

Abstract
Short application of the volatile anesthetic isoflurane at reperfusion after ischemia exerts strong protection of the heart against injury. Mild depolarization and acidification of the mitochondrial matrix are involved in the protective mechanisms of isoflurane, but the molecular basis for these changes is not clear. In this study, mitochondrial respiration, membrane potential, matrix pH, matrix swelling, ATP synthesis and -hydrolysis, and H2O2 release were assessed in isolated mitochondria. We hypothesized that isoflurane induces mitochondrial depolarization and matrix acidification through direct action on both complex I and ATP synthase. With complex I-linked substrates, isoflurane (0.5 mM) inhibited mitochondrial respiration by 28±10%, and slightly, but significantly depolarized membrane potential and decreased matrix pH. With complex II- and complex IV-linked substrates, respiration was not changed, but isoflurane still decreased matrix pH and depolarized mitochondrial membrane potential. Depolarization and matrix acidification were attenuated by inhibition of ATP synthase with oligomycin, but not by inhibition of mitochondrial ATP- and Ca2+-sensitive K+ channels or uncoupling proteins. Isoflurane did not induce matrix swelling and did not affect ATP synthesis and hydrolysis, but decreased H2O2 release in the presence of succinate in an oligomycin- and matrix pH-sensitive manner. Isoflurane modulated H+ flux through ATP synthase in an oligomycin-sensitive manner. Our results indicate that isoflurane-induced mitochondrial depolarization and acidification occur due to inhibition of the electron transport chain at the site of complex I and increased proton flux through ATP synthase. K+ channels and uncoupling proteins appear not to be involved in the direct effects of isoflurane on mitochondria.
Keywords: isoflurane, mitochondria, mitochondrial potassium channels, uncoupling proteins, ATP-synthase
1. Introduction
Studies have revealed that mitochondria participate in the initiation of the cardiac protection elicited by volatile anesthetics such as isoflurane before ischemia (anesthetic preconditioning) (De Hert et al., 2005; Hirata et al., 2011; Stowe and Kevin, 2004). Moreover, and clinically more relevant, they also contribute to the immediate protection when the anesthetic is applied at the time of reperfusion (anesthetic postconditioning) (Chiari et al., 2005). This occurs by lowering the mitochondrial matrix pH through interaction of volatile anesthetics with electron transport chain complexes and/or mitochondrial cation channels (Hanley et al., 2002b; Ljubkovic et al., 2007; Pravdic et al., 2010; Sedlic et al., 2009). Changes in the matrix pH modulate reactive oxygen species production and opening of mitochondrial permeability transition pore (Halestrap, 2009; Lambert and Brand, 2004). Inhibition of mitochondrial respiration and membrane potential depolarization may contribute to isoflurane-induced changes of the mitochondrial matrix pH (Pravdic et al., 2010), but the molecular targets on the mitochondrial level by which a short application of the volatile anesthetic isoflurane exerts strong protection of the heart are controversial. We and others have shown that isoflurane inhibits the electron transport chain at the level of complex I, (Hanley et al., 2002b) which may lead to a small rise in reactive oxygen species production in the triggering phase of anesthetic preconditioning (Hirata et al., 2011; Kevin et al., 2003). Isoflurane-induced reactive oxygen species formation in turn can further inhibit mitochondrial respiration (Riess et al., 2005). The small burst of rea originating from mitochondria was reported to be mandatory for activating pro-survival signaling pathways (Kevin et al., 2003; Penna et al., 2006). Isoflurane was shown to activate mitochondrial cardiac ATP-sensitive K+ (mitoKATP) channels in cardiomyocytes and when reconstituted in artificial lipid bilayers (Jiang et al., 2007). Activation of mitochondrial inner membrane K+ channels and subsequent K+ influx have been proposed to elicit cellular protection through matrix swelling and/or mild uncoupling and depolarization by affecting matrix Ca2+ homeostasis, reactive oxygen species production and mitochondrial permeability transition pore opening (Da Silva et al., 2003; Ljubkovic et al., 2007). However, the molecular identity of mitochondrial K+ channels is unknown and unspecific side effects of all drugs used to target those channels have been reported (Hanley and Daut, 2005; Hanley et al., 2002a). Similarly to K+ influx, an increased mitochondrial H+ leak may lead to mild mitochondrial depolarization, and protection against ischemia and reperfusion injury (Nadtochiy et al., 2006). Interestingly, the general anesthetic 2,6-diisopropylphenol increases proton permeability in mitochondria by acting on ATP synthase, (Rigoulet et al., 1996) and the anesthetics chloroform and halothane inhibit ATP synthesis (Rottenberg, 1983). Overall, while several mitochondrial targets for isoflurane have been identified, the molecular basis for isoflurane-induced depolarization and matrix pH change is not clear. Based on previously identified potential isoflurane targets in mitochondria we hypothesized that complex I of the electron transport chain and ATP synthase mediate depolarization and matrix acidification through isoflurane. Uncoupling proteins and mitochondrial K+ channels were also included as potential targets.
2. Materials and Methods
2.1. Animals
We used male adult Wistar rats (200–250 g) that were housed in accordance with the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (Milwaukee, Wisconsin) and experiments were conducted in accordance with U.S. National Institutes of Health standards (NIH Publication 95-23, revised 1996).
2.2. Isolation of rat heart mitochondria
Heart mitochondria were isolated as previously described (Pravdic et al., 2009). Hearts were washed in the isolation buffer [200 mM mannitol, 50 mM sucrose, 5 mM KH2PO4, 5 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 1 mM glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 0.1% bovine serum albumin (BSA), pH 7.3 at 25°C adjusted with potassium hydroxide (KOH)], and minced into small pieces. The suspension was homogenized in the presence of 5 U/ml of protease (from Bacillus licheniformis) with a T 25 disperser (IKA-Werke, Staufen, Germany), and the mitochondria were isolated by differential centrifugation. The final mitochondrial pellet was resuspended in the isolation buffer at a concentration of 10–20 mg/ml, stored on ice and used for experiments within four hours. Protein concentration was determined by a modified Lowry assay kit (Bio-Rad, Hercules, CA).
2.3. Mitochondrial oxygen consumption measurements
Oxygen consumption was measured polarographically with a Clark oxygen electrode in a closed, temperature-controlled vessel equipped with magnetic stirring (Hansatech Instruments, Norfolk, UK) at 30°C. Mitochondria (0.5 mg/ml) were suspended in mitochondrial respiration buffer (130 mM KCl, 5 mM K2HPO4, 20 mM MOPS, 2.5 mM EGTA, 1 μM Na4P2O7 and 0.1% BSA, and pH 7.4 adjusted with KOH). Respiration (state 2) was initiated by addition of complex I substrates 5 mM pyruvate and 5 mM malate, complex II substrate succinate (5 mM) in the presence of complex I inhibitor rotenone (1 μM), or complex IV substrate ascorbate (2.5 mM) in combination with tetramethylphenylene diamine (TMPD; 0.25 mM). The adenosine diphosphate (ADP)-stimulated respiration (state 3) was measured in the presence of 250 μM ADP, and state 4 respiration was measured after all ADP was phosphorylated to ATP. Respiration was also measured in the presence of mitochondrial uncoupler carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 1 μM) and oligomycin (5 μg/ml), an inhibitor of ATP synthase, to exclude changes in respiration due to an effect of isoflurane on ATP synthase. Mitochondria used for experiments exhibited a respiration control ratio (state 3/state 4) higher than 5 (with pyruvate/malate as substrates).
2.4. Mitochondrial membrane potential measurement
Measurements of mitochondrial membrane potential (ΔΨm) were performed in a stirred cuvette in respiration buffer containing 0.5 mg/ml of mitochondria at room temperature using a QM-8 spectrofluorometer (Photon Technology International, Inc., Birmingham, NJ). ΔΨm changes were measured by following the changes in fluorescence intensity of rhodamine 123 (Invitrogen, Carlsbad, CA) added at a concentration of 50 nM. The probe was excited at 503 nm, and emission was collected at 525 nm. ΔΨm was expressed as a relative value of rhodamine 123 fluorescence whereby the depolarization induced by addition of 1 μM FCCP was set as 100%. To test the role of mitoKATP and large conductance Ca2+-activated K+ (mitoBKCa) channels, 5-hydroxydecanoic acid (5-HD; 100 μM, Sigma Aldrich) and paxilline (10 μM, Sigma Aldrich) were used (Heinen et al., 2007; Wojtovich and Brookes, 2009). In addition, experiments were conducted in K+-free media by replacing potassium chloride with choline chloride. To test the role of uncoupling proteins, we used the uncoupling protein inhibitor guanosine-diphosphate (GDP; 1 mM; Sigma Aldrich) (Brookes et al., 2008).
2.5. Mitochondrial matrix pH determination
Matrix pH was measured with 2′-7′-bis(carboxyethyl)-5(6)-carboxyfluorescein-acetoxymethyl ester (BCECF-AM) fluorescent indicator (8 μM; Invitrogen). Dye loading was performed in mitochondrial isolation buffer for 20 min at room temperature as described previously (Andrukhiv et al., 2006). After incubation, mitochondrial suspension was diluted 10-fold with mitochondrial isolation buffer and spun at 8,000 g for 10 min to remove excess probe. The change in the fluorescence excitation ratio of 2′-7′-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF), corresponding to pH change, was monitored at 490 nm/440 nm with the emission at 510 nm. Changes in the matrix pH were expressed as the percentage of the pH change induced by addition of 1 μM FCCP (set as 100%). Appropriate corrections for autofluorescence were made. The role of mitoKATP, mitoBKCa, and uncoupling proteins was tested as described for ΔΨm measurements.
2.6. Production of reactive oxygen species
H2O2 release was measured using Amplex Red (12.5 μM; Invitrogen) and horseradish peroxidase (0.1 U/ml, Sigma Aldrich), which produce the fluorescent product resorufin upon reaction with H2O2. Mitochondria were energized with 5 mM succinate. Excitation and emission wavelengths were set to 530 nm and 583 nm, respectively (Ljubkovic et al., 2007). The slope of resorufin fluorescence was used to calculate the relative change in H2O2 production. Some experiments were performed in the presence of rotenone (1 μM). Rotenone inhibits complex I (NADH:ubiquinone oxidoreductase) near the binding site for ubiquinol (Chen et al., 2003), the electron acceptor for complex I. This decreases superoxide production at complex I due to reverse electron transport (Adam-Vizi and Chinopoulos, 2006). FCCP (1 μM) was used to depolarize ΔΨm completely and thereby stop H2O2 production (Heinen et al., 2007). To study the pH-dependence of reactive oxygen species modulation by isoflurane, we used ammonium chloride (NH4Cl, 2 mM) to induce a transient increase in the matrix pH (Wiederkehr et al., 2009).
2.7. Mitochondrial swelling assay
Mitochondrial K+-influx was measured by swelling/light scattering assay within 1.5 h of mitochondrial isolation (Silic-Benussi et al., 2009; Wojtovich and Brookes, 2009). Increase in mitochondrial volume (swelling) was detected as a decrease in light scattering at 540 nm. Mitochondria were suspended in 100 mM KCl, 10 mM 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (HEPES), 2 mM succinate (or 5 mM pyruvate and malate), 2 mM MgCl2, 2 mM KH2PO4, 2.5 mM EGTA, 0.1% BSA, and pH 7.2 adjusted with KOH and supplemented with 2.5 μg/ml oligomycin. Adenosine-5′-triphosphate (ATP, 1 mM) was used to inhibit mitoKATP opening (Wojtovich and Brookes, 2009). Where indicated, KCl in the buffer was replaced with choline chloride and other potassium containing salts by the corresponding sodium salts (potassium free buffer). In some experiments, after baseline recording isoflurane (0.5 mM) was added and at the end of the recording the K+ ionophore valinomycin (2 nM) was added to induce maximal swelling.
2.8. Measurement of mitochondrial ATP synthesis
Mitochondrial ATP synthesis rate was determined with a chemiluminescence-based method utilizing the reaction of firefly luciferase and luciferin with ATP. Reaction solution contained respiration buffer, 0.2 μM diadenosine pentaphosphate, 30 μM ADP, 5 μg/ml mitochondria, 0.1 mg/ml luciferin, and 1.25 μg/ml luciferase (Pravdic et al., 2010). The reaction was initiated by the addition of 5 mM pyruvate and 5 mM malate or 5 mM succinate. The blank was obtained in the absence of substrate. Chemiluminescence was measured in a Modulus luminometer (Turner Biosystems, Sunnyvale, CA) at room temperature for 120 s. The standard curve was obtained with defined ATP concentrations, from which the rate of mitochondrial ATP production was calculated.
2.9. Preparation of submitochondrial particles
Submitochondrial particles (SMP) were prepared by sonicating the mitochondria on ice (10 burst of 10 sec, 20 watts). The sonicated suspension was centrifuged at 10 000 g for 10 min (4°C). The supernatant was then centrifuged for 1 h at 100 000 g at 4°C, the pellet resuspended in the mitochondrial isolation buffer and the protein amount determined. This SMP suspension was used for the measurement of ATP hydrolysis.
2.10. Measurement of ATP hydrolysis in SMPs
ATPase activity was quantified using the EnzChek Phosphate Assay Kit (Invitrogen) in the presence of 5 μg/ml SMPs and 2 mM ATP by following the release of phosphate. The formation of 2-amino-6-mercapto-7-methylpurine from 2-amino-6-mercapto-7-methylpurine riboside was measured spectrophotometrically at 360 nm for 10 min at room temperature. Oligomycin (2.5 μg/ml) served as control for the specificity of mitochondrial ATPase and values in the presence of oligomycin were subtracted from the results as background. A standard curve was generated using phosphate standards from 0.1 to 100 μM (final concentration).
2.11. ATP-driven proton-pumping activity in SMPs
The proton-pumping activity coupled to the ATP hydrolysis of submitochondrial particles was determined from the quenching of 9-amino-6-chloro-2 methoxyacridine (ACMA) fluorescence (Invitrogen) (Zanotti et al., 2009). Measurements were performed in respiration buffer (but without MOPS) containing 0.5 mg submitochondrial particles and 1 μM ACMA. The reaction was started by the addition of 1 mM ATP. The assay was carried out at room temperature using a spectrofluorometer with excitation and emission set at 419 and 483 nm, respectively. Isoflurane was applied after ATP induced ACMA fluorescence quenching in the absence or presence of oligomycin (5 μg/ml). Changes of buffer pH were expressed as the percentage of the ACMA fluorescence quenching induced by addition of ATP (set as 100%).
2.12. Application of isoflurane
Isoflurane was dispersed in dimethyl sulfoxide and added to the mitochondria at the indicated concentrations. Dimethyl sulfoxide without isoflurane was added to control mitochondria and was always kept below 0.002 % (v/v). Isoflurane concentrations were measured in experimental buffer by gas chromatography.
2.13. Statistics
Results are presented as means±S.D., where n represents the number of separate mitochondrial preparations from different rats. The significance between groups was assessed using one-way analysis of variance. If F values were significant (P<0.05), Bonferroni’s post hoc test was used to test differences among groups. If P value was less than 0.05 differences were considered as significant.
3. Results
3.1. Inhibition of complex I activity by isoflurane
To determine the effect of isoflurane on electron transport chain complexes, we measured mitochondrial respiration when respiration was supported with complex I, complex II, and complex IV substrates. Maximal uncoupled respiration was measured in the presence of FCCP and oligomycin to prevent ADP/ATP turnover and thus exclude effects of isoflurane on ATP synthase. Isoflurane decreased oxygen consumption with complex I substrates in a concentration-dependent manner (Fig. 1A, B). Significant inhibition of oxygen consumption was observed at 0.5 mM or higher concentrations of isoflurane. At the highest concentration tested (1.5 mM), isoflurane decreased the rate of oxygen consumption to 65.2±13.7% (n = 7) of control. When succinate or ascorbate and TMPD were used as substrates, the rate of oxygen consumption was not decreased (Fig. 1C–F).
Figure 1.
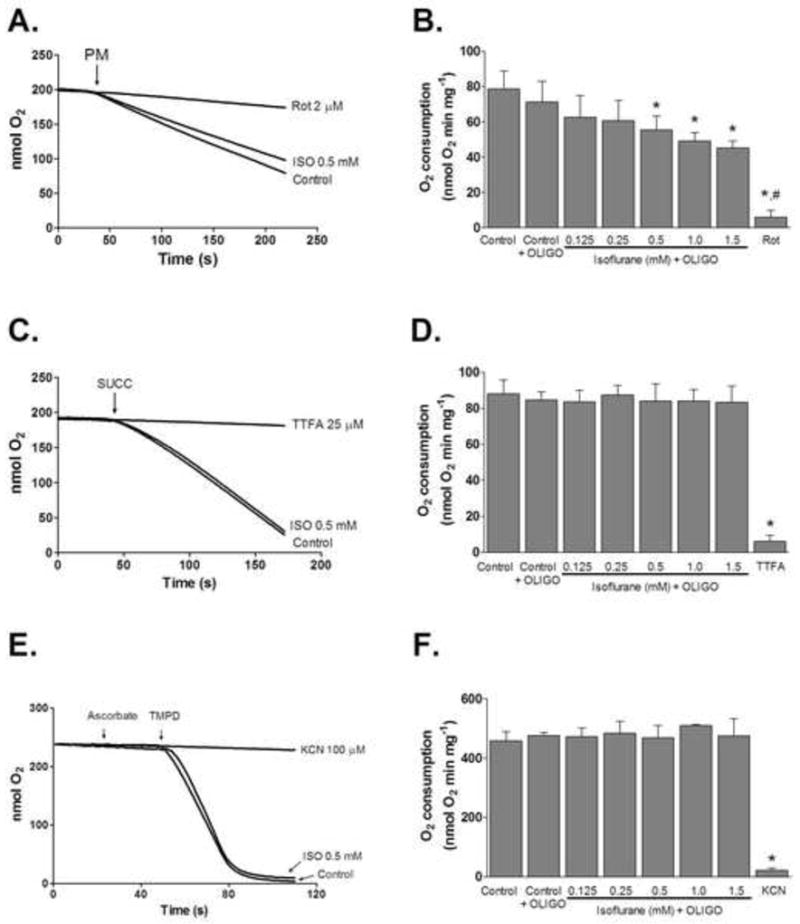
Effect of isoflurane on mitochondrial respiration. (A) Representative respiratory traces of mitochondria respiring on complex I-linked substrates pyruvate plus malate (PM) in the presence of FCCP (1 μM) and oligomycin (OLIGO, 5 μg/ml). Where indicated isoflurane (0.5 mM) or rotenone (Rot, 2 μM) were present in media. (B) Summary data showing dose-dependent reduction of oxygen consumption with isoflurane (ISO). (C and E) Representative respiratory traces of mitochondria respiring on complex II-linked substrate succinate (SUCC) plus rotenone (2 μM) and complex IV-linked substrates ascorbate (2.5 mM) plus TMPD (tetra methyl phenylene diamine, 0.25 mM), respectively. Complex II inhibitor 2-thenoyltrifluoroacetone (TTFA; 25 μM) and complex IV inhibitor potassium cyanide (KCN; 100 μM) were used as positive controls. (D and F) Summarized results demonstrating the lack of an effect of isoflurane on complex II- and IV-supported respiration. All values are means±S.D., n=5, *P<0.05 versus control; #P<0.05 versus isoflurane.
3.2. Depolarization of mitochondrial membrane potential (ΔΨm)
Administration of isoflurane significantly depolarized ΔΨm during pyruvate/malate and succinate-supported respiration in a dose-dependent fashion (Fig. 2A–C). The effect of isoflurane was significantly depended on the substrates used, with higher depolarization observed in the presence of pyruvate/malate. To investigate the role of mitoKATP, mitoBKCa, and uncoupling proteins, 300 μM 5-HD, 10 μM paxilline, and 1 mM GDP were applied, respectively. In addition, experiments were performed in K+-free buffer, where KCl was replaced with choline chloride. As shown in Fig. 2B–C, under these conditions isoflurane-induced depolarization of ΔΨm was not prevented. In contrast, administration of oligomycin (5 μg/ml) significantly attenuated depolarization of ΔΨm by isoflurane (Fig. 2B–C).
Figure 2.
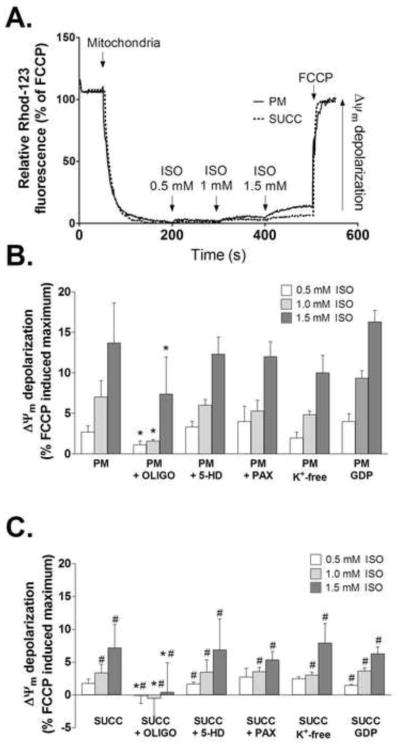
Depolarization of ΔΨm by isoflurane. (A) Representative traces showing isoflurane-induced depolarization of ΔΨm with both complex I (PM) and complex II (SUCC) substrates. FCCP was used to induce maximal depolarization. (B and C) Summary data showing the changes in ΔΨm relative to FCCP-induced maximum with (B) pyruvate and malate or (C) succinate plus rotenone as substrates. Isoflurane-induced depolarization was reversed by oligomycin. ISO = isoflurane; OLIGO = oligomycin; 5-HD = 5-hydroxydecanoic acid; PAX = paxilline; GDP = guanosine diphosphate; FCCP = carbonylcyanide-p-trifluoromethoxyphenylhydrazone. Data are presented as means±S.D., n=7, *P<0.05 versus control; #P<0.05 versus PM.
3.3. Matrix pH measurements
To further investigate whether observed depolarization is due to increase in H+ concentration in mitochondrial matrix or K+ influx, we measured mitochondrial matrix pH with the fluorescent dye BCECF. The representative trace in Fig. 3A shows that BCECF fluorescence, and corresponding mitochondrial pH increased upon addition of substrate due to H+ pumping outside of the mitochondrial matrix. Addition of 250 μM ADP caused transient acidification due to H+ influx through ATP synthase. At the end of the recording, 1 μM FCCP was added to induce maximal matrix acidification by dissipating the H+ gradient. When mitochondria were exposed to the same experimental conditions in the absence of BCECF, changes in the fluorescence ratio (440/490) due to autofluorescence were negligible (not shown). As presented in Fig. 3B, addition of isoflurane caused a slight acidification of the matrix pH. A pH decrease was observed with both, complex I and complex II substrates, but the decrease was greater with pyruvate/malate as substrates than with succinate. Oligomycin decreased the effect of isoflurane while 5-HD, paxilline and GDP failed to prevent the observed acidification. This further supports our hypothesis that K+ influx and uncoupling proteins are not responsible for the observed matrix pH changes and ΔΨm depolarization in isolated mitochondria.
Figure 3.
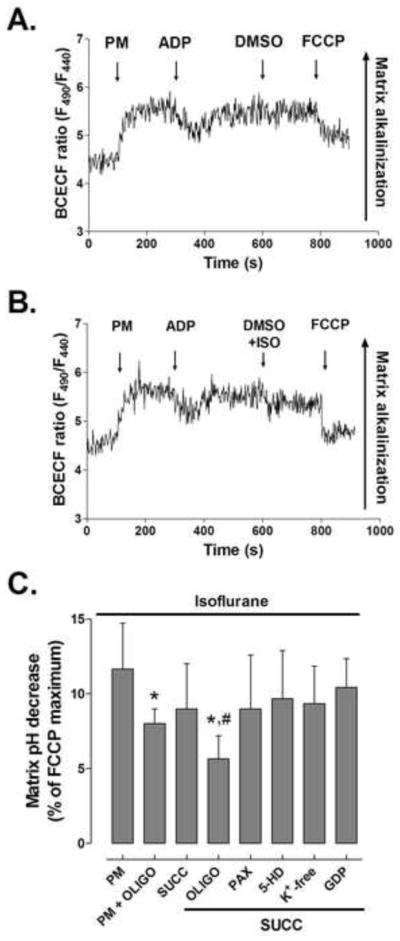
Acidification of mitochondrial matrix by isoflurane. (A) Representative matrix pH trace of BCECF-loaded mitochondria respiring on pyruvate and malate (PM). Addition of substrates raises matrix pH, while ADP induces transient matrix acidification. FCCP abolishes the proton gradient and causes maximal acidification. (B) Isoflurane (ISO) causes a slight decrease in matrix pH. (C) Magnitude of matrix pH decrease relative to FCCP-induced maximum. SUCC = succinate; OLIGO = oligomycin; 5-HD = 5-hydroxydecanoic acid; PAX = paxilline; GDP = guanosine diphosphate. FCCP = carbonylcyanide-p-trifluoromethoxyphenylhydrazone. Values are means±S.D., n=5–7, *P<0.05 versus control, #P<0.05 versus PM + OLIGO.
3.4. MitoKATP channel activity
MitoKATP channel opening has been proposed to increase steady-state matrix volume and to depolarize mitochondria (Andrukhiv et al., 2006). To test the response of mitoKATP channel to isoflurane, K+ influx was followed by the light scattering assay, assessing swelling of mitochondria. The validity of the method was confirmed by applying 1 mM ATP in the presence of oligomycin (5 μg/ml) to block ATP hydrolysis to ADP. Fig. 4A shows that ATP completely blocked swelling of mitochondria. Further, as summarized in Fig. 4B, the inhibitory effect of ATP was not observed in K+-free choline buffer (Wojtovich and Brookes, 2009). The results (Fig. 4B) show that isoflurane (0.5–1.5 mM) did not increase swelling in mitochondria respiring either on succinate or on pyruvate/malate as substrates, respectively. In addition, isoflurane did not change the rate of swelling in the absence of ATP and oligomycin. These results confirm that increased K+ influx is not the mechanism by which isoflurane depolarizes ΔΨm in isolated mitochondria. Application of K+ ionophore valinomycin (1 nM) induced maximal swelling (Fig. 4A, inset).
Figure 4.

Effect of isoflurane on mitochondrial swelling. (A) Representative light scattering traces of mitochondria (0.5 mg/ml) in K+ media supplied with 2 mM succinate. Where indicated, 1 mM ATP or 1 mM ATP plus 0.5 mM isoflurane (Iso) were present in the media from the beginning of the recording. Inset depicts maximum swelling induced by valinomycin (Val). (B) Magnitude of swelling, compared to control, determined as decrease in light scattering at 540 nm during 60 sec. Experimental conditions are listed below the graph. Values are means±S.D., n=4, *P<0.05 versus control.
3.5. ATP synthesis and ATP hydrolysis
Changes in production and hydrolysis of adenine nucleotides can affect the matrix pH. Monitoring the rate of mitochondrial ATP production revealed that at a concentration at which isoflurane depolarized mitochondria no significant inhibition of ATP synthesis was observed with either pyruvate/malate or succinate as substrates. A tendency towards lower activity was only noticed when complex I substrates were used at higher isoflurane doses (Fig. 5A). Similarly, the rate of ATP hydrolysis in SMPs was not affected in the presence of isoflurane (Fig. 5B), suggesting that isoflurane does not affect directly the catalytic center of the ATP synthase.
Figure 5.

Effect of isoflurane on rate of ATP production and ATP hydrolysis. (A) Summary data showing the effect of increasing isoflurane concentrations on ATP synthesis with substrates for complex I or II in isolated mitochondria. (B) Effect of isoflurane on ATP hydrolysis measured as phosphate release in submitochondrial particles. Oligomycin (Oligo) was used as a positive control. PM = pyruvate/malate. All values are means±S.D., n=6, *P<0.05 versus 0 mM isoflurane.
3.6. H+-pumping activity
ACMA fluorescence is quenched when an H+ gradient forms at membranes, and was utilized to assess directly the effect of isoflurane on H+-pumping of ATP synthase. Figure 6A shows a representative trace of ACMA fluorescence quenching induced by ATP hydrolysis in SMPs. The fluorescence quenching induced by ATP was reversed partially by isoflurane, but not by DMSO in the absence of oligomycin (Figure 6B and 6C). Oligomycin almost completely abolished the ATP-induced quenching. DMSO and isoflurane had no effect on ACMA fluorescence in the presence of oligomycin (Figure 6D and 6E). These results, summarized in Figure 6F, suggest that isoflurane modulates H+ flow through ATP synthase.
Figure 6.

Effect of isoflurane (ISO, 0.5mM) on the proton-pumping activity coupled to ATP hydrolysis in submitochondrial particles. (A) Fluorescence quenching of ACMA, indicative of a proton gradient, was induced by addition of 1mM ATP. (B) DMSO had a little effect on ATP-induced ACMA quenching, but (C) ISO decreased quenching significantly. Oligomycin (Oligo) almost completely restored the initial ACMA fluorescence. There was no effect of (D) DMSO and (E) ISO on ACMA fluorescence in the presence of Oligo. (F) Summary data of ACMA fluorescene, expressed relatively to maximal quenching by ATP, are means±S.D.. n=5/group *P<0.05 versus corresponding DMSO group.
3.7. H2O2 generation from complex I during reverse electron transport
When mitochondria use succinate as substrate (in the absence of rotenone), most of the superoxide production occurs in complex I due to reverse electron transport. This reactive oxygen species production is more sensitive to the pH gradient than to the membrane potential (Lambert and Brand, 2004). The rate of H2O2 production from mitochondria respiring on succinate is shown in Fig. 7. In the presence of isoflurane, the rate of H2O2 production was significantly attenuated to 46±19 a.u./min compared to 184±37 a.u./min without isoflurane (P<0.05). A strong decrease in H2O2 production was obtained also with complex I inhibitor rotenone (13±7 a.u./min, P<0.05), suggesting a blockade of reverse electron flow through the complex I. To examine the effect of a matrix pH change induced by isoflurane on H2O2 production, we applied ammonium chloride to increase the matrix pH (Wiederkehr et al., 2009). Under this condition, isoflurane- (0,5 mM), but not rotenone-induced reduction of superoxide production with succinate as substrate was partially reversed (74±30 a.u./min, P<0.05), suggesting that the isoflurane effect was, at least in part, mediated by matrix acidification. In the presence of oligomycin, the effect of isoflurane was also significantly attenuated (67±16 a.u./min; P<0.05).
Figure 7.

Effect of isoflurane on H2O2 production. Summary data of H2O2 release of control mitochondria respiring on succinate (5 mM) with and without isoflurane (0.5 mM). The effect of matrix alkalinization or ATP synthase inhibition was tested by addition of in the ammonium chloride (2 mM) or oligomycin (5 μg/ml). Rotenone (1 μM) was used as a positive control. All data are means±S.D., n=6, *P<0.05 versus corresponding baseline; #P<0.05 versus isoflurane with no addition.
4. Discussion
The present study shows that electron transport chain isoflurane-induced depolarization of ΔΨm and acidifaction of mitochondrial matrix is dependent on action on complex I of the electron transport chain and ATP synthase. Isoflurane decreases reactive oxygen species production from complex I during reverse electron transport in a pH-dependent manner. These changes could underlie the triggering and effecting of anesthetic pre- and postconditioning and thus, protection from ischemia and reperfusion injury.
While a contribution of mitochondria to anesthetic-induced cardioprotection is well established, the precise mechanism of the changes in mitochondrial function is still a matter of controversy. We have previously demonstrated that isoflurane reversibly increased flavoprotein oxidation in isolated cardiac myocytes at clinically relevant doses (Ljubkovic et al., 2007). This was attributed to depolarization of ΔΨm and mitoKATP channel opening (Ljubkovic et al., 2007; Sedlic et al., 2009). In addition to flavoprotein oxidation, isoflurane also increased the NAD(P)H/NAD(P)+ ratio, a possible indicator for inhibition of electron transport chain at the site of complex I (Sedlic et al., 2010). Indeed, when measuring activity of respiratory complexes, we detected inhibition of complex I but not of other electron transport chain complexes (Hirata et al., 2011). Accordingly, our observation that isoflurane reduced mitochondrial oxygen consumption in intact mitochondria respiring on complex I substrates, but not on complex II or IV substrates is consistent with this interpretation. A mild inhibition of the respiratory chain by isoflurane during reperfusion (anesthetic postconditioning) would result in a gradual recovery of mitochondrial function, which has been demonstrated to be protective against reperfusion injury (Burwell et al., 2009).
Depolarization of ΔΨm is another proposed cardioprotective mechanism of isoflurane action on mitochondria (Ljubkovic et al., 2007). Inhibition of the electron transport chain can produce depolarization of ΔΨm. Interestingly, we observed a depolarization of ΔΨm with both substrates, for complex I and complex II while, as discussed above, isoflurane did not inhibit respiration on succinate. The larger isoflurane-induced depolarization that we observed with pyruvate and malate compared with succinate as substrates might be explained by the inhibition of the electron transport chain at the level of complex I combined with a proton leak through ATP synthase. Contrary, isoflurane-induced depolarization in the presence of succinate is mainly mediated by the isoflurane effect on ATP synthase. Previously, opening of mitoKATP channel has been proposed to mediate depolarization of ΔΨm by isoflurane in isolated cardiac myocytes (Ljubkovic et al., 2007; Sedlic et al., 2009), and to mediate isoflurane-induced cardioprotection (Krolikowski et al., 2005). This was mostly based on the reversibility of protection in the presence of 5-HD, but the specificity of this putative mitoKATP inhibitor is questionable (Hanley et al., 2002a). In our experiments on isolated mitochondria, neither blockade of mitoKATP nor of mitoBKCa channel altered the response of ΔΨm to isoflurane. In addition, our observation that isoflurane depolarized ΔΨm in K+-free solution is also consistent with a K+ flux-independent mechanism. Another approach to investigate the effect of isoflurane on K+ flux is to follow mitochondrial swelling that occurs concomitantly to K+ influx (Facundo et al., 2007; Wojtovich and Brookes, 2009). Studies using various known stimuli of preconditioning on mitochondria yielded contradictory results regarding the effect on K+ flux (Ozcan et al., 2002; Wojtovich and Brookes, 2009). In agreement with our other results, we were unable to detect changes in mitochondrial volume even when higher doses of isoflurane were applied. However, we cannot rule out the possibility that activation of mitoKATP by isoflurane may require certain cytosolic signaling proteins such as protein kinase C (Pravdic et al., 2009), and therefore is not observed in isolated mitochondria. Opening of uncoupling proteins has also been implicated as protective mechanism in ischemia and reperfusion injury, possibly by modulating mitochondrial proton leak and decreasing ΔΨm (Nadtochiy et al., 2006). However, we did not observe a reduction in ΔΨm depolarization evoked by isoflurane in the presence of uncoupling protein inhibitor GDP. In contrast, we found the inhibitor of ATP synthase, oligomycin, reversed isoflurane-induced depolarization both with complex I and complex II substrates. This suggests an increased H+ flux through the F0 subunit of the ATP synthase.
We recently reported that cardiac protection at reperfusion by isoflurane is dependent on pH (Pravdic et al., 2010). However, intracellular pH was not affected by isoflurane, suggesting that the protection was not mediated by intracellular acidosis but rather at the level of mitochondrial matrix pH. Here, we provided direct evidence that, similar to ΔΨm depolarization, the decrease of matrix pH is not due to K+ influx or uncoupling protein activation (Dlaskova et al., 2006; Lee et al., 2005; Selivanov et al., 2008). The oligomycin sensitivity of this pH decrease suggests participation of the ATP synthase in this effect. The greater matrix acidification observed with complex I compared to complex II substrate again supported an additional role of complex I in pH regulation.
It is possible that depolarization of ΔΨm and reduction of matrix pH contribute to isoflurane-induced protection. During reperfusion, reverse electron flow-induced reactive oxygen species generation may be an important contributor to injury since during ischemia, succinate concentration rises from low physiological values from 0.2–0.4 mM to 4–7 mM (Heinen et al., 2007; Liu et al., 2002). This, in turn, generates a high ΔΨm and proton gradient during early reoxygenation which promotes reactive oxygen species generation from complex I. The semiquinone radicals associated with electron transport chain complex I and III participate in superoxide anion generation (Ohnishi et al., 2005). The concentration of the semiquinone radical increases with increased pH, as proton binding is proposed to be a limiting step in the reduction of the semiquinone radical to ubiquinol (Selivanov et al., 2008). Therefore, mitochondrial matrix pH affects reactive oxygen species production (Lambert and Brand, 2004; Selivanov et al., 2008), and mitochondrial acidification leads to reduced mitochondrial reactive oxygen species production from complex I during reverse electron transport with succinate as substrate (Lambert and Brand, 2004). Here, the presence of isoflurane reduced reactive oxygen species production form complex I when succinate was used as substrate. This effect was blunted when matrix pH was increased by ammonium chloride, suggesting that matrix acidification by isoflurane can modulate reactive oxygen species production during reverse electron flow. Thus, the effect of isoflurane on H2O2 generation is not dependent on the observed drop in ΔΨm. This is in agreement with our previous observation that isoflurane-induced protection of cardiomyocytes at reoxygenation was abolished at increased extracellular pH (Pravdic et al., 2010). The fact that oligomycin significantly blunted the isoflurane-induced reduction of reactive oxygen species production from complex I during reverse electron transport suggests that an effect of isoflurane on the ATP synthase is one of the underlying mechanisms for ΔΨm and pH change and subsequent reactive oxygen species generation. A protonophoric property of isoflurane is unlikely due to its chemical structure and the fact that ΔΨm and pH change were oligomycin sensitive. However the mechanisms how ATP synthase is involved in this process are not completely understood. We did not observe significant inhibition of ATP-synthesis or stimulation ATP hydrolysis in the presence of isoflurane at doses that produced ΔΨm and pH changes, in contrast to earlier findings in rat liver mitochondria with halothane and chloroform (Rottenberg, 1983). We speculate that isoflurane may increase proton permeability through the F0 part of the ATP synthase, most likely through alterations of the membrane fluidity and without affecting significantly ATP synthesis. This is in agreement with our observation that isoflurane decreases the proton gradient created by ATP hydrolytic activity of ATP synthase in SMPs, whereby this effect was reversed by oligomycin (Fig. 6).
The observed tendency towards lower ATP synthesis activity in the presence of higher isoflurane doses with pyruvate and malate as substrates might be related to complex I inhibition, as in the presence of succinate such trend was not observed (Fig. 5). Interestingly, Bains et al. (Bains et al., 2009) concluded from membrane potential measurements on synaptosomes that sevoflurane but not propofol caused a reversal of ATP synthase function. According to these findings, reversal of ATP synthase compensates for mitochondrial depolarization resulting from electron transport chain inhibition. In contrast, our data in cardiac mitochondria and submitochondrial particles (the later exclude effects dependent on the electron transport chain) indicate that isoflurane directly causes a proton leak through ATP synthase. Either tissue difference (neuronal versus cardiac mitochondria), or, more likely, the experimental model (cells versus isolated mitochondria) may account for this discrepancy. While important signaling pathways are absent when working with isolated mitochondria our study allows dissecting direct mitochondrial effects of isoflurane from those mediated by more complex signaling pathways.
In summary, we report that isoflurane has two distinct direct effects on rat heart mitochondria responsible for ΔΨm depolarization and matrix pH decrease: inhibition of respiratory chain at the site of complex I and an oligoymycin-sensitive H+ leak, possibly by modification of ATP synthase. K+ channels and uncoupling proteins are likely not involved in these direct effects of isoflurane on isolated mitochondria. Matrix pH decrease and subsequent reduction in reactive oxygen species generation may contribute to the isoflurane-induced reduction of mitochondrial injury at reperfusion. Our data suggest that isoflurane when applied at the onset of reperfusion acts rapidly with distinct actions on mitochondrial bioenergetics before pro-survival signaling pathways are activated.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants [P01GM066730, R01HL034708 (to ZJB) and R01HL098490 (to MB)], Bethesda, Maryland. We thank Mary B. Ziebell, Research Technologist, for isoflurane measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci. 2006;27:639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Andrukhiv A, Costa AD, West IC, Garlid KD. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol. 2006;291:H2067–2074. doi: 10.1152/ajpheart.00272.2006. [DOI] [PubMed] [Google Scholar]
- Bains R, Moe MC, Vinje ML, Berg-Johnsen J. Sevoflurane and propofol depolarize mitochondria in rat and human cerebrocortical synaptosomes by different mechanisms. Acta Anaesthesiol Scand. 2009;53:1354–1360. doi: 10.1111/j.1399-6576.2009.02047.x. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Parker N, Buckingham JA, Vidal-Puig A, Halestrap AP, Gunter TE, Nicholls DG, Bernardi P, Lemasters JJ, Brand MD. UCPs--unlikely calcium porters. Nat Cell Biol. 2008;10:1235–1237. doi: 10.1038/ncb1108-1235. author reply 1237–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut-down and gradual wake-up. J Mol Cell Cardiol. 2009;46:804–810. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of Reactive Oxygen Species by Mitochondria. Journal of Biological Chemistry. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology. 2005;102:102–109. doi: 10.1097/00000542-200501000-00018. [DOI] [PubMed] [Google Scholar]
- Da Silva MM, Sartori A, Belisle E, Kowaltowski AJ. Ischemic preconditioning inhibits mitochondrial respiration, increases H2O2 release, and enhances K+ transport. Am J Physiol Heart Circ Physiol. 2003;285:H154–162. doi: 10.1152/ajpheart.00955.2002. [DOI] [PubMed] [Google Scholar]
- De Hert SG, Turani F, Mathur S, Stowe DF. Cardioprotection with volatile anesthetics: mechanisms and clinical implications. Anesth Analg. 2005;100:1584–1593. doi: 10.1213/01.ANE.0000153483.61170.0C. [DOI] [PubMed] [Google Scholar]
- Dlaskova A, Spacek T, Skobisova E, Santorova J, Jezek P. Certain aspects of uncoupling due to mitochondrial uncoupling proteins in vitro and in vivo. Biochim Biophys Acta. 2006;1757:467–473. doi: 10.1016/j.bbabio.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Facundo HT, de Paula JG, Kowaltowski AJ. Mitochondrial ATP-sensitive K+ channels are redox-sensitive pathways that control reactive oxygen species production. Free Radic Biol Med. 2007;42:1039–1048. doi: 10.1016/j.freeradbiomed.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Daut J. K(ATP) channels and preconditioning: a re-examination of the role of mitochondrial K(ATP) channels and an overview of alternative mechanisms. J Mol Cell Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. K(ATP) channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002a;542:735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002b;544:687–693. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen A, Aldakkak M, Stowe DF, Rhodes SS, Riess ML, Varadarajan SG, Camara AK. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am J Physiol Heart Circ Physiol. 2007;293:H1400–1407. doi: 10.1152/ajpheart.00198.2007. [DOI] [PubMed] [Google Scholar]
- Hirata N, Shim YH, Pravdic D, Lohr NL, Pratt PF, Jr, Weihrauch D, Kersten JR, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane differentially modulates mitochondrial reactive oxygen species production via forward versus reverse electron transport flow: implications for preconditioning. Anesthesiology. 2011;115:531–540. doi: 10.1097/ALN.0b013e31822a2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang MT, Nakae Y, Ljubkovic M, Kwok WM, Stowe DF, Bosnjak ZJ. Isoflurane activates human cardiac mitochondrial adenosine triphosphate-sensitive K+ channels reconstituted in lipid bilayers. Anesth Analg. 2007;105:926–932. doi: 10.1213/01.ane.0000278640.81206.92. table of contents. [DOI] [PubMed] [Google Scholar]
- Kevin LG, Novalija E, Riess ML, Camara AK, Rhodes SS, Stowe DF. Sevoflurane exposure generates superoxide but leads to decreased superoxide during ischemia and reperfusion in isolated hearts. Anesth Analg. 2003;96:949–955. doi: 10.1213/01.ANE.0000052515.25465.35. table of contents. [DOI] [PubMed] [Google Scholar]
- Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, Kersten JR, Pagel PS. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: the role of mitochondrial KATP channels. Anesth Analg. 2005;101:1590–1596. doi: 10.1213/01.ANE.0000181288.13549.28. [DOI] [PubMed] [Google Scholar]
- Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KU, Lee IK, Han J, Song DK, Kim YM, Song HS, Kim HS, Lee WJ, Koh EH, Song KH, Han SM, Kim MS, Park IS, Park JY. Effects of Recombinant Adenovirus-Mediated Uncoupling Protein 2 Overexpression on Endothelial Function and Apoptosis. Circ Res. 2005;96:1200–1207. doi: 10.1161/01.RES.0000170075.73039.5b. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–1590. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395:611–618. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi ST, Ohnishi T, Muranaka S, Fujita H, Kimura H, Uemura K, Yoshida K, Utsumi K. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J Bioenerg Biomembr. 2005;37:1–15. doi: 10.1007/s10863-005-4117-y. [DOI] [PubMed] [Google Scholar]
- Ozcan C, Bienengraeber M, Dzeja PP, Terzic A. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H531–539. doi: 10.1152/ajpheart.00552.2001. [DOI] [PubMed] [Google Scholar]
- Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, Losano G, Pagliaro P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol. 2006;101:180–189. doi: 10.1007/s00395-006-0584-5. [DOI] [PubMed] [Google Scholar]
- Pravdic D, Mio Y, Sedlic F, Pratt PF, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane protects cardiomyocytes and mitochondria by immediate and cytosol-independent action at reperfusion. Br J Pharmacol. 2010;160:220–232. doi: 10.1111/j.1476-5381.2010.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravdic D, Sedlic F, Mio Y, Vladic N, Bienengraeber M, Bosnjak ZJ. Anesthetic-induced preconditioning delays opening of mitochondrial permeability transition pore via protein Kinase C-epsilon-mediated pathway. Anesthesiology. 2009;111:267–274. doi: 10.1097/ALN.0b013e3181a91957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riess ML, Kevin LG, McCormick J, Jiang MT, Rhodes SS, Stowe DF. Anesthetic preconditioning: the role of free radicals in sevoflurane-induced attenuation of mitochondrial electron transport in Guinea pig isolated hearts. Anesth Analg. 2005;100:46–53. doi: 10.1213/01.ANE.0000139346.76784.72. [DOI] [PubMed] [Google Scholar]
- Rigoulet M, Devin A, Averet N, Vandais B, Guerin B. Mechanisms of inhibition and uncoupling of respiration in isolated rat liver mitochondria by the general anesthetic 2,6-diisopropylphenol. Eur J Biochem. 1996;241:280–285. doi: 10.1111/j.1432-1033.1996.0280t.x. [DOI] [PubMed] [Google Scholar]
- Rottenberg H. Uncoupling of oxidative phosphorylation in rat liver mitochondria by general anesthetics. Proc Natl Acad Sci U S A. 1983;80:3313–3317. doi: 10.1073/pnas.80.11.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlic F, Pravdic D, Hirata N, Mio Y, Sepac A, Camara AK, Wakatsuki T, Bosnjak ZJ, Bienengraeber M. Monitoring mitochondrial electron fluxes using NAD(P)H-flavoprotein fluorometry reveals complex action of isoflurane on cardiomyocytes. Biochim Biophys Acta. 2010;1797:1749–1758. doi: 10.1016/j.bbabio.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlic F, Pravdic D, Ljubkovic M, Marinovic J, Stadnicka A, Bosnjak ZJ. Differences in production of reactive oxygen species and mitochondrial uncoupling as events in the preconditioning signaling cascade between desflurane and sevoflurane. Anesth Analg. 2009;109:405–411. doi: 10.1213/ane.0b013e3181a93ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selivanov VA, Zeak JA, Roca J, Cascante M, Trucco M, Votyakova TV. The role of external and matrix pH in mitochondrial reactive oxygen species generation. J Biol Chem. 2008;283:29292–29300. doi: 10.1074/jbc.M801019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silic-Benussi M, Cannizzaro E, Venerando A, Cavallari I, Petronilli V, La Rocca N, Marin O, Chieco-Bianchi L, Di Lisa F, D’Agostino DM, Bernardi P, Ciminale V. Modulation of mitochondrial K(+) permeability and reactive oxygen species production by the p13 protein of human T-cell leukemia virus type 1. Biochim Biophys Acta. 2009;1787:947–954. doi: 10.1016/j.bbabio.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Stowe DF, Kevin LG. Cardiac preconditioning by volatile anesthetic agents: a defining role for altered mitochondrial bioenergetics. Antioxid Redox Signal. 2004;6:439–448. doi: 10.1089/152308604322899512. [DOI] [PubMed] [Google Scholar]
- Wiederkehr A, Park KS, Dupont O, Demaurex N, Pozzan T, Cline GW, Wollheim CB. Matrix alkalinization: a novel mitochondrial signal for sustained pancreatic beta-cell activation. Embo J. 2009;28:417–428. doi: 10.1038/emboj.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009;104:121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti F, Gnoni A, Mangiullo R, Papa S. Effect of the ATPase inhibitor protein IF1 on H+ translocation in the mitochondrial ATP synthase complex. Biochem Biophys Res Commun. 2009;384:43–48. doi: 10.1016/j.bbrc.2009.04.046. [DOI] [PubMed] [Google Scholar]