70:857–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigue KM, Kennedy KM, Devous MD, Sr, et al. beta-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology 2012;78:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker JA, Hedden T, Carmasin J, et al. Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol 2011;69:1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jack CR, Jr, Wiste HJ, Vemuri P, et al. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain 2010;133:3336–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci USA 2009;106:6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kantarci K, Lowe V, Przybelski SA, et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology 2012;78:232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vemuri P, Wiste HJ, Weigand SD, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol 2010;67:308–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 2009;461:916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain 2009;132:1310–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hyman BT. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol 2011;68:1062–1064 [DOI] [PubMed] [Google Scholar]
- 38.Villemagne VL, Ong K, Mulligan RS, et al. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J Nucl Med 2011;52:1210–1217 [DOI] [PubMed] [Google Scholar]
- 39.Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med 2011;17:1060–1065 [DOI] [PubMed] [Google Scholar]
- 40.Sperling RA, Jack CR, Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med 2011;3:111cm33. [DOI] [PMC free article] [PubMed] [Google Scholar]