

Abstract
In marked contrast to the variety of strategies available for oxidation and nucleophilic functionalization of methylene groups adjacent to amines, relatively few approaches for modification of this position with electrophilic reaction partners have been reported. In the course of an investigation of the reactions of photogenerated α-amino radicals with electrophiles, we made the surprising observation that the efficiency of radical photoredox functionalization of N-aryl tetrahydroisoquinolines is dramatically increased in the presence of a Brønsted acid co-catalyst. Optimized conditions provide high yields and efficient conversion to radical addition products for a range of structurally modified tetrahydroisoquinolines and enones using convenient household light sources and commercially available Ru(bpy)3Cl2 as a photocatalyst. Our investigations into the origins of this unexpected additive effect have demonstrated that the carbon–carbon bond-forming step is accelerated by TFA and is a rare example of Brønsted acid catalysis in radical addition reactions. Moreover, a significant conclusion arising from these studies is the finding that product formation is dominated by radical chain processes and not by photocatalyst turnover. Together, these findings have important implications for the future design and mechanistic evaluation of photocatalytic radical processses.
Introduction
The α-functionalization of amines has been a challenge of enduring interest to synthetic chemists (Figure 1).1 It has long been known that a variety of amines undergo facile one-electron oxidation and that C–H groups adjacent to the resulting amine radical cations (2) are significantly acidified. Deprotonation affords α-amino radicals (3), which can undergo a second one-electron oxidation to produce iminium ions (4) that are susceptible to attack by a variety of nucleophiles.2,3 The oxidation of amines to iminium ions has been accomplished electrochemically,3a,b using stoichiometric oxidants,3c–e photochemically,3f,g and most recently via transition metal catalyzed oxidation.3d In a seminal contribution, Stephenson demonstrated that transition metal photoredox catalysts could effectively oxidize tetrahydroisoquinolines to the corresponding iminium electrophiles, enabling nucleophilic substitution of the position adjacent to the amine.4,5,6 We became curious if interception of the putative α-amino radical intermediate might enable the complementary electrophilic functionalization of the same position under similar photocatalytic conditions.7
Figure 1.

Nucleophilic and electrophilic reactivity accessible from oxidation of amines.
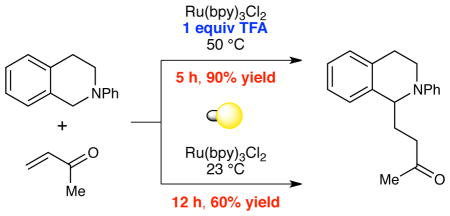
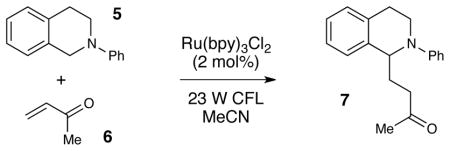
Indeed, while we were conducting our initial explorations of this hypothesis, Pandey and Reiser reported that the α-amino radical reactivity of tetrahydroisoquinolines was accessible using photoredox catalysis (Figure 2).8 In their study, the authors observed that when a mixture of tetrahydroisoquinoline 5 and methyl vinyl ketone 6 is irradiated with high-intensity blue LED lights for 20–24 h in the presence of 5 mol% [Ir(ppy)2(dtb-bpy)]PF6 (8•PF6), the radical addition product 7 can be isolated in 68% yield.9 The same reaction conducted using commercially available Ru(bpy)3Cl2 (9•Cl2), however, affords a somewhat lower yield of 7 (58%).
Figure 2.

Reactions of photocatalytically generated α-aminoradicals (Pandey and Reiser, reference 7).
These results reported by Pandey and Reiser were in accord with parallel observations made in our own laboratory at the time. In the process of optimizing for better yields and shorter reaction times, we have discovered that a Brønsted acid additive provides a dramatic improvement in the efficiency of this transformation. As a result, we have been able to design a photocatalytic radical amine functionalization reaction that can be conducted using commercially available Ru(bpy)3Cl2 in place of Ir(ppy)2(dtb-bpy)PF6 and convenient household light sources rather than LEDs. Further, our efforts to understand the origins of the beneficial effect of the Brønsted acid additive have resulted in fundamental insights relevant to the catalysis of radical reactions and to the mechanisms of photocatalytic processes.
Results and Discussion
Our exploratory investigations are summarized in Table 1. Consistent with the Pandey–Reiser report,8 the photoreaction of 5 and 6 in the presence of Ru(bpy)3Cl2 was sluggish and stalled before complete consumption of 5 (entry 1). We speculated that increasing the ionic strength of the solution might support formation of the charged radical cation intermediate;10 however, addition of various electrolytes failed to significantly improve the yield of the reaction (e.g., entry 2). Similarly, we hoped that base co-catalysts might promote deprotonation of the amine radical cation,11 but to our surprise, we observed little effect on the efficiency of the transformation (entry 3). On the other hand, Brønsted acidic co-catalysts had a significant but complex impact on the reaction (entries 4–6), and in particular, the addition of 1 equiv of TFA enabled the reaction to proceed to completion within 12 h (entry 5). Upon further optimization, we found that the equivalents of methyl vinyl ketone could be lowered (entry 7), and that conducting the reaction at 50 °C provided excellent yield of the radical coupling product in just 5 h (entry 8). Control experiments verified the importance of each reaction component. When the reaction is conducted at 50 °C without TFA, the yield decreases dramatically (entry 9). Experiments conducted in the absence of either photocatalyst or light fail to generate any product (entries 10 and 11).
Table 1.
Optimization and control studiesa
| |||||
|---|---|---|---|---|---|
| entry | additive | equiv 6 | temp | time | yieldb |
| 1 | none | 4 | 23 °C | 12 h | 60% |
| 2 | NaBF4 | 4 | 23 °C | 12 h | 68% |
| 3 | NaHCO3 | 4 | 23 °C | 12 h | 70% |
| 4 | HCO2H | 4 | 23 °C | 12 h | 50% |
| 5 | CF3CO2H | 4 | 23 °C | 12 h | 88% |
| 6 | TsOH | 4 | 23 °C | 12 h | 38% |
| 7 | CF3CO2H | 2 | 23 °C | 12 h | 98% |
| 8 | CF3CO2H | 2 | 50 °C | 5 h | 96% |
| 9 | none | 2 | 50 °C | 5 h | 28% |
| 10c | CF3CO2H | 2 | 50 °C | 5 h | 0% |
| 11d | CF3CO2H | 2 | 50 °C | 5 h | 0% |
Unless otherwise noted, reactions were conducted using 2 mol% Ru(bpy)3Cl2 and 1 equiv of additive in degassed MeCN (0.25 M) and were irradiated using a 23 W compact fluorescent light bulb at a distance of 30 cm.
Yields determined by 1H NMR using an internal standard.
Reaction conducted in the absence of Ru(bpy)3Cl2.
Reaction conducted in the dark.
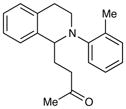
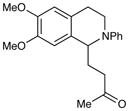
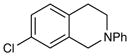
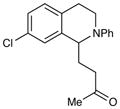
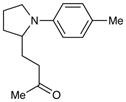
An exploration of the scope of the radical coupling under these conditions (Table 2) suggests that N-aryl tetrahydroisoquinolines are particularly well suited to this chemistry, in agreement with the scope of the oxidative functionalization reactions reported by Stephenson.4 Substrates bearing N-aryl moieties of varying electronic nature react in high yields (entries 1–4), although ortho substitued N-arenes significantly retard the rate of reaction (entry 5). The isoquinoline ring system, similarly, can tolerate both electron-donating and electron-withdrawing substitution (entries 6 and 7). However, N-alkyl substituted isoquinolines do not react under these conditions, and other heterocyclic N-aryl amines exhibit diminished reactivity (entry 8).
Table 2.
Reactions of structurally varied amines wtih 6.a
| entry | amine | product | time | yieldb |
|---|---|---|---|---|
|
|
|||
| 1 | R = H | 5 h | 90% | |
| 2 | R = Me | 3 h | 97% | |
| 3 | R = OMe | 24 h | 94% | |
| 4 | R = Cl | 18 h | 99% | |
| 5 |
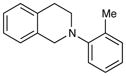
|
|
72 h | 12%c |
| 6 |
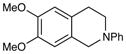
|
|
24 h | 97% |
| 7 |
|
|
18 h | 96% |
| 8 |
|
|
36 h | 30%c |
Unless otherwise noted, reactions were conducted using 2 mol% Ru(bpy)3Cl2, 2 equiv of MVK, and 1 equiv of TFA in degassed MeCN (0.25 M) at 50 °C and were irradiated using a 23 W compact fluorescent light bulb at a distance of 30 cm.
Values represent the averaged isolated yields of two reproducible experiments unless otherwise noted.
Yield determined by 1H NMR using an internal standard.
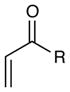
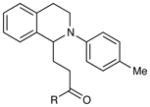
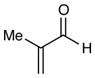
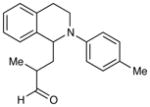
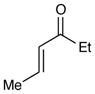
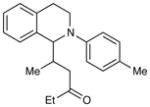
Experiments exploring the generality of the reaction with respect to the Michael acceptor are summarized in Table 3. Both aliphatic and aromatic enones react efficiently (entries 1 and 2). Acrylate esters also participate but with poor efficiency, consistent with their lower electrophilicity (entry 3). Aldehydes are excellent reaction partners and provide very high yields of the product in low reaction times (entries 4–5). Unfortunately, reactions with substituted enones provide little diastereocontrol (entry 5), and β-substituted enones react at significantly diminished rates (entry 6).
Table 3.
Reactions of Michael acceptors with tetrahydroisoquinoline 5.a
| entry | enone | product | time | yieldb (d.r.)c |
|---|---|---|---|---|
|
|
|||
| 1 | R = Et | 4 h | 99% | |
| 2 | R = Ph | 2 h | 91% | |
| 3 | R = OMe | 1 h | 12%d | |
| 4 | R = H | 1 h | 96% | |
| 5 |
|
|
5 h | 93% (2:1 d.r.) |
| 6 |
|
|
72 h | 46%d (2:1 d.r.) |
Unless otherwise noted, reactions were conducted using 2 mol% Ru(bpy)3Cl2, 2 equiv of MVK, and 1 equiv of TFA in degassed MeCN (0.25 M) at 50 °C and were irradiated using a 23 W compact fluorescent light bulb at a distance of 30 cm.
Values represent the averaged isolated yields of two reproducible experiments unless otherwise noted.
Diastereomer ratios determined by 1H NMR.
Yield determined by 1H NMR using an internal standard.
Collectively, these results indicate that the use of the TFA additive confers several practical benefits. The photocatalytic radical functionalization of tetrahydroisoquinolines is markedly more efficient in the presence of the Brønsted acid co-catalyst, providing uniformly higher yields with shorter reaction times. In addition, these conditions utilize a commercially available Ru(bpy)32+ photocatalyst in place of the more precious iridium chromophore and a standard household light bulb in place of a high-intensity monochromatic blue LED strip.
The most intriguing aspect of this reaction, however, is the unexpected beneficial effect of TFA. In addition to providing higher yields of the desired product, this additive also accelerated the radical coupling reaction and reduced the formation of undesired side-products. To better understand these results, we elected to study the origins of this dramatic Brønsted acid effect in greater detail.
Mechanistic studies
We first performed a qualitative analysis of the progress of the room-temperature reaction between tetrahydroisoquinoline 5 and enone 10, both in the presence and absence of the TFA additive. These data are summarized in Figure 3. In the presence of TFA, the reaction is complete within 6 h, and the starting tetrahydroisoquinoline is cleanly converted to the radical addition product in high yield. On the other hand, the reaction conducted in the absence of TFA appeared to proceed at a slower initial rate and stall after approximately 8 h of irradiation. Moreover, 1H NMR analysis of the reaction mixture revealed the formation of a number of side products that were not observed in the TFA-promoted reaction. Thus, the TFA additive appears to improve the initial rate of product formation, prevent the formation of undesirable side products, and avoid a catalyst deactivation pathway.
Figure 3.
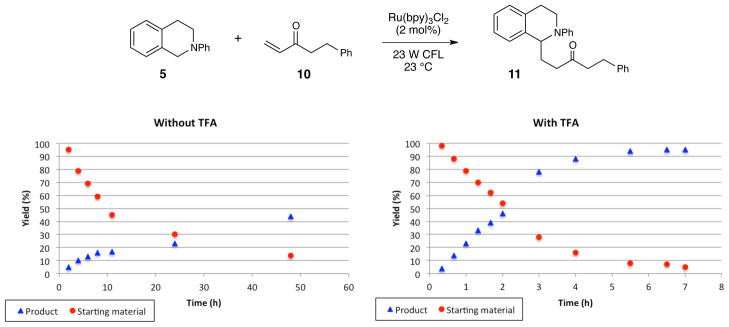
Reaction progress in the absence and presence of TFA.
We next investigated the relationship between the pKa of the Brønsted acid additive and the yield of the reaction (Figure 4). The formation of 7 was most efficient within a relatively narrow range of acidity (pKa ~ 1). Additives of both greater and weaker acidity were less effective, and very strong acids inhibited the reaction completely. In the high pKa regime, the observation that stronger acids result in higher yields is consistent with the interpretation that the Brønsted acid is a co-catalyst for the formation of 7. Brønsted acid inhibition in the low pKa range is also reasonably easy to rationalize. The formation of the key α-amino radical requires the availability of free amine; in the presence of strong acids such as TfOH, the protonated ammonium cannot be photoxidized by Ru*(bpy)32+ to initiate the reaction. On the other hand, we propose that weaker acid co-catalysts results in an equilibrium concentration of free amine sufficient to participate in reductive quenching of Ru*(bpy)32+.
Figure 4.

Relationship between co-catalyst acidity and efficiency of the radical coupling of 5 and 6.a
aReactions conducted using 2 mol% Ru(bpy)3Cl2, 2 equiv of MVK, and 1 equiv of acid in degassed MeCN (0.25 M) at room temperature under irradiation by a 23 W compact fluorescent bulb at 30 cm.
In support of this hypothesis, we examined the reaction of 5 and 6 in the presence of varying concentrations of TFA and found that the efficiency of these reactions were comparable within a dynamic range of 0.5 to 2 equiv of TFA, while higher concentrations of acid completely inhibited product formation.12 Consistent with these results, we performed a titration of isoquinoline 5 with TFA and observed that 1H NMR analysis showed saturation behavior, which presumably corresponds to quantitative formation of the unreactive ammonium TFA salt (Figure 5).
Figure 5.

1H NMR Titration of 5 with TFA.
Finally, we investigated the effect of isotopic labeling of the each of the coupling partners (Figure 6). To facilitate measurement of kinetic isotope effects (KIE) by GC, we examined labeled and unlabeled reactions of isoquinoline 5 with 2-phenylethyl enone 10. In the absence of the TFA co-catalyst, the KIE associated with amine 5 was negligible; however, we observed an inverse second-order KIE with respect to the enone. Addition of TFA to the reaction conditions, on the other hand, gave significantly different results. We measured a first-order KIE with respect to the amine and a normal second-order KIE with respect to the enone. Interpretation of the magnitude of these isotope effects is challenging due to the presence of multiple equilibria and because there is presumably some contribution of the uncatalyzed reaction to the formation of products even in the presence of TFA. Nevertheless, these data strongly suggest that the rate determining step changes upon addition of the Brønsted co-acid catalyst.
Figure 6.

Kinetic Isotope Effects
We interpreted the results of these experiments in the context of the mechanistic hypothesis proposed in Scheme 1, which is quite similar to the mechanism proposed by Pandey and Reiser. Oxidation of the amine by the excited photocatalyst Ru*(bpy)32+ would produce radical cation 12. Subsequent deprotonation of this intermediate would form the key α-amino radical 13, which can then react with the Michael acceptor to form radical adduct 16. The observed product 7 would be generated from the α-keto radical species 16 by one of two possible pathways: either a chain-propagating hydrogen atom abstraction (path IV) of tetrahydroisoquinoline 5 or chain-terminating electron transfer from the reduced Ru(bpy)3+ photocatalyst (path V). Several of these steps could potentially be rate-limiting, and we considered the effect Brønsted acid additives could have on each of them in turn.
Scheme 1.

First, we evaluated the step involving oxidation of the amine (step I in Scheme 1). The rates of reductive quenching of Ru*(bpy)32+ by a variety of tertiary anilines have been studied. These are generally quite fast and can be close to the diffusion-controlled limit.13 This step is thus unlikely to be responsible for the slow rate of the overall reaction. Moreover, the observation that the formation of the ammonium salt inhibits product formation is also inconsistent with the contention that Brønsted acids could accelerate of this step.
Similarly, we also considered the deprotonation of the tetrahydroisoquinoline radical cation (step II). Amine radical cations are generally considered to be quite acidic, although various reports have raised doubts about this common assumption.14 Nevertheless, it does not seem reasonable to suppose that the Brønsted acid accelerates this deprotonation step. We can also rule out the possibility that TFA accelerates this step via a solvent polarity effect, because the addition of ionic electrolytes has a negligible effect on the reaction (e.g., Table 1, entry 2). Moreover, we observe a kinetic isotope effect with respect to the enone under both acid-catalyzed and acid-free conditions; it must be the case that the enone is involved in the rate-determining step of this transformation under both sets of conditions.
The data suggest that the addition of the acid co-catalyst results in a change of the rate-determining step. When reactions are performed without added TFA, the data are most consistent with rate-limiting addition of α-amino radical 13 to the enone (step III). The observation of an inverse secondary KIE upon deuteration of the enone under these conditions supports this interpretation. When the reaction is conducted in the presence of TFA, however, the normal secondary KIE with respect to the enone and the primary KIE with respect to the tetrahydroisoquinoline suggests that the rate-limiting step involves chain-propagating hydrogen atom abstraction from 5 by α-keto radical 16 (step IV). Rate-limiting photocatalyst turnover (step V) cannot be the dominant pathway of product formation because it would not result in the observed primary KIE for 5.
This interpretation also explains the observation that tetrahydroisoquinoline dimer 14 is an observable by-product only in the absence of TFA. A consequence of rate-limiting addition into the enone would be buildup of the concentration of α-amino radical 13, at which point the rate of dimerization to 14 would become non-negligible. This termination step is problematic for the photocatalytic process because it provides no opportunity for turnover of the reduced photocatalyst, which would accumulate in its photochemically inactive Ru(bpy)3+ oxidation state. The addition of TFA therefore benefits this reaction both by accelerating the slow carbon–carbon bond-forming step and by keeping the concentration of 13 so low that this catalyst-deactivating dimerization step becomes insignificant.
These findings have a number of important implications. The first is that TFA is capable of lowering the kinetic barrier for addition of the nucleophilic α-amino radical to the enone, to the point that step III becomes no longer rate-limiting in this process. Although the ability of Lewis acids to accelerate the addition of nucleophilic radicals to Michael acceptors has been of significant recent interest,15 only a few reports have suggested the ability of Brønsted acids to catalyze similar radical additions.16 We expect that this observation could have important implications in the design of other radical addition reactions.
A second implication of this study is that chain propagation, and not catalyst turnover, is the process dominating the kinetics of product formation.17 The importance of radical chain mechanisms in photoinduced radical addition reactions has been well documented by Fagnoni18 and Hoffmann19 using organic photosensitizers. We believe that chain processes may also be a common feature of many of the recent reports of free radical transformations initiated by photoredox catalysis.20 Scheme 1 suggests that Ru(bpy)3+ is generated by reductive quenching of the excited photocatalyst with 5 to generate the corresponding amine radical cation. Thus, the total concentration of all radical species in solution at any given point should be no greater than the concentration of Ru(bpy)3+, which in turn can be no greater than the initial concentration of the Ru(bpy)32+ photocatalyst. In reactions using catalytic loadings of the photocatalyst, therefore, the rate of bimolecular interaction of Ru(bpy)3+ and α-keto radical 16 will be constrained by the low concentrations of these two species, and it is reasonable to hypothesize that radical chain propagation might be the dominant pathway for product formation for many mechanistically analogous photoredox reactions. Indeed, the participation of productive chain processes may account for the low loadings of photocatalyst that can be used in many reported photoredox processes.
Conclusion
We may draw several conclusions from the observation that Brønsted acid additives can provide a significant improvement in the efficiency of the radical functionalization of the α-amino C–H bond of a variety of tetrahydroisoquinolines. From a practical synthetic perspective, we have shown that the rates and yields of this process can be significantly improved, and that these conditions enable excellent conversion using commercially available Ru(bpy)32+ in place of a precious third-row transition metal photocatalyst, and a household light bulb instead of a high-intensity monochromatic LED light source. From this study and many others, it is apparent that there is a synergistic benefit of combining various co-catalysts with photoredox catalysts.21
Our investigations into the origin of this dramatic additive effect have demonstrated that the key carbon–carbon bond-forming step is accelerated by TFA and is a rare example of Brønsted acid catalysis in radical addition reactions. We anticipate that the rational application of this concept in other contexts will facilitate the discovery of new synthetically useful radical addition reactions. Our investigations also document the participation of chain-propagation events in this transformation and suggest that the mechanism of other radical reactions initiated by photoredox catalysis may be more complicated than generally appreciated. We are continuing to probe these possibilities in ongoing studies in our laboratory.
Experimental Section
2-(o-Tolyl)-1,2,3,4-tetrahydroisoquinoline (Table 2, entry 5)
A 25 mL round-bottomed flask was charged with Pd2(dba)3 (20.6 mg, 0.02 mmol), BINAP (28.6 mg, 0.05 mmol), and 3 mL toluene. The resulting solution was degassed by sparging with argon for 10 min before being heated to 110 °C for 15 min. The reaction mixture was allowed to cool to room temperature before NaOt-Bu (100.5 mg, 1.1 mmol), 2-bromotoluene (191.4 mg, 1.1 mmol), and 1,2,3,4,-tetrahydroisoquinoline (300 mg, 2.2 mmol) were added. The resulting mixture was heated to reflux for 20 h before being cooled to room temperature and filtered through a pad of Celite. Solvents were removed under reduced pressure, and the residue was purified by chromatography on silica gel using 30:1 hexanes:EtOAc as the eluent to afford the product as a yellow oil (136 mg, 27% yield). IR (thin film): 3020, 2918, 1492, 1446, 1379, 1107 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.16 (m, 5H), 7.07 (m, 2H), 6.99 (td, J = 7.4, 1.2 Hz, 1H), 4.10 (s, 2H), 3.19 (t, J = 5.8 Hz, 2H), 2.99 (t, J = 5.8 Hz, 2H), 2.33 (s, 3H); 13C NMR (125 MHz, CDCl3) 151.5, 135.4, 134.6, 132.9, 131.1, 128.9, 126.6, 126.4, 126.1, 125.7, 123.2, 119.3, 54.2, 50.3, 29.7, 18.0; HRMS (ESI+) calcd for [C16H17N+H]+ requires m/z 224.1434, found m/z 224.1434.
2-Phenyl-3,4-dihydroisoquinolin-1(2H)-one
After three cycles of evacuation and back-filling with dry nitrogen, an oven dried Schlenk tube equipped with a magnetic stirring bar was charged with 3,4-dihydroisoquinolin-1(2H)-one22 (700 mg, 4.8 mmol), CuI (38 mg, 0.2 mmol), K2PO4 (7.8 g, 1.7 mmol) and iodobenzene (0.8 ml, 3.9 mmol). The tube was evacuated and back-filled with nitrogen, and then 2,2,6,6-tetramethyl-3,5-heptanedione (72 mg, 0.39 mmol) and anhydrous degassed toluene (4 mL) were added under a stream of nitrogen by syringe at room temperature. The tube was sealed under a positive pressure of nitrogen, stirred and heated to 130 °C for 24 h. After cooling to room temperature, the reaction was diluted with 100 mL CH2Cl2 and washed twice with water. The aqueous phases were extracted five times with dichloromethane. The organic layers were combined, dried over MgSO4, filtered, and concentrated under vacuum. Purification of the residue by chromatography on silica gel using 4:1 hexanes:EtOAc as the eluent afforded the product as a white solid (780 mg, 75% yield). IR (thin film): 3057, 1652, 1415, 1221, 741 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 7.8 Hz, 1H), 7.47 (td, J = 7.6, 1.3 Hz, 1H), 7.40 (m, 5H), 7.25 (m, 2H), 4.00 (t, J = 6.6 Hz, 2H), 3.15 (t, J = 6.6 Hz, 2H); 13C NMR (125 MHz, CDCl3) 164.2, 143.1, 138.3, 132.0, 129.7, 128.9, 128.8, 127.2, 126.9, 126.2, 125.3, 49.4, 28.6; HRMS (EI+) calcd for [C15H13NO]+ requires m/z 223.0992, found m/z 223.0989. (mp = 102 °C).
1,1-Dideutero-2-phenyl-1,2,3,4-tetrahydroisoquinoline. (5-d2)
To a stirred suspension of LiAlD4 (82 mg, 2.0 mmol) in 20 mL THF was added 2-phenyl-3,4-dihydroisoquinolin-1(2H)-one under an argon atmosphere. After refluxing for 2 h, the mixture was cooled to 0 °C and quenched with water. The product was extracted with eight portions of Et2O. The combined organics were then washed with brine, dried over MgSO4 and concentrated in vacuo. Purification by chromatography on silica gel using 4:1 hexanes:EtOAc as the eluent afforded the product as a white solid (360 mg, 95% yield). IR (thin film): 3023, 2963, 1660, 1598, 1109, 1049 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.28 (m, 2H), 7.16 (m, 4H), 6.98 (dt, J = 7.9, 0.9 Hz, 2H), 6.82 (tt, J = 7.2, 1.1 Hz, 1H), 3.56 (t, J = 5.8 Hz, 2H), 2.98 (t, J = 5.8 Hz, 2H); 13C NMR (125 MHz, CDCl3) 149.5, 133.9, 133.3, 128.2, 127.5, 125.5, 125.3, 125.0, 117.6, 114.1, 49.0 (t, J = 21.0 Hz), 45.4, 28.1; HRMS (ESI+) calcd for [C15H13D2N+H]+ requires m/z 212.1413, found m/z 212.1407. (mp = 45 °C).
1,1,2-Trideutero-5-phenylpent-1-en-3-one. (10-d3)
To a solution of 4-phenyl-1-(triphenylphosphoranylidene)butan-2-one (2 g, 4.9 mmol) in 10 mL of DCM was added 20% formaldehyde-d2 solution in D2O (3.9 mL, 24.5 mmol). The reaction mixture was stirred at room temperature overnight and extracted twice with DCM. The combined organics were then washed with brine, dried over MgSO4 and concentrated in vacuo. Purification of the residue by chromatography on silica gel using 20:1 hexanes:EtOAc as the eluent afforded the product as a yellow oil (342 mg, 52% yield). IR (thin film): 3030, 2927, 2363, 1681, 1551, 1496, 1451 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 2H), 7.21 (m, 3H), 2.94 (m, 4H); 13C NMR (125 MHz, CDCl3) 199.8, 141.1, 136.0 (t, J = 24.3 Hz), 128.5, 128.4, 127.6 (q, J = 24.5 Hz), 127.4, 127.2, 126.2, 41.2, 29.7; HRMS (ESI+) calcd for [C15H13D2N+H]+ requires m/z 163.1071, found m/z 163.1073.
General procedure for photochemical reactions
A dry 25 mL Schlenk tube was charged with the amine (1 equiv), Michael acceptor (2 equiv), Ru(bpy)3Cl2•6H2O (0.02 equiv), TFA (1 equiv), and acetonitrile (0.25 M) and degassed using three freeze/pump/thaw cycles under nitrogen in the dark. The reaction was then heated in an oil bath at 50 °C and allowed to stir while being irradiated by a 23 W (1280 lumen) compact fluorescent lamp at a distance of 30 cm. Upon completion of the reaction, the mixture was allowed to cool to room temperature, was neutralized with K2CO3 (2 equiv), and filtered through a small plug of silica with Et2O. Removal of the solvent in vacuo afforded analytically pure product.
4-(2-Phenyl-1,2,3,4-tetrahydroisoquinolin-1-yl)butan-2-one. (Table 2, entry 1)
Colorless oil. Experiment 1: 120 mg (0.43 mmol, 90% yield). Experiment 2: 116 mg (0.42 mmol, 89% yield). IR (thin film): 3060, 2923, 1712, 1597, 1503, 1159 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.21 (m, 2H), 7.14 (m, 3H), 7.08 (m, 1H), 6.87 (d, J = 8.4 Hz, 2H), 6.73 (t, J = 7.0 Hz, 1H), 4.72 (dd, J = 9.1, 5.5 Hz, 1H), 3.59 (m, 1H), 3.54 (m, 1H), 2.98 (m, 1H), 2.74 (dt, J = 16.3, 4.6 Hz, 1H), 2.55 (t, J = 7.1 Hz, 2H), 2.21 (m, 1H), 2.07 (s, 3H), 2.04 (m, 1H); 13C NMR (125 MHz, CDCl3) 149.8, 138.3, 134.9, 129.3, 128.8, 127.2, 126.5, 125.9, 117.7, 114.6, 57.9, 41.5, 40.3, 30.3, 30.2, 26.3; HRMS (ESI+) calcd for [C19H21NO+H]+ requires m/z 280.1696, found m/z 280.1704.
4-(2-(p-Tolyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)butan-2-one. (Table 2, entry 2)
White solid. Experiment 1: 128 mg (0.44 mmol, 98% yield). Experiment 2: 124 mg (0.42 mmol, 97% yield). IR (thin film): 3022, 2919, 2247, 1712, 1616, 1517, 909 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.13 (m, 3H), 7.05 (d, J = 6.9 Hz, 1H), 7.01 (d, J = 8.3 Hz, 2H), 6.79 (dt, J = 9.0, 2.9 Hz, 2H), 4.63 (dd, J = 9.5, 5.3 Hz, 1H), 3.56 (m, 1H), 3.50 (m, 1H), 2.95 (m, 1H), 2.67 (dt, J = 16.2, 4.2 Hz, 1H), 2.55 (t, J = 6.9 Hz, 2H), 2.23 (s, 3H), 2.18 (m, 1H), 2.07 (s, 3H), 2.03 (m, 1H); 13C NMR (125 MHz, CDCl3) 208.6, 147.9, 138.5, 134.9, 129.8, 128.8, 127.4, 127.3, 126.4, 125.9, 115.4, 58.1, 41.8, 40.4, 30.5, 30.3, 26.0, 20.3; HRMS (ESI+) calcd for [C20H23NO+H]+ requires m/z 294.1853, found m/z 294.1853.
4-(2-(4-Methoxyphenyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)butan-2-one. (Table 2, entry 3)
White solid. Experiment 1: 120 mg (0.39 mmol, 93% yield). Experiment 2: 124 mg (0.40 mmol, 96% yield). IR (thin film): 2997, 2950, 1711, 1509, 1037 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.16 (m, 2H), 7.13 (m, 1H), 7.06 (d, J = 7.0 Hz, 1H), 6.84 (dt, J = 8.6, 2.5 Hz, 2H), 6.78 (dt, J = 9.2, 2.3 Hz, 2H), 4.51 (dd, J = 10.2, 4.4 Hz, 1H), 3.73 (s, 3H), 3.48 (dd, J = 7.6, 4.4 Hz, 2H), 2.89 (m, 1H), 2.63 (dt, J = 16.3, 4.2 Hz, 1H), 2.55 (td, J = 7.3, 2.1 Hz, 2H), 2.17 (m, 1H), 2.08 (s, 3H), 2.03 (m, 1H); 13C NMR (125 MHz, CDCl3) 208.8, 152.9, 144.6, 138.4, 134.9, 128.9, 127.2, 126.3, 125.9, 118.2, 114.6, 58.7, 55.6, 43.0, 40.4, 30.6, 30.3, 25.8; HRMS (ESI+) calcd for [C20H23NO2+H]+ requires m/z 310.1802, found m/z 310.1817.
4-(2-(4-Chlorophenyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)butan-2-one. (Table 2, entry 4)
White solid. Experiment 1: 133 mg (0.42 mmol, 99% yield). Experiment 2: 133 mg (0.42 mmol, 99% yield). IR (thin film): 2923, 2847, 1712, 1594, 1496, 1159 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.15 (m, 5H), 7.10 (m, 1H), 6.79 (dt, J = 9.5, 3.2 Hz, 2H), 4.66 (dd, J = 9.4, 4.9 Hz, 1H), 3.54 (dd, J = 7.2, 4.9 Hz, 2H), 2.96 (m, 1H), 2.77 (dt, J = 16.2, 4.7 Hz, 1H), 2.53 (t, J = 7.1 Hz, 2H), 2.20 (m, 1H), 2.09 (s, 3H), 2.02 (m, 1H); 13C NMR (125 MHz, CDCl3) 208.4, 137.9, 134.7, 129.1, 128.8, 127.2, 126.7, 126.0, 122.4, 115.6, 58.0, 41.8, 40.2, 30.2, 30.2, 26.3; HRMS (ESI+) calcd for [C19H20ClNO+H]+ requires m/z 314.1307, found m/z 314.1322.
4-(6,7-Dimethoxy-2-phenyl-1,2,3,4-tetrahydroisoquinolin-1-yl)butan-2-one. (Table 2, entry 6)
Colorless oil. Experiment 1: 120 mg (0.35 mmol, 95% yield). Experiment 2: 123 mg (0.36 mmol, 98% yield). IR (thin film): 2999, 2935, 2252, 1710, 1597, 1249 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.21 (td, J = 7.7, 1.8 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 6.73 (t, J = 7.4 Hz, 1H), 6.67 (s, 1H), 6.56 (s, 1H), 4.65 (dd, J = 9.8, 4.9 Hz, 1H), 3.87 (s, 3H), 3.82 (s, 3H), 3.65 (dt, J = 13.1, 4.6 Hz, 1H), 3.50 (m, 1H), 2.91 (m, 1H), 2.58 (m, 3H), 2.16 (m, 1H), 2.09 (s, 3H), 2.04 (m, 1H); 13C NMR (125 MHz, CDCl3) 208.8, 150.1, 147.6, 147.3, 130.3, 129.3, 126.7, 118.0, 115.1, 111.5, 110.2, 57.6, 56.0, 55.8, 41.3, 40.3, 30.3, 30.3, 25.5; HRMS (ESI+) calcd for [C21H25NO3+H]+ requires m/z 340.1908, found m/z 340.1920.
4-(7-Chloro-2-phenyl-1,2,3,4-tetrahydroisoquinolin-1-yl)butan-2-one. (Table 2, entry 7)
Colorless oil. Experiment 1: 123 mg (0.39 mmol, 96% yield). Experiment 2: 122 mg (0.38 mmol, 95% yield). IR (thin film): 2924, 1711, 1599, 1507, 749, 693 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.21 (m, 3H), 7.10 (dd, J = 8.2, 1.8 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.85 (d, J = 8.0 Hz, 2H), 6.75 (t, J = 7.2 Hz, 1H), 4.68 (dd, J = 9.2, 5.4 Hz, 1H), 3.64 (dt, J = 13.3, 4.2 Hz, 1H), 3.49 (tt, J = 10.4, 4.6 Hz, 1H), 2.92 (m, 1H), 2.64 (dt, J = 16.3, 3.7 Hz, 1H), 2.57 (t, J = 6.7 Hz, 2H), 2.18 (m, 1H), 2.09 (s, 3H), 2.05 (m, 1H); 13C NMR (125 MHz, CDCl3) 208.3, 149.7, 140.2, 133.2, 131.4, 130.2, 129.4, 127.1, 126.6, 118.3, 115.1, 57.7, 41.1, 40.2, 30.3, 25.4; HRMS (ESI+) calcd for [C19H20ClNO+H]+ requires m/z 314.1307, found m/z 314.1303.
1-(2-(p-Tolyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)pentan-3-one. (Table 3, entry 1)
Colorless oil. Experiment 1: 135 mg (0.44 mmol, 98% yield). Experiment 2: 137 mg (0.45 mmol, 99% yield). IR (thin film): 3023, 2975, 2249, 1710, 1615, 1517, 909 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.14 (m, 2H), 7.11 (m, 1H), 7.05 (d, J = 7.0 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.78 (dt, J = 8.7, 3.1 Hz, 2H), 4.64 (dd, J = 9.8, 5.4 Hz, 1H), 3.55 (m, 1H), 3.51 (m, 1H), 2.94 (m, 1H), 2.66 (dt, J = 16.8, 4.3 Hz, 1H), 2.52 (t, J = 6.7 Hz, 2H), 2.35 (m, 2H), 2.22 (s, 3H), 2.18 (m, 1H), 2.06 (m, 1H), 1.01 (t, 3H); 13C NMR (125 MHz, CDCl3) 211.4, 147.8, 138.5, 134.9, 129.8, 128.8, 127.3, 126.4, 125.8, 115.4, 58.2, 41.7, 39.1, 36.1, 30.5, 26.0, 20.3, 7.8; HRMS (ESI+) calcd for [C21H25NO+H]+ requires m/z 308.2009, found m/z 308.2004.
1-Phenyl-3-(2-(p-tolyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)propan-1-one. (Table 3, entry 2)
White solid. Experiment 1: 143 mg (0.40 mmol, 90% yield). Experiment 2: 146 mg (0.41 mmol, 92% yield). IR (thin film): 3061, 3025, 2247, 1682, 1615, 1517, 999 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 8.1 Hz, 2H), 7.21 (d, J = 6.7 Hz, 1H), 7.14 (m, 2H), 7.06 (d, J = 6.7 Hz, 1H), 6.97 (d, J = 9.0 Hz, 2H), 6.78 (d, J = 8.1 Hz, 2H), 4.75 (dd, J = 9.4, 4.3 Hz, 1H), 3.57 (m, 2H), 3.09 (m, 2H), 2.96 (m, 1H), 2.69 (dt, J = 16.5, 5.3 Hz, 1H), 2.35 (m, 1H), 2.24 (m, 1H), 2.20 (s, 3H); 13C NMR (125 MHz, CDCl3) 200.1, 147.9, 138.5, 137.1, 134.9, 132.8, 129.7, 128.8, 128.5, 128.0, 127.4, 126.4, 125.9, 115.6, 58.4, 42.0, 35.5, 31.0, 26.1, 20.3; HRMS (ESI+) calcd for [C25H25NO+H]+ requires m/z 356.2009, found m/z 356.2012.
3-(2-(p-Tolyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)propanal. (Table 3, entry 4)
White solid. Experiment 1: 117 mg (0.42 mmol, 94% yield). Experiment 2: 121 mg (0.43 mmol, 97% yield). IR (thin film): 3063, 2920, 2246, 1719, 1616, 1517, 909 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.70 (t, J = 1.4 Hz, 1H), 7.14 (m, 3H), 7.06 (d, J = 6.9 Hz, 1H), 7.02 (d, J = 8.2 Hz, 2H), 6.79 (d, J = 8.5 Hz, 2H), 4.61 (dd, J = 10.1, 5.3 Hz, 1H), 3.52 (m, 2H), 2.92 (m, 1H), 2.65 (dt, J = 16.0, 4.5 Hz, 1H), 2.52 (m, 2H), 2.28 (m, 1H), 2.23 (s, 3H), 2.11 (m, 1H); 13C NMR (125 MHz, CDCl3) 201.7, 147.7, 137.9, 135.0, 129.8, 128.9, 128.2, 127.2, 126.5, 126.0, 116.3, 58.4, 42.5, 41.1, 29.8, 25.9, 20.3; HRMS (ESI+) calcd for [C19H21NO+H]+ requires m/z 280.1696, found m/z 3280.1706. (mp = 66 °C).
2-Methyl-3-(2-(p-tolyl)-1,2,3,4-tetrahydroisoquinolin-1-yl)propanal. (Table 3, entry 5)
Inseparable 1:1.5 mixture of diastereomers, colorless oil. Experiment 1: 120 mg (0.41 mmol, 91% yield). Experiment 2: 124 mg (0.43 mmol, 94% yield). IR (thin film): 2965, 2922, 2247, 1719, 1615, 1513, 1038 cm−1; 1H NMR (500 MHz, CDCl3): Major diastereoisomer δ 9.58 (d, J = 1.1 Hz, 1H), 6.99–7.18 (m, 6H), 6.76 (dt, J = 8.7, 2.9 Hz, 2H), 4.69 (dd, J = 9.8, 4.7 Hz, 1H), 3.49 (m, 2H), 2.86 (m, 1H), 2.43–2.66 (m, 3H), 2.22 (s, 3H), 2.15 (m, 1H), 1.14 (d, J = 6.9 Hz, 3H). Minor diastereomer d 9.57 (d, J = 2.5 Hz, 1H), 6.99–7.18 (m, 6H), 6.80 (dt, J = 8.7, 2.7 Hz, 2H), 4.61 (dd, J = 10.8, 4.6 Hz, 1H), 3.49 (m, 2H), 2.86 (m, 1H), 2.43–2.66 (m, 2H), 2.22 (s, 3H), 2.21 (m, 1H), 1.74 (dt, J = 1.45, 4.9 Hz, 1H), 1.11 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) 203.8, 203.4, 147.7, 138.0, 137.8, 135.0, 134.9, 129.8, 129.7, 129.1, 129.0, 128.8, 128.8, 127.2, 126.5, 126.4, 126.0, 126.0, 117.3, 117.3, 57.5, 55.5, 44.8, 43.5, 43.0, 42.4, 39.9, 38.6, 25.4, 25.0, 20.4, 14.6, 13.5; HRMS (ESI+) calcd for [C20H23NO+H]+ requires m/z 294.1853, found m/z 294.1864.
1-Phenyl-5-(2-phenyl-1,2,3,4-tetrahydroisoquinolin-1-yl)pentan-3-one. (11)
White solid. The yields represented here are 1H-NMR yields, calculated from the crude against an internal standard. A clean sample of the product was obtained by using a 1:1 mixture of starting material and acceptor. The data for that sample is shown below. Experiment 1: 94% yield. Experiment 2: 97% yield. IR (thin film): 3030, 2927, 2360, 1713, 1601, 1501, 1398 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.27-7.07 (m, 11H), 6.86 (d, J = 7.9 Hz, 2H), 6.73 (t, J = 6.7 Hz, 1H), 4.71 (dd, J = 9.6, 5.7 Hz, 1H), 3.56 (m, 2H), 2.98 (m, 1H), 2.85 (t, J = 8.3 Hz, 2H), 2.73 (dt, J = 16.6, 4.7 Hz, 1H), 2.65 (m, 2H), 2.51 (t, J = 6.7 Hz, 2H), 2.24 (m, 1H), 2.05 (m, 1H); 13C NMR (125 MHz, CDCl3) 209.6, 149.8, 141.1, 138.3, 134.9, 129.4, 128.8, 128.5, 128.3, 127.3, 126.5, 126.1, 125.9, 117.8, 114.6, 58.0, 44.6, 41.4, 39.7, 30.4, 29.7, 26.3; HRMS (ESI+) calcd for [C26H27NO+H]+ requires m/z 370.2166, found m/z 370.2177. (mp = 114 °C).
Supplementary Material
Acknowledgments
The authors thank Prof. Bob Bergman and Prof. Clark Landis for helpful discussions concerning the mechanism of this process. The authors are grateful for funding from the NIH (GM095666) and the Sloan Foundation. EMW is the recipient of a Hilldale Fellowship from UW–Madison. The NMR facilities at UW–Madison are funded by the NSF (CHE-1048642) and a generous gift from Paul J. Bender.
Footnotes
Supporting Information. Experimental details for kinetic and NMR titration studies, and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews, see: Campos KR. Chem Soc Rev. 2007;36:1069. doi: 10.1039/b607547a.Li CJ. Acc Chem Res. 2009;42:335. doi: 10.1021/ar800164n.Jones KM, Klussmann M. Synlett. 2012:149.Mitchell EA, Peschiulli A, Lefevre N, Meerpoel L, Maes BUW. Chem Eur J. 2012;18:10092. doi: 10.1002/chem.201201539.
- 2.For reviews of amine oxidation, see: Chow YL, Danen WC, Nelsen SF, Rosenblatt DH. Chem Rev. 1978;78:243.Yoon UC, Mariano PS. Acc Chem Res. 1992;25:233. doi: 10.1021/ar010004o.Murahashi SI. Angew Chem Int Ed Engl. 1995;34:2443.Li CJ. Acc Chem Res. 2009;42:335. doi: 10.1021/ar800164n.
- 3.For selected examples, see: Chiba T, Takata Y. J Am Chem Soc. 1977;42:2973.Shono T, Matsumura Y, Tsubata K. J Am Chem Soc. 1981;103:1172.Shu XZ, Xia XF, Yang YF, Ji KG, Liu XY, Liang YM. J Org Chem. 2009;74:7464. doi: 10.1021/jo901583r.Chu L, Qing FL. Chem Commun. 2010;46:6285. doi: 10.1039/c0cc01073a.Allen JM, Lambert TH. J Am Chem Soc. 2011;133:1250. doi: 10.1021/ja109617y.Santamaria J, Herlem D, Kyuong-Huu F. Tetrahedron. 1977;33:2389.Pandey G, Reddy PY, Bhalerao UT. Tetrahedron Lett. 1991;32:5147.Li Z, Li CJ. J Am Chem Soc. 2004;126:11810. doi: 10.1021/ja0460763.Li Z, Li CJ. J Am Chem Soc. 2005;127:3672. doi: 10.1021/ja050058j.Li Z, Bohle DS, Li CJ. Proc Natl Acad Sci USA. 2006;103:8928. doi: 10.1073/pnas.0601687103.Murahahi SI, Nakae T, Terai H, Koyima N. J Am Chem Soc. 2008;130:11005. doi: 10.1021/ja8017362.
- 4.(a) Condie AG, Gonzalez-Gomez JC, Stephenson CRJ. J Am Chem Soc. 2010;132:1464. doi: 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]; (b) Freeman DB, Furst L, Condie AG, Stephenson CRJ. Org Lett. 2012;14:94. doi: 10.1021/ol202883v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For a recent review of photoredox amine functionalization see: Shi L, Xia W. Chem Soc Rev. 2012;41:7687. doi: 10.1039/c2cs35203f.
- 6.(a) Zeitler K. Angew Chem Int Ed. 2009;48:9785. doi: 10.1002/anie.200904056. [DOI] [PubMed] [Google Scholar]; (b) Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; (c) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (d) Telpý F. Collect Czech Chem Commun. 2011;76:859. [Google Scholar]; (e) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]; (f) Xuan J, Xiao WJ. Angew Chem Int Ed. 2012;51:6828. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]; (g) Maity S, Zheng N. Synlett. 2012;23:1851. doi: 10.1055/s-0032-1316592. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. doi: 10.1021/cr300503r. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For representative examples of photogenerated α-amino radical chemistry, see: Cookson RC, Hudec J, Mirza NA. J Chem Soc, Chem Commun. 1967:824.Lewis FD, Ho TI. J Am Chem Soc. 1977;99:7991.Ohashi M, Miyake K, Tsujimoto K. Bull Chem Soc Jpn. 1980;53:1683.Pienta NJ, McKimmey JE. J Am Chem Soc. 1982;104:5501.Das S, Dileep Kumar JS, Thomes KG, Shivaramayya K, Goerge MV. J Org Chem. 1994;59:628.
- 8.Kohls P, Jadhav D, Pandey G, Reiser O. Org Lett. 2012;14:672. doi: 10.1021/ol202857t. [DOI] [PubMed] [Google Scholar]
- 9.Conceptually similar reactions of α-amino radicals have recently been reported by Nishibayashi and Rueping and also required iridium photocatalysts and illumination with LEDs: Miyake Y, Nakajima K, Nishibayashi Y. J Am Chem Soc. 2012;134:3338. doi: 10.1021/ja211770y.Zhu S, Das A, Bui L, Zhou H, Curran DP, Rueping M. J Am Chem Soc. 2013;135:1823. doi: 10.1021/ja309580a.
- 10.Zhang X, Yeh SR, Hong S, Freccero M, Albini A, Falvey DE, Mariano PS. J Am Chem Soc. 1994;116:4211. [Google Scholar]
- 11.(a) Sinha A, Bruice TC. J Am Chem Soc. 1984;106:7291. [Google Scholar]; (b) Dinnocenzo JP, Banach TE. J Am Chem Soc. 1989;111:8646. doi: 10.1021/ja00279a078. [DOI] [PubMed] [Google Scholar]
- 12.For details, see the Supporting Information.
- 13.Bock CR, Connor JA, Gutierrez AR, Meyer TJ, Whitten DG, Sullivan BP, Nagle JK. J Am Chem Soc. 1979;101:4815. [Google Scholar]
- 14.Nelsen SF, Ippoliti JT. J Am Chem Soc. 1986;108:4879. [Google Scholar]
- 15.For recent reviews, see: Renaud P, Gerster M. Angew Chem Int Ed. 1998;37:2562. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2562::AID-ANIE2562>3.0.CO;2-D.Sibi MP, Manyem S, Zimmerman J. Chem Rev. 2003;103:3263. doi: 10.1021/cr020044l.
- 16.For example, see: Luo R, Chen Y, Sen A. J Polym Sci A Polym Chem. 2008;46:5499.Eisenbach CD, Sperlich B. Macromolecules. 1996;29:7748.
- 17.Based upon a suggestion by a reviewer, we monitored the progress of a reaction conducted in alternating hour-long intervals of light and dark, and we observed product formation only during the periods of illumination (see Supporting Information). It is important to note that this behavior is not evidence against a chain propagation mechanism; it merely indicates the existence of a chain-terminating process that occurs at a non-negligible rate.
- 18.Mosca R, Fagnoni M, Mella M, Albini A. Tetrahedron. 2001;57:10319. [Google Scholar]
- 19.Hoffmann N, Bertrand S, Marinković S, Pesch J. Pure Appl Chem. 2006;78:2227. [Google Scholar]
- 20.Barton and Stephenson have previously suggested the importance of radical chain processes in transition metal photocatalyzed atom transfer radical addition reactions: Barton DHR, Csiba MA, Jaszberenyi JC. Tetrahedron Lett. 1994;35:2869.Wallentin CJ, Nguyen JD, Finkbeiner P, Stephenson CRJ. J Am Chem Soc. 2012;134:8875. doi: 10.1021/ja300798k.
- 21.For examples with Lewis acid, Brønsted acid, transition metal, and chiral organic catalysts, see: Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f.Lu Z, Shen M, Yoon TP. J Am Chem Soc. 2011;133:1162. doi: 10.1021/ja107849y.Du J, Ruiz Espelt L, Guzei IA, Yoon TP. Chem Sci. 2011;2:2115. doi: 10.1039/C1SC00357G.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566. doi: 10.1021/ja208068w.Ye Y, Sanford MS. J Am Chem Soc. 2012;134:9034. doi: 10.1021/ja301553c.Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976.Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875. doi: 10.1021/ja9053338.
- 22.(a) Shi J, Manolikakes G, Yeh CH, Guerrero CA, Shenvi RA, Shigehisa H, Baran PS. J Am Chem Soc. 2011;133:8014. doi: 10.1021/ja202103e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mohamed MA, Yamada K, Tomioka K. Tetrahedron Lett. 2009:3436. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.