

Abstract
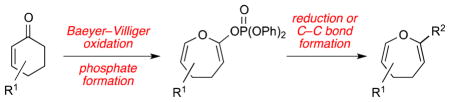
A three-step sequence to access functionalized 4,5-dihydrooxepines from cyclohexenones has been developed. This approach features a regioselective Baeyer–Villiger oxidation and subsequent functionalization via the corresponding enol phosphate intermediate.
4,5-Dihydrooxepines are featured as structural motifs within various natural products, ranging from sesquiterpenes, such as miscandenin1 and endiadric acid derivative beilshmiedin, 2 to polyketides, such as conioxepinol A3 (Figure 1). This framework is also found in some of the most interesting members of the epidithiodiketopiperazine family as represented by aranotin 4 (Figure 1). Consequently, a number of approaches have been developed to access this structural motif. These include acid-catalyzed cyclization, 5 Rh-catalyzed cycloisomerization, 6 ring-closing metathesis, 7 [4+2] cycloaddition/epoxidation/retro [4+2] cycloaddition, 8 Cope rearrangement, 9 fragmentation, 10 and Criegee rearrangement.11
Figure 1.

Selected natural products containing the 4,5-dihydrooxepine structural motif.
Despite this progress, the synthesis of related natural products in which the dihydrooxepine unit is highly functionalized remains challenging, in part because the scope and generality of existing methods are rather limited. Post-functionalization of pre-formed dihydrooxepines is also difficult due to the sensitive nature of these structural moieties. Therefore, a general approach through which substrates with a diverse array of substitution patterns can be reliably transformed into functionalized dihydrooxepines is highly desirable.
As part of our continuing efforts toward the total synthesis of members of the dihydrooxepine epidithiodiketopiperazine family,8a,12 we opted to develop a method to synthesize 4,5-dihydrooxepines from cyclohexenones. Such a strategy would benefit from the ready availability of functionalized cyclohexenones, thus allowing access to a broad range of dihydrooxepine structures. We reasoned that ring expansion of the cyclohexenone could be achieved through a regioselective Baeyer–Villiger oxidation. Further functionalization of the resulting enol lactone through either reduction or C–C bond formation would give rise directly to the bis-enol ether moiety found in 4,5-dihydrooxepines (Figure 2).13
Figure 2.
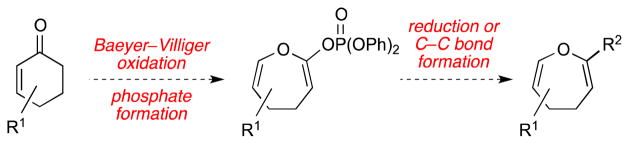
Proposed synthesis of functionalized 4,5-dihydrooxepines from the corresponding cyclohexenones.
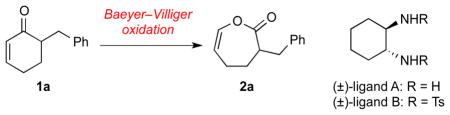
Our experimentation began with the Baeyer–Villiger oxidation of enone 1a (Table 1). Thus, reaction of 1a with mCPBA gave the desired enol lactone as a single regioisomer, albeit in low conversion (Table 1, entry 1). Attempts to use stronger oxidants such as CF3CO3H led to partial decomposition of the product (entry 2). We then reasoned that substrate activation by a suitable Lewis acid would improve conversion under milder reaction conditions that would avoid product decomposition. Indeed, the combination of SnCl4 and bis(trimethylsilyl)peroxide (BTSP), in the presense of trans-1,2-diaminocyclohexane (ligand A), generated the desired product 2a in 83% yield (entry 3).14 The use of this ligand proved to be critical as it successfully tempered the Lewis acidity of SnCl4. Neither SnCl4 itself nor its combination with other ligands tested, such as trans-1,2-di(tosylamino)cyclohexane (ligand B), led to comparable yields (entries 4–6). The presence of dry molecular sieves was essential for the success of this reaction, as in its absence, only trace amounts of the product was observed (entry 7).
Table 1.
Study of the Baeyer–Villiger Oxidation of Enonesa
| ||
|---|---|---|
| entry | conditions | yield (%)b |
| 1c | mCPBA, CH2Cl2 | 15 |
| 2c | UHP, TFAA, CH2Cl2 | decomp. |
| 3c | BTSP, SnCl4, ligand A, 4 Å MS, CH2Cl2 | 83 |
| 4d | BTSP, SnCl4, 4 Å MS, CH2Cl2 | trace |
| 5d | BTSP, SnCl4, ligand B, 4 Å MS, CH2Cl2 | 22 |
| 6d | BTSP, SnCl4, pyridine, 4 Å MS, CH2Cl2 | 32 |
| 7d | BTSP, SnCl4, ligand A, CH2Cl2 | trace |
Reactions were carried out on 0.25 mmol scale.
1H NMR yield.
Reactions were carried out at 0.1 M concentration with 0.5 equiv of SnCl4, 0.5 equiv of ligand A, 3.0 equiv of BTSP and 50 mg 4 Å MS at 25 °C.
Reactions were carried out under the identical conditions in entry 3 with changes indicated in the table.
mCPBA = meta-chloroperoxybenzoic acid, UHP = urea hydrogen peroxide, TFAA = trifluoroacetic anhydride, BTSP = bis(trimethylsilyl)peroxide.
Conversion of the Baeyer–Villiger product 2a to the corresponding enol phosphate 3a went smoothly under our previously developed conditions (Scheme 1).15 The phosphate group was chosen over the more common triflate group because the former is well-known to be more stable than the latter.15
Scheme 1.
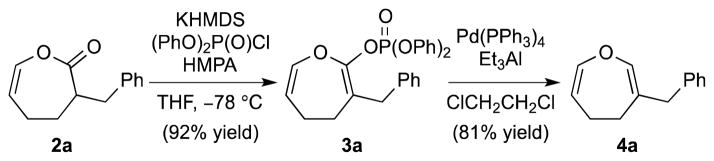
Enol Phosphate Formation and Pd-Catalyzed Reduction to 4,5-Dihydrooxepine
Pd-catalyzed reduction of the diphenyl phosphate 3a proved to be unsuccessful when using either Ph2SiH2 or nBu3SnH as the reducing agent. After extensive screening, Et3Al turned out to be the optimal reducing agent, giving the desired 4,5-dihydrooxepine 4a in 81% yield (Scheme 1). However, when applying the same reduction conditions to phosphate 3b (Table 2), we obtained an inseparable mixture of the desired product 4b and the ethylated product 5b in ca. 3:2 ratio (Table 2, entry 1). This result reflects the competition between β-hydride elimination and reductive elimination of the ethylated intermediate (see Table 2). 16 Attempts to optimize the reduction of 3b by changing the solvent (entry 2) or using the diethyl phosphate 3b′ (entry 3) gave a mixture of 4b and 5b, albeit in different ratios (see Table 2). We then turned to some other reducing agents and found that LiBH4 proved to be the best, giving exclusively the benzodihydrooxepine 4b in good yield [entries 4 (66% yield) and 5 (67% yield)].
Table 2.
Optimization of Enol Phosphate Reductiona
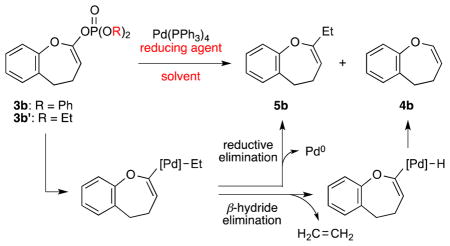
| |||||
|---|---|---|---|---|---|
| entry | R | reducing agent | solvent | yield (%)b (4b+5b) | 4b:5bc |
| 1 | Ph | Et3Al | ClCH2CH2Cl | 58 | 60:40 |
| 2 | Ph | Et3Al | CH2Cl2 | 62 | 37:63 |
| 3 | Et | Et3Al | ClCH2CH2Cl | 70 | 90:10 |
| 4 | Et | LiBH4 | THF | 66 | 4b only |
| 5 | Ph | LiBH4 | THF | 67 | 4b only |
Reactions were run on 0.1 mmol scale at 0.05 M concentration with 0.2 equiv of Pd(PPh3)4 and 2.5 equiv of Et3Al at 25 °C or 10 equiv of LiBH4 at 0 °C.
1H NMR yield.
Ratios determined by 1H NMR spectroscopic analysis.
With the developed optimized conditions in hand, we then proceeded to assess the generality and scope of this three-step procedure to functionalized dihydrooxepines. As shown in Table 3, a variety of substrates with diverse substitution patterns and functional groups could be reliably transformed into the corresponding 4,5-dihydrooxepines. Cyclohexenones with either a methyl group on the olefinic bond (entries 3 and 5) or gem-dimethyl groups on the 4-position (entry 4) are good substrates for these transformation, although the latter exhibits lower reactivity in the first and third steps as compared to the others. Functional groups such as an isolated olefinic bond, an electron-rich arene, a TBS-protected secondary alcohol, or a ketal group are all tolerated in these procedures (entries 5–8). Most notably, the current method is also applicable to relatively complex structures, including the protected Wieland-Miescher ketone 1h and the cholesterol derivative 1i (entries 8 and 9, respectively). Thus, application of the present method to these substrates allows rapid access to the relatively complex dihydrooxepines 4h and 4i, respectively.
Table 3.
Scope and Generality of the 4,5-Dihydrooxepine-Forming Sequencea
| entry | substrate | lactone (% yield)b | enol phosphate (% yield)b | 4,5-dihydrooxepine (% yield)b |
|---|---|---|---|---|
| 1 |
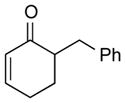 1a |
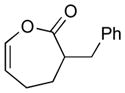 2a (83) |
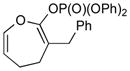 3a (92) |
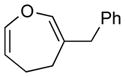 4a (81)c |
| 2 |
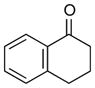 1b |
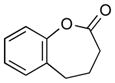 2b (70) |
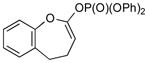 3b (93) |
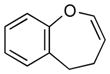 4b (67)d,e |
| 3 |
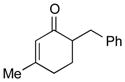 1c |
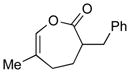 2c (72) |
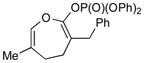 3c (89) |
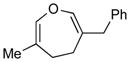 4c (90)c |
| 4 |
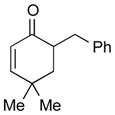 1d |
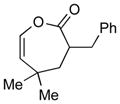 2d (70) |
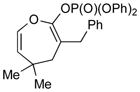 3d (55) |
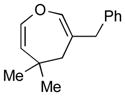 4d (81)c |
| 5 |
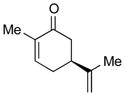 1e |
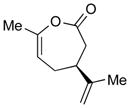 2e (74)e |
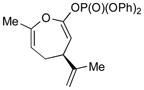 3e (84) |
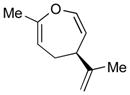 4e (61)d,e |
| 6 |
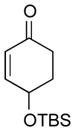 1f |
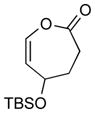 2f [34 (46 brsm)]f |
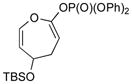 3f (75) |
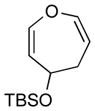 4f (64)d,e |
| 7 |
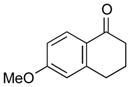 1g |
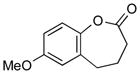 2g (79) |
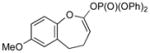 3g (83) |
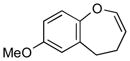 4g (86)d |
| 8 |
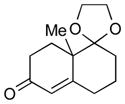 1h |
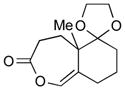 2h (54) |
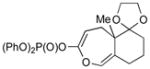 3h (90) |
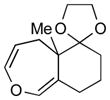 4h (71)d |
| 9 |
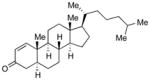 1i |
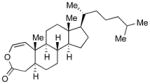 2i (99) |
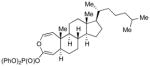 3i (57) |
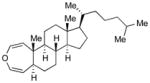 4i (82)d |
Lactone formation: reactions were carried out on 1.0 mmol scale at 0.1 M concentration in CH2Cl2 with 0.5 equiv of SnCl4, 0.5 equiv of ligand A, 3.0 equiv of BTSP and 200 mg 4 Å MS at 25 °C; enol phosphate formation: reactions were carried out on 0.5 mmol scale at 0.1 M concentration in THF with 2.0 equiv of KHMDS, 2.0 equiv of (PhO)2P(O)Cl, 3.0 equiv of HMPA at −78 °C; dihydrooxepine formation (method A): reactions were carried out on 0.2 mmol scale at 0.05 M concentration in ClCH2CH2Cl with 0.2 equiv of Pd(PPh3)4 and 2.5 equiv of Et3Al; dihydrooxepine formation (method B): reactions were carried out on 0.2 mmol scale at 0.05 M concentration in THF with 0.2 equiv of Pd(PPh3)4 and 10 equiv of LiBH4 at 0 °C.
Isolated yield unless otherwise noted.
Using method A.
Using method B.
Due to the volatility of the product, the yield refers to 1H NMR yield.
Anhydrous K2CO3 (200 mg) was added.
brsm = based on recovered starting material.
In addition to the above Pd-catalyzed reduction, the enol phosphate intermediate also provides a platform for a series of C–C bond forming reactions, thereby allowing further functionalization of the dihydrooxepine system. Thus, as demonstrated in Scheme 2, 3b could be successfully engaged in Ni-catalyzed Negishi (conditions a) and Kumada couplings (conditions b), leading to the corresponding alkyl-substituted products 5b and 6b, respectively, without competition from the β-hydride elimination pathway. Introduction of phenyl (conditions c), 3-thienyl (conditions d) and alkylnyl (conditions f) substituents can also be achieved in high yields using PdCl2(dppf) as the catalyst (products 7b, 8b and 10b, respectively). The same catalyst is also effective in converting the phosphate into an ester group (conditions e), albeit in moderate yield (product 9b).
Scheme 2.

Functionalization of the 4,5-Dihydrooxepine Structural Motif via the Corresponding Enol Phosphate
In summary, we have developed a three-step approach for the synthesis of functionalized dihydrooxepines from readily available cyclohexenones. This sequence features a regioselective Baeyer–Villiger oxidation, subsequent enol phosphate formation and Pd-catalyzed functionalization. The large variety of available cyclohexenones provides the basis for the generality of this approach, while the mildness of reaction conditions ensures their reliable transformation to functionalized dihydrooxepines with minimal loss due to facile decomposition. The current method holds considerable promise for application to the synthesis of bioactive natural products and their analogs.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the National Science Foundation (grant CHE-1243661) and the National Institutes of Health, USA (grant AI 055475).
Footnotes
Supporting Information Available: Experimental procedures, characterization and spectroscopic data for new compounds. This material is free of charge via the Internet at http://pubs.acs.org.
References
- 1.Herz W, Subramaniam PS, Santhanam PS, Aota K, Hall AL. J Org Chem. 1970;35:1453. doi: 10.1021/jo00830a043. [DOI] [PubMed] [Google Scholar]
- 2.Chouna JR, Nkeng-Efouet PA, Lenta BN, Wensi JD, Kimbu SF, Sewald N. Phytochemistry Lett. 2010;3:13. [Google Scholar]
- 3.Wang Y, Zheng Z, Liu S, Zhang H, Li E, Guo L, Che Y. J Nat Prod. 2010;73:920. doi: 10.1021/np100071z. [DOI] [PubMed] [Google Scholar]
- 4.Nagarajan R, Huckstep LL, Lively DH, Delong DC, Marsh MM, Neuss N. J Am Chem Soc. 1968;90:2980. [Google Scholar]
- 5.(a) Peng J, Clive DLJ. J Org Chem. 2009;74:513. doi: 10.1021/jo802344t. [DOI] [PubMed] [Google Scholar]; (b) Peng J, Clive DLJ. Org Lett. 2007;9:2939. doi: 10.1021/ol071147z. [DOI] [PubMed] [Google Scholar]
- 6.Codelli JA, Puchlopek ALA, Reisman SE. J Am Chem Soc. 2012;134:1930. doi: 10.1021/ja209354e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Gross U, Nieger M, Bräse S. Chem Eur J. 2010;16:11624. doi: 10.1002/chem.201001169. [DOI] [PubMed] [Google Scholar]; (b) Fustero S, Sánchez-Roselló M, Jiménez D, Sanz-Cervera JF, del Pozo C, Aceña JL. J Org Chem. 2006;71:2706. doi: 10.1021/jo0525635. [DOI] [PubMed] [Google Scholar]
- 8.(a) Nicolaou KC, Lu M, Totokotsopoulos S, Heretsch P, Giguère D, Sun YP, Sarlah D, Nguyen TH, Wolf IC, Smee DF, Day CW, Bopp S, Winzeler EA. J Am Chem Soc. 2012;134:17320. doi: 10.1021/ja308429f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marx JN, Ajlouni A. Nat Prod Commun. 2010;5:5. [PubMed] [Google Scholar]
- 9.(a) Xu X, Hu WH, Zavalij PY, Doyle MP. Angew Chem Int Ed. 2011;50:11152. doi: 10.1002/anie.201105557. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shimizu M, Fujimoto T, Liu X, Takeda Y, Hiyama T. Heterocycles. 2008;76:329. [Google Scholar]; (c) Shimizu M, Fujimoto T, Liu X, Hiyama T. Chem Lett. 2004;33:438. [Google Scholar]; (d) Chou WN, White JB, Smith WB. J Am Chem Soc. 1992;114:4658. [Google Scholar]; (e) Clark DL, Chou WN, White JB. J Org Chem. 1990;55:3975. [Google Scholar]; (f) Aitken RA, Cadogan JIG, Gosney I, Hamill BJ, McLaughlin LM. J Chem Soc, Chem Commun. 1982:1164. [Google Scholar]
- 10.Leyhane AJ, Snapper ML. Org Lett. 2006;8:5183. doi: 10.1021/ol061682j. [DOI] [PubMed] [Google Scholar]
- 11.(a) Goodman RM, Kishi Y. J Org Chem. 1994;59:5125. [Google Scholar]; (b) Lu T, Vargas D, Fischer NH. Phytochemistry. 1993;34:737. doi: 10.1016/0031-9422(94)00625-4. [DOI] [PubMed] [Google Scholar]
- 12.(a) Nicolaou KC, Giguère D, Totokotsopoulos S, Sun Y. Angew Chem, Int Ed. 2012;51:728. doi: 10.1002/anie.201107623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Totokotsopoulos S, Giguère D, Sun Y. J Am Chem Soc. 2011;133:8150. doi: 10.1021/ja2032635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.In their elegant synthesis of acetylaranotin, Tokuyama et al. prepared the 4,5-dihydrooxepine structural motif of the molecule from a lactone via the corresponding enol triflate; see: Fujiwara H, Korugi T, Okaya S, Okano K, Tokuyama H. Angew Chem, Int Ed. 2012;51:13062. doi: 10.1002/anie.201207307.
- 14.Götlich R, Yamakoshi K, Sasai H, Shibasaki M. Synlett. 1997:971. [Google Scholar]
- 15.Nicolaou KC, Shi GQ, Gunzner JL, Gärtner P, Yang Z. J Am Chem Soc. 1997;119:5467. [Google Scholar]
- 16.Sato M, Takai K, Oshima K, Nozaki H. Tetrahedron Lett. 1981;22:1609. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.