

Abstract
Activation-induced deaminase (AID) initiates diversity of immunoglobulin genes through deamination of cytosine to uracil. Two opposing models have been proposed for the deamination of DNA or RNA by AID. Although most data support DNA deamination, there is no physical evidence of uracil residues in immunoglobulin genes. Here we demonstrate their presence by determining the sensitivity of DNA to digestion with uracil DNA glycosylase (UNG) and abasic endonuclease. Using several methods of detection, we identified uracil residues in the variable and switch regions. Uracil residues were generated within 24 h of B cell stimulation, were present on both DNA strands and were found to replace mainly cytosine bases. Our data provide direct evidence for the model that AID functions by deaminating cytosine residues in DNA.
After encountering foreign antigen, B cells diversify their immunoglobulin genes by somatic hypermutation (SHM), gene conversion (GC) and class-switch recombination (CSR). SHM introduces mutations into variable (V)-region genes, and the mutant proteins are then selected by antigen to cause affinity maturation. GC modifies the V-region gene by recombining related gene sequences to encode different proteins. CSR allows antibodies to change their constant (C)-region gene from immunoglobulin M (IgM) to C-region genes of other isotypes to produce antibodies with different effector functions. This phenomenon of genomic mutagenesis is initiated by the enzyme activation-induced deaminase (AID).
AID is a member of the APOBEC family of polynucleotide deaminases that catalyze the conversion of cytosine to uracil in RNA and DNA. On the basis of its sequence similarity to the RNA-editing enzyme APOBEC1, AID was initially hypothesized to function as a RNA deaminase1. The RNA-editing model proposes that AID mutates RNA transcripts by deaminating cytidine to uridine to change codon specificity. The edited RNA might then encode an endonuclease that specifically cleaves DNA in immunoglobulin genes to initiate SHM, GC and CSR2. In contrast, the DNA-deamination model contends that AID mutates DNA, on the basis of the finding that uracil DNA glycosylase (UNG) is required for CSR and GC and influences the mutation spectrum of SHM3–8. UNG binds to U:G mispairs in DNA to remove the uracil base and allow further processing of the remaining abasic nucleotide by the apurinic-apyrimidinic endonuclease APE1. APE1 cleaves the abasic site to produce a single-strand break, and adjacent single-strand breaks on both DNA strands are substrates for CSR. Thus, both UNG and APE1 are particularly integral for CSR if deamination occurs in DNA, whereas they are dispensable if deamination occurs in RNA.
There are conflicting reports on the roles of UNG and APE1 in CSR. The fact that Ung−/− B cells are deficient in CSR suggests that uracil is present in DNA4. However, mutant forms of UNG with impaired glycosylase activity in vitro are proficient in CSR, which suggests that UNG has a nonenzymatic role, such as acting as a scaffolding protein9,10. Indeed, the identification of formation of foci of the histone variant γ-H2AX and the presence of DNA strand breaks in Ung−/− cells suggested a role for UNG in repairing DNA breaks rather than in forming them11. However, biochemical characterization of the UNG mutants has called those results into question because the mutants may retain enough catalytic activity to initiate switching in vivo12,13. There is also disagreement over the role of APE1, with conflicting reports that deficiency in APE1 either results in less CSR14 or does not affect CSR15. Apart from the role of UNG and APE1 enzymes in B cells, examination of AID activity in Escherichia coli and biochemical characterization of AID protein have supported the idea that it deaminates DNA substrates16–18. Nonetheless, controversy remains, as the RNA-editing enzyme APOBEC1 can also function as a DNA deaminase in bacteria19, and recombinant AID binds to RNA substrates18. Collectively, evidence supporting the proposal that AID functions as a DNA deaminase is strong but is not universally accepted, in part because of lack of direct evidence for deamination of either DNA or RNA.
In addition to resolving the mechanism question, the ability to detect uracil residues would show when and where AID deamination occurs in B cells. Traditionally, AID activity has been examined by investigation of the mutational pattern in immunoglobulin genes from mice deficient in UNG and mismatch-repair proteins20–22. Such studies have secondarily identified deamination events by looking at C-to-T transitions that result from the recognition of uracil as thymine by the DNA replication complex. A drawback to this approach is that it requires that the secondary step of replication make the transitions and therefore does not allow detailed examination of when deamination occurs. In this report, we track primary events by physically identifying uracil residues through the sensitivity of genomic DNA to in vitro digestion with UNG and APE1. The method provides an absolute measure of uracil content independently of processing by DNA replication into mutations. This technique has been used before to locate uracil residues in plasmids from bacteria expressing AID23, and we have now modified it to identify them in immunoglobulin genes from B cells.
RESULTS
Experimental strategy
Our strategy to detect the presence of uracil was to isolate genomic DNA from Ung−/− B cells at various stages of activation and then treat the DNA in vitro with UNG to remove uracil. Because recombinant UNG is very specific for the removal of uracil, with no detectable activity in removing the structurally similar bases cytosine or thymine24, we proposed that UNG sensitivity would be a direct measure of uracil content. The resulting abasic site from UNG processing was converted to a nick by the addition of APE1, which produced fragmented DNA. Therefore, the presence of uracil could be measured as either a loss of signal of the intact DNA or a gain of signal from the presence of more fragment ends (Fig. 1).
Figure 1.
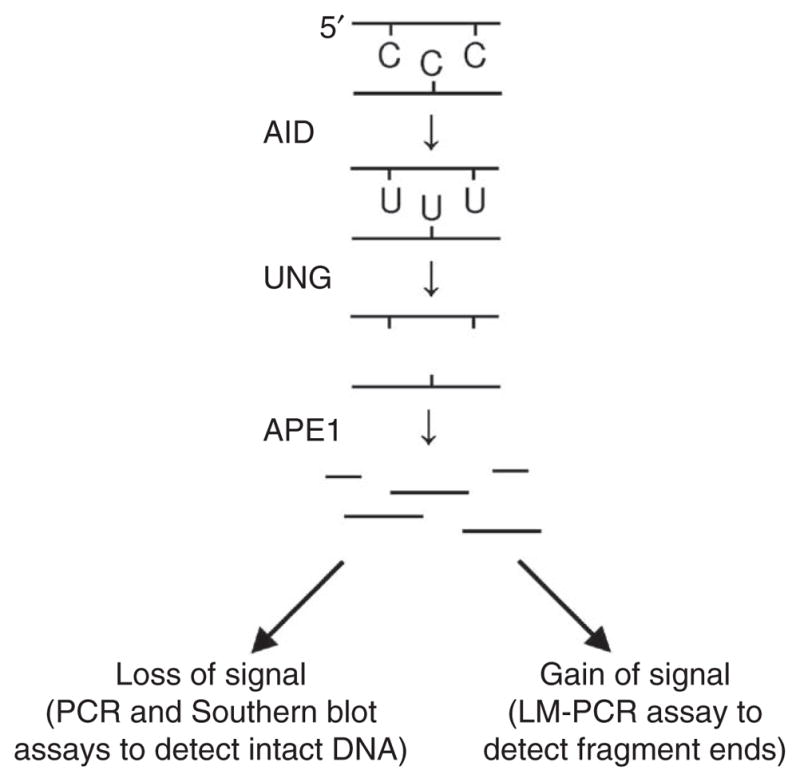
Strategy for identifying uracil residues in DNA. Genomic DNA is isolated from activated Ung−/− B cells and treated with UNG enzyme to remove the uracil bases. The DNA is then digested with APE1 enzyme to convert abasic sites into single-strand breaks. The fragmented DNA is measured in assays that detect either loss of intact DNA or gain of fragment ends. LM-PCR, ligation-mediated PCR.
Uracils in the chicken immunoglobulin λ-chain locus during SHM
We used the DT40 chicken B cell line, which continuously undergoes GC and SHM25,26, to detect the accumulation of uracil residues in the immunoglobulin λ-light chain locus (Igl). To promote abundant generation and retention of uracil residues and to inhibit GC, we used UNG-deficient cells containing a transgene to over-express chicken AID8. In DT40 cells, one allele of Igl is maintained in an unrearranged and inactive configuration, whereas the second is rearranged and actively undergoes GC and SHM. Therefore, we were able to do Southern blot analysis with the unrearranged allele as a loading control for DNA (Fig. 2a). We obtained DNA from cells grown for 2 weeks, digested the DNA with the restriction enzymes SacI and SpeI, then incubated it with or without UNG, followed by treatment of all samples with APE1. The band for the rearranged allele from the UNG-treated sample had lower intensity than that from the untreated sample (Fig. 2b). When standardized to the intensity of the band for the unrearranged allele to control for loading differences, the band for the rearranged allele had a significant decrease in intensity of 25% after treatment. This was dependent on AID, as we found no difference in DNA from AID-deficient (Aicda−/−) cells.
Figure 2.

Uracil residues in the rearranged Igl allele from UNG-deficient DT40 cells. (a) Unrearranged and rearranged alleles. Digestion with the restriction enzymes SacI (Sa) and SpeI (Sp) produces fragments of 6.4 and 4.6 kb. Dotted oval indicates the scope of SHM; gray bar shows the position of the Cλ probe; double-ended arrows indicate the area amplified for quantitative PCR analysis. (b) Southern blot analysis (top) of genomic DNA obtained from cells containing a transgene for overexpression of chicken AID (cAIDR) and from Aicda−/− clones, then treated with APE1 with (+) or without (−) UNG treatment, separated by electrophoresis though an alkaline gel and blotted with the Cλ probe. U, unrearranged; R, rearranged. Below, quantification of band intensity. *P = 0.0001 (two-tailed t-test). Data are representative of six experiments with individual isolation of DNA in each (error bars, s.e.m.). (c) Expression of human AID (top) by DT40 clones transfected with vector encoding wild-type human AID (hAIDR; n = 6 clones) or a catalytically inactive mutant of human AID (hAIDR(E58A); n = 4 clones) or empty vector (Aicda−/−; n = 5 clones), presented relative to the expression of chicken β-actin. ND, not detectable. Below, quantitative PCR analysis of the amplification of VJλ in genomic DNA from treated and untreated samples, presented relative to the amplification of Cλ. *P = 0.0001 (two-tailed t-test). Data are representative of three experiments (average and s.e.m.).
To determine if the sensitivity depended on AID’s deaminase activity, rather than its functioning as a scaffold, we generated Ung−/− Aicda−/− cell lines that expressed human AID. By this strategy, we simultaneously examined lines transfected with either wild-type human AID or a catalytically inactive mutant of human AID27. We selected DT40 clones with similar expression of both the wild-type and mutant constructs (Fig. 2c, top), then grew them for an additional 2 weeks in culture and then assessed the uracil content by quantitative PCR assay to compare the variable–λ-chain joining region (VJλ) with the λ-chain constant region (Cλ).
By quantitative PCR analysis, the amplification of VJλ relative to that of Cλ was 57% lower in treated samples from cells transfected with vector encoding wild-type AID than in untreated samples (Fig. 2c, bottom). Consistent with biochemical studies27, DNA from clones expressing the active-site mutant did not have less amplification, similar to Aicda−/− clones transfected with vector only.
Uracil residues in mouse VH and Vκ regions during SHM
Although the initiation of SHM in DT40 cells and mice is very similar, differences exist in mutation frequency and spectra. Therefore, we extended the uracil analysis to mouse cells undergoing SHM in vivo and used quantitative PCR to specifically identify V and C regions. We isolated DNA from germinal center B cells from the spleens of mice immunized with keyhole limpet hemocyanin (KLH) in adjuvant complex and evaluated fragmentation of the V and C regions of the immunoglobulin heavy-chain (Igh) and immunoglobulin κ-chain (Igk) loci (Fig. 3a). By quantitative PCR analysis, UNG-treated DNA from Ung−/− cells showed lower cycling threshold values that corresponded to a 53% lower value for the VDJ intron and 24% lower value for the VJ intron than for those of untreated DNA, whereas the Cμ and Cκ genes did not have lower values (Fig. 3b). This decrease was due to AID, as there was no difference in amplification from Ung−/−Aicda−/− DNA.
Figure 3.
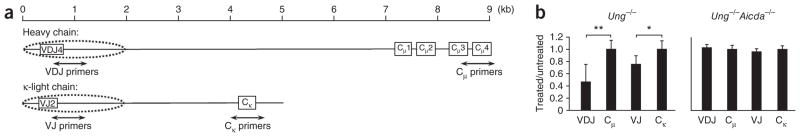
Uracil residues in VH and Vκ regions from mouse germinal center B cells. (a) Rearranged mouse Igh and Igk loci. Primers were designed to amplify 500-bp fragments spanning introns downsteam of V-gene segments rearranged to JH4 and Jκ2 gene segments, and genes encoding Cμ, and Cκ. Dotted ovals indicate the range of SHM on both loci. (b) Quantitative PCR analysis of intact DNA isolated from B220+GL7+ spleen cells of Ung−/− and Ung−/−Aicda−/− mice (immunized with KLH in adjuvant), then treated with UNG and APE1; results are presented as the difference in amplification of treated versus untreated DNA, relative to Gapdh amplification. *P = 0.008 and **P = 0.001 (two-tailed t-test). Data are from six experiments (error bars, s.e.m.).
Uracil residues in the μ-chain switch region during CSR
The third AID-dependent mechanism in B cells is the initiation of CSR. Mutations and strand breaks were present in a 4-kilobase (kb) region encompassing the μ-chain switch region (Sμ)22,28,29 (Fig. 4a) and preceded nonhomologous end joining to recombine Sμ with downsteam S regions. To measure the introduction of uracil residues during CSR, we isolated splenic B cells from Ung−/− mice and cultured them for 3 d ex vivo with lipopolysaccharide (LPS) and interleukin 4 (IL-4). To eliminate apoptotic DNA fragmentation during culture, we collected the cells with a Histopaque gradient to recover live cells. In these cells, we detected AID by immunoblot analysis as early as day 1, and it increased during subsequent days (Fig. 4b). We then collected DNA on days 0–3, and analyzed it by quantitative PCR, comparing regions of Sμ and the reference gene Gapdh of about ~850 base pairs (bp; Supplementary Fig. 1a). The amplification of Sμ from treated DNA was 23% lower on day 1 than that of Sμ from treated DNA (Fig. 4c). This modest sensitivity to UNG treatment may have been due to the stimulation conditions, as only 30% of splenic B cells are activated by LPS in culture30. The decrease in amplification of ~20% could therefore have reflected a 66% decrease in the stimulated population (20% sensitivity/30% stimulated cells). By days 2–3, the DNA was less sensitive to digestion with UNG, even though AID expression continued to increase.
Figure 4.
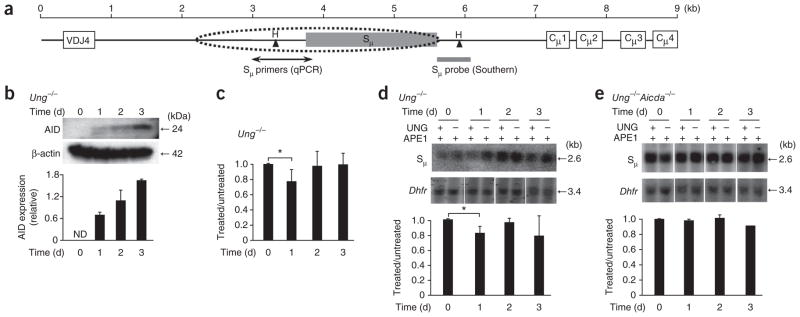
Uracil residues in the Sμ region after ex vivo stimulation of mouse splenic B cells with LPS and IL-4. (a) Map of the Sμ region. Dotted oval indicates the scope of SHM; Sμ primers amplify 858 bp by quantitative PCR; gray bar indicates position of the probe to hybridize a 2.6-kb fragment generated by digestion with HindIII (H). (b) Immunoblot analysis (top) of AID expression in Ung−/− mouse splenic B cells treated with LPS and IL-4. Below, quantification of band intensity, presented relative to that of β-actin. Data are representative of three experiments (error bars, s.e.m.). (c) Quantitative PCR analysis of the amplification of Sμ in intact DNA from Ung−/− cells (presented as in Fig. 3b). *P = 0.02 (two-tailed t-test). Data are representative of four experiments (error bars, s.e.m.). (d,e) Southern blot analysis (top) of intact DNA isolated from Ung−/− cells (d) or Ung−/−Aicda−/− cells (e), digested with HindIII, treated with APE1 with or without UNG, separated by electrophoresis through in alkaline agarose gels and blotted with probes to Sμ and Dhfr. Below, quantification of uracil in treated samples, standardized to Dhfr results and presented relative to that of untreated samples. *P = 0.02 (two-tailed t-test). Data are from three independent experiments (error bars, s.e.m.).
To confirm the phenotype reported above, we used Southern blot analysis to measure the sensitivity of DNA to treatment with UNG. After digesting DNA with HindIII, UNG and APE1, we separated the DNA by electrophoresis through a denaturing gel and transferred it to a membrane. We quantified the remaining 2.6-kb intact band hybridization with a probe targeting the 3′ end of the HindIII-digested fragment, which does not undergo mutation, to ensure complete annealing of the probe. As a control, we measured uracil residues in Dhfr (encoding dihydrofolate reductase; Supplementary Fig. 1b). The Sμ band of treated DNA had lower intensity on day 1 than did the Sμ band of untreated DNA, whereas the Dhfr band had no detectable sensitivity to treatment (Fig. 4d). When we normalized the Sμ values to Dhfr values, we found a significant decrease in intensity of 18% by day 1 (P = 0.02). Consistent with the PCR analysis, DNA collected on days 2–3 did not show substantially less sensitivity, which suggested that uracil residues were less abundant. This phenotype was dependent on AID, as cultured cells from Ung−/−Aicda−/− mice showed no sensitivity to UNG treatment (Fig. 4e).
Balance between AID activity and DNA replication
The loss of UNG sensitivity on day 2 corresponded to the time frame during which ex vivo cells begin to replicate, as identified by incorporation of thymidine analog EdU (5-ethynyl-2′-deoxyuridine; data not shown). To address the effect of DNA replication on uracil content during CSR, we blocked replication to assess whether the frequency of uracil residues would increase; for this, we incubated Ung−/− B cells on day 0 with LPS and IL-4 plus the DNA-replication inhibitor aphidicolin (Fig. 5a). Comparison of B cells cultured for 72 h in the presence or absence of aphidicolin showed a distinct difference in dilution of the cytosolic dye CFSE (carboxyfluorescein diacetate succinimidyl ester), which indicated inhibition of cellular division after the addition of aphidicolin (Supplementary Fig. 2a). Notably, the presence of aphidicolin did not inhibit the expression of AID, which continued to increase over the 2-day culture (Supplementary Fig. 2b). We then examined DNA from cells cultured in aphidicolin for sensitivity to treatment with UNG and APE1 by quantitative PCR and Southern blot analysis to measure the loss of intact DNA in the Sμ region. Similar to the results obtained with cells cultured in the absence of aphidicolin, the DNA showed an initial decrease in band intensity of 20% by day 1 (Fig. 5b,c). However, by day 2, the DNA had a decrease in intensity of ~40%, which suggested that AID continually functioned to increase the uracil content.
Figure 5.

DNA replication decreases the uracil content. (a) Model of the effect of DNA replication on uracil content, with the presumption that AID functions within the first 24 h of stimulation. The U:G mispair would then be replicated so that one daughter cell receives a U:A pair and the other receives a C:G pair, thus diminishing the uracil content by half. When replication is blocked by aphidicolin, uracil residues accumulate. (b) Quantitative PCR analysis of the amplification of Sμ in DNA from Ung−/− B cells (presented as in Fig. 3b). *P = 0.04 and **P = 0.000038 (two-tailed t-test). Data are from four independent experiments (error bars, s.e.m.). (c) Southern blot analysis (top) of DNA from Ung−/− B cells treated with APE1 with or without UNG and separated by electrophoresis through alkaline agarose gels. Below, quantification of uracil content in treated samples, standardized to Dhfr intensity and presented relative to that of untreated samples. *P = 0.02 and **P = 0.0001 (two-tailed t-test). Data are from at least two independent experiments (error bars, s.e.m.).
Uracil residues on both DNA strands
The S regions contain a high density of deoxycytidine tracts on the transcribed strand, which form R-loops during transcription31. One characteristic of R-loop structures is that the nontranscribed strand will be single stranded, whereas the transcribed strand will be in complex with newly formed RNA molecules. Therefore, the nontranscribed strand may be targeted by AID at a higher frequency than the transcribed strand is, as AID requires single-stranded DNA. To test that assumption, we measured the uracil content of both strands in Ung−/− mouse B cells undergoing CSR by Southern blot analysis with strand-specific RNA probes. As a control, we examined DNA from unstimulated cells with each probe and found no sensitivity to treatment with UNG (day 0; Fig. 6a). We then incubated DNA from B cells for 1 d in LPS and IL-4 and assessed its sensitivity to treatment with UNG. Hybridization to a double-stranded DNA probe was 16% lower on day 1 than day 0; hybridization to the nontranscribed probe was 19% lower on day 1 than day 0; and hybridization to the transcribed probe was 14% lower on day 1 than day 0 (Fig. 6b). These results suggested that there was a slight bias for the presence of uracil residues on the nontranscribed strand, which accounted for 58% of the total sensitivity detected with the double-stranded probe, compared with the transcribed strand, which had 42% of the sensitivity (Table 1).
Figure 6.
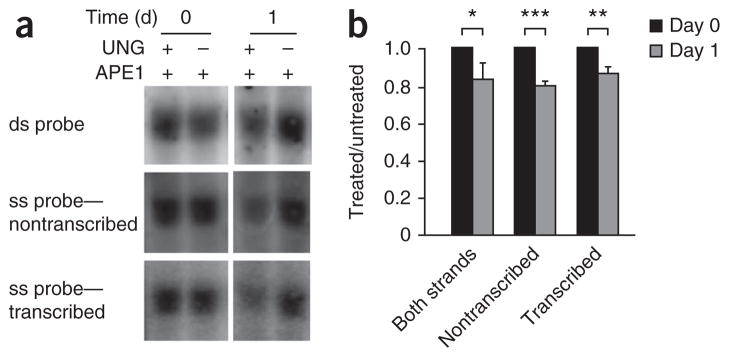
Uracil residues are present on both DNA strands in Sμ after ex vivo stimulation. (a) Southern blot analysis of DNA obtained from Ung−/− splenic B cells and treated with APE1 with or without UNG, then separated by electrophoresis through alkaline agarose gels and hybridized to a double-stranded (ds) probe (both strands) and single-stranded (ss) probes (nontranscribed or transcribed strands); the 2.6 kb band (HindIII digestion) is presented here. (b) Quantification of the uracil content of treated samples in a, presented relative to that of untreated samples. *P = 0.04, **P = 0.008 and ***P = 0.004 (two-tailed t-test). Data are from three independent experiments (error bars, s.e.m.).
Table 1.
Strand contribution to uracil content
| Untreated/treated (day 1) | UNG-sensitive sites | |
|---|---|---|
| Both strands | 84% | 100% |
| Nontranscribed | 81% | 58% |
| Transcribed | 86% | 42% |
Analysis of the contribution of each strand to the total uracil content in both strands, based on the data in Figure 6. Data are the average of three experiments.
Uracil residues replace mainly cytosine residues
To locate the deamination events, we used ligation-mediated PCR to find UNG-dependent DNA breaks in the heavy-chain joining region 4 (JH4) intron. We isolated genomic DNA from germinal center B cells from the spleens of mice immunized with KLH in adjuvant and treated the DNA with UNG and APE1 or with APE1 alone. We then used a mutant form of DNA polymerase-β, which lacks polymerase activity but has lyase activity, to remove the sugar phosphate and leave a single nucleotide gap, for processing of the breaks. We then subjected DNA to a single round of extension by a high-fidelity polymerase, ligated it to an asymmetric linker and amplified it by PCR (Fig. 7a). We separated the products by electrophoresis and identified them by Southern blot analysis with a JH probe. Thus, bands of varying sizes would reflect uracil residues at different positions in the sequence. There were a few faint bands in treated DNA from wild-type C57BL/6 and Ung−/−Aicda−/− mice, as well in some untreated samples. We used many methods to isolate genomic DNA with the least shearing and were unsuccessful in our attempts to clone bands from untreated DNA, which suggested that these bands represented the background limits of the PCR assay. In contrast, UNG-treated DNA from Ung−/− mice and Ung−/− mice also deficient in DNA-mismatch repair; (Ung−/− Msh2−/− mice) showed strongly hybridizing bands (Fig. 7b). Most of these bands were smaller in size because they amplified more efficiently than larger fragments did.
Figure 7.

Detection of uracil residues by ligation-mediated PCR. (a) Experimental design: genomic DNA (100 ng) is treated with UNG, APE1 and DNA polymerase β-lyase to create a blunt 5′ end, then is extended from a primer (arrow) with polymerase to produce a double strand. The product is ligated to a linker and amplified with primers (arrows) for the linker and JH4 intron. (b) Southern blot analysis of DNA obtained from C57BL/6, Ung −/−Aicda−/−, Ung−/− and Ung −/−Msh2−/− B cells, treated as in a (below lanes), amplified by extension initiated ~400 bp downstream of the JH4 gene segment along the nontranscribed strand, separated by gel electrophoresis and hybridized to a JH4 probe. Below, ethidium bromide staining of Gapdh (control for the DNA input used for the initial extension). Data are representative of 50 experiments with amplification of DNA from six to nine mice per strain.
We then cloned and sequenced the bands to locate the position of the DNA breaks. The first missing nucleotide in the ligation site represents the base removed during UNG treatment and is referred to as the ‘linker site’. A map of the linker sites showed that the bands originated from a diverse pool of breaks in the JH4 intron (Fig. 8a and Supplementary Fig. 3a). Although we cloned some large fragments, most of the bands were under 300 bp, probably because of the greater amplification and cloning efficiency of smaller DNA fragments. There was significant over-representation of cytosine at the linker sites in Ung−/− clones (Fig. 8b). To determine if the breaks found at the other three bases were due to mismatch-repair activity, we sequenced DNA from Ung−/−Msh2−/− clones. There was no difference between the two genotypes in the percentage of linkers, which suggested that these breaks occurred during DNA isolation. Another assumption from biochemical studies32–34 and mutational studies35–38 is that AID is more likely to deaminate cytosine in the WGC sequence motif (where ‘W’ is A or T). We therefore compared the total number of cytosine break points in WGC with the percentage of motifs in the sequence (Supplementary Fig. 3b,c). In both the Ung−/− and Ung−/−Msh2−/− samples, we found linkers in the WGC motif at a twofold higher frequency than estimated by chance, although this value was not statistically significant because of the low number of events analyzed.
Figure 8.

Uracil residues preferentially replace cytosine residues. (a) Linker sites identified after sequencing of the PCR products in Figure 7. Downward triangles, linker location; rectangles, JH4 gene segment; arrow, primer for extension. (b) Bases adjacent to the linker ligation position for Ung−/− cells (n = 69) and Ung−/−Msh2−/− cells (n = 48). Size of and numbers in circles indicate proportion and percent, respectively, of total linkers at each nucleotide; data are corrected for nucleotide composition on the nontranscribed strand (C = 16%, G = 27%, T = 31%, A = 26%). *P = 0.00016 for Ung−/− and 0.02 for Ung−/−Msh2−/− (linkers at C versus total cytosine content; Fisher’s exact test). Data are derived from 50 experiments.
Lack of dUTP incorporation during error-prone repair
In addition to the generation of uracil residues by AID deamination, uracil could be misincorporated into DNA during repair synthesis, in which it is inserted opposite template A to generate a U:A mismatch. It has been proposed that a DNA polymerase might use this non-canonical base from the nucleotide pool to fill gaps produced during SHM39,40. To test that hypothesis, we first did DNA-replication assays on a primer-template substrate (Supplementary Fig. 4a) followed by digestion with UNG and APE1. If uracil were incorporated, replication products would be truncated after uracil excision. This analysis showed that polymerase-η used dUTP at a high frequency, whereas polymerase-β and polymerase-η did not (Supplementary Fig. 4b). In fact, polymerase-η, which has a distinct role in producing mutation of A:T pairs during SHM, efficiently incorporated dUTP with kinetics similar to those of its incorporation of dTTP, even when dTTP was in excess (Supplementary Fig. 4c,d). To determine if this polymerase influenced the uracil content of B cells, we next examined DNA from mice deficient in polymerase-η (Polh−/−). Ligation-mediated PCR sequencing analysis showed no difference between Ung−/− and Ung−/−Polh−/− samples in the amount of linkers at T bases (Supplementary Fig. 4e), which suggested that the incorporation of dUTP by polymerase-η does not contribute substantially to SHM. A published study with dUTPase reached the similar conclusion that integration of dUTP does not affect A-to-T mutations41.
DISCUSSION
Rearranged V-gene segments are an important chief target of AID, as the resulting mutations can generate high-affinity antibodies that effectively bind pathogens. V-gene segments can also generate diversity by GC in some species. To detect the presence of uracil residues in cells undergoing both these processes, we analyzed DNA from the DT40 chicken B cell line. The rearranged allele from the Igl locus was sensitive to treatment with UNG and APE1, whereas the unrearranged allele was not, consistent with studies showing that only the rearranged VJ gene undergoes SHM and GC25,26. Additionally, the deamination events required an intact active site in AID, which showed that AID enzymatic activity is required for deamination of DNA. To track deamination events in cells undergoing SHM in vivo, we examined DNA from mouse germinal centers after immunization with KLH in adjuvant. Quantitative PCR analysis with gene-specific primers showed that DNA encoding VH and Vκ regions had 53% and 24% sensitivity to treatment with UNG, respectively, whereas Cμ and Cκ regions had no sensitivity. The partial sensitivity of the V regions might reflect the transient presence of uracil residues, which could be diluted or fixed into mutations after replication. Thus, the PCR assays provided physical evidence that AID actively deaminates DNA in vivo in V regions but not C regions.
A second target of AID is the S regions of the Igh locus, which provide the structural basis for CSR for the production of antibodies with different isotypes. CSR can be readily induced ex vivo in mouse splenic B cells, so the mechanism of deamination can be determined in synchronized cells. Indeed, by doing a time-course study, we found greater sensitivity to UNG and APE1 in the Sμ region after the first 24 h of stimulation but not during subsequent days in culture. However, published time-related studies have shown that double- strand breaks28, foci of the DNA-damage sensor NBS1 (ref. 42), Sμ mutations43 and isotype switching42 do not occur until day 2. Our results have shown that uracil residues were introduced hours before they were processed into strand breaks, mutations and switching. This delay in repair events may increase the number of uracil-containing substrates before cell division to promote switching and mutagenesis while potentially inhibiting the ability to faithfully repair the uracil residues. To determine whether replication does have a role in decreasing the uracil content, we added aphidicolin to cell cultures, which induces an intra-S-phase checkpoint. UNG sensitivity was increased considerably to 40% on day 2 in the absence of cell division, which showed that AID continually deaminates cytosine residues in the Sμ region after the first 24 h. Thus, DNA replication might diminish the load of mutagenic uracil residues to minimize the risk of further breaks and possible translocations.
How many uracil residues are there in the Sμ region after ex vivo activation? The sensitivity to UNG on day 1 was consistent with the proposal that most of the stimulated cells contained at least one uracil residue in a 850-bp region, or ~0.8 uracil residues per kilobase, in Sμ. This is a minimum estimate, as this technique cannot quantify the difference between one or multiple breaks to cleave intact DNA. The amount of uracil in Ung−/− mouse embryo fibroblast cells has been estimated at 2,000 residues per genome44, or about 0.001 per kilobase. Therefore, AID generates ~800-fold more uracil residues in Sμ than is found during normal cellular metabolism. However, it must be noted that this value is calculated from Ung−/− cells, and the total amount of uracil residues at a single time will presumably be lower in wild-type cells.
Other characteristics of AID, beyond its focused targeting to V and S regions, is that it deaminates only single-stranded DNA and has a propensity for WGC hotspot motifs32–34. To compare those biochemical data with the actual activity of AID in B cells, investigators have analyzed the patterns of mutation in V and S regions. However, mutations are the end product over time after multiple processing events associated with DNA replication and repair and may not reflect the primary deamination events. We therefore examined these two characteristics of AID activity by identifying uracil residues in cells.
The first characteristic is that AID is active on both DNA strands with a similar frequency. This is particularly enigmatic in the S regions, which form R-loops that should favor the nontranscribed strand as a single-strand substrate for AID. In fact, in simple in vitro systems, AID has a strong propensity to deaminate cytosine residues on the nontranscribed strand in the context of transcription45–47. However, in Ung−/−Msh2−/− B cells, in which C-to-T transitions represent replication past uracil, there was no evidence for preferential deamination on the nontranscribed strand in Sμ (ref. 22). This suggests that either AID has equal access to both strands in vivo or DNA replication is different in the leading and lagging strands48. To determine if the mutations accurately reflected AID activity, we calculated the percentage of uracil content per strand by Southern blot analysis. The data confirmed that both strands indeed had similar sensitivity to UNG. Thus, despite the secondary structure of Sμ, both the top and bottom strands must be temporarily single stranded long enough to allow AID access for deamination. It is unclear at present how AID can function on both strands, although several models have been proposed, such as supercoiling of DNA during transcription49 or destabilization of the R-loops by RNase50. The second characteristic is that AID mostly deaminates cytosine residues located in the WGC motif. We therefore mapped the location of uracil residues by ligation-mediated PCR and found that most UNG-associated breaks were at cytosine residues, consistent with deamination of DNA. Furthermore, the uracil residues were present in the WGC sequence at a twofold higher frequency than would be expected by random deamination. The structural basis for why AID interacts with WGC sequences has been identified; it is due to an 11–amino acid loop in the protein that interacts with the DNA51,52. Thus, for both characteristics, the uracil residues identified are in agreement with the mutational studies and support the hypothesis that C-to-T transitions are a signature of AID.
In summary, mutagenic uracil residues in immunoglobulin genes are processed by either an error-prone pathway to generate mutations or an error-free process to remove the rogue uracil by base excision or mismatch repair. Notably, we did not detect uracil residues in DNA from wild-type mice, which have both repair pathways. This suggests that the U:G mispair is quickly processed by repair or replication. It remains to be determined if uracil residues persist in B cells from people with compromised DNA repair due to aging or genetic defects, in which uracil residues could remain and become targets for cancer or genome instability.
ONLINE METHODS
Mice and immunization
Ung−/−, Ung−/−Aicda−/− and Ung−/−Msh2−/− mice have been described29,53, and the Ung−/−Polh−/− strain was generated in the mouse colony of P.J.G. (National Institutes of Health). Mice were used at 3–6 months of age. Some mice were immunized intraperitoneally with KLH (50 μg/ml; Calbiochem) in adjuvant complex (Sigma-Aldrich), and spleens were removed 14 d after immunization. All animal procedures were reviewed and approved by the Animal Care and Use Committee of the National Institute on Aging or the Institutional Animal Care and Use Committee of Yale University.
Cell culture and isolation
For experiments with DT40 cells, human AID was amplified from the vectors pMSCV2.2*hAID-eGFP and pMSCV2.2*hAID(E58A)-eGFP27 with the primers human AID-NheI-Fwd (5′-acagaggctagcCCACCATGGACAGCCTCTTGATGAAC-3′) and human AID-BglII-Rev (5′-acagagagatctTCAAAGTCCCAAAGTACGAAATGCG-3′), where lowercase letters indicate the NheI and BglII sites flanking the human AID sequence. The resulting PCR products were digested and ligated into linearized pAID-Express Puro2 vector54 to replace the sequence encoding chicken AID with that encoding human AID. Empty vector plasmid was created by blunt-end cloning of linearized vector with T4 polymerase to fill digestion overhangs. Ung−/−Aicda−/− DT40 cells were transfected by electro-poration of 20 μg linearized plasmid DNA with a Bio-Rad Gene Pulser (580 V, 25 microfarads and infinite resistance). Stable transfectants were selected with puromycin (0.2 μg/ml) and were analyzed by flow cytometry for their expression of IgM and green fluorescent protein (GFP). Quantitative RT-PCR was used for analysis of the expression of human AID in DT40 cells as described27. For mouse experiments, resting splenic B cells were isolated as described29. Cells were cultured in RPMI medium supplemented with 10% (vol/vol) FBS, 1% (vol/vol) penicillin-streptomycin and 50 μM β-mercaptoethanol, with 1% (vol/vol) chicken serum added for DT40 cultures. For ex vivo stimulation, B cells were stimulated with LPS (5 μg/ml; Escherichia coli serotype 0111:B4; Sigma-Aldrich) and IL-4 (5 ng/ml; BD Biosciences). Aphidicolin (1 μg/ml; Sigma-Aldrich) was added for inhibition of DNA replication. Live cells from ex vivo cultures were isolated by separation on Histopaque-1077 density- gradient separation medium (Sigma). For isolation of germinal center B cells, spleens were removed 14 d after immunization and cells were stained with phycoerythrin-labeled antibody to B220 (RA3-6B2; eBioscience) and rat antibody to GL7 (GL-7; eBioscience) and then were stained with fluorescein- labeled antibody to rat IgM (HIS40; eBioscience). A MoFlo cell sorter (Beckman Coulter) was used for sorting of the B220+GL7+ population.
Treatment with UNG and APE1
UNG reactions were done in BER reaction buffer (50 mM HEPES pH 7.5, 20 mM KCl and 2 mM dithiothreitol supplemented with 4 mM EDTA). Reactions were initiated with or without the addition of 10 nM UNG (provided by J. Stivers) in UNG dilution buffer (50% (vol/vol) glycerol, 50 mM Tris, pH 7.5, 100 mM NaCl, 0.1 mM EDTA, 1 mM dithiothreitol and 0.1% (vol/vol) Triton X-100) and were incubated for 30 min at 37 °C. Reactions were then diluted 1:1 with APE1 reaction mix containing BER reaction buffer, 10 mM MgCl2, 5 ng APE1 and 10 ng polymerase β-lyase (provided by S. Wilson) and were incubated for 1 h at 37 °C.
PCR
Primers (Supplementary Table 1) were designed in house and were synthesized by Invitrogen. For quantitative PCR, 10 ng treated genomic DNA in a reaction volume of 25 μl was amplified with PerfeCTa SYBR Green SuperMix reagents according to the manufacturer’s instructions (Quanta BioSciences). The ratio of intact treated genomic DNA to intact untreated genomic DNA was calculated by the Pfaffl method with Cμ or Gapdh (encoding glyceralde-hyde phosphate dehydrogenase) as the reference gene and untreated DNA as the control55. For ligation-mediated PCR, 125 ng treated DNA was subjected to a single round of PCR extension with FideliTaq enzyme (USB) and the oligonucleotide 3′JH3116 by incubation for 5 min at 95 °C, 5 min at 52 °C and 5 min at 68 °C. The extension-reaction products were then purified with PCR purification columns (Qiagen), followed by ligation for 12 h at 16 °C with T4 DNA ligase (Invitrogen) and 250 nM preannealed linker. Samples were then purified (30 μl) and examined for DNA content by amplification of Gapdh with 1 μl and 2 μl template in a standard rTaq (Takara) reaction with GAPDH-F and GAPDH-R primers. Samples were adjusted for DNA content, determined by Gapdh amplification, and were used in touchdown PCR reactions (volume, 20 μl) with rTaq enzyme and primers 3′JH2906 and LL4. The touchdown program consisted of an initial incubation at 95 °C for 3 min followed by 10 cycles of 95 °C for 30 s, 58–55 °C for 30 s and 72 °C for 45 s; the initial three cycles dropped the annealing temperature 1 °C per cycle until a temperature of 55 °C was reached for the remainder of the reaction. After completion, 7 μl of the reaction volume was used for standard rTaq PCR (volume, 25 μl) with the primers 3′JH2826Bam and LL2. For visualization of amplified products, 10 μl of the final PCR product was separated by electrophoresis through a 1% agarose gel, transferred to a HybondN+ membrane (GE Healthcare) and hybridized to 32P-end-labeled oligonucleotide 3′JH2732. Bands were cloned by digestion of the products of final-round reactions with EcoRI and BamHI. Digestion products were then ligated into pBluescript SK+ vector and sequenced with the T7 promoter primer.
Alkaline Southern blot analysis
Genomic DNA (20 μg) was treated and then separated by electrophoresis at 30 V for 24 h through a 1% alkaline agarose gel (50 mM NaOH and 1 mM EDTA). DNA was visualized by staining for 45 min with ethidium bromide in a neutralizing solution (0.5 M Tris pH 7.4 and 1.5 M NaCl). Gels were then blotted overnight to a HybondXL membrane (GE Healthcare) by the alkaline transfer technique with 0.4 M NaOH. DNA was immobilized on the membrane by crosslinking with ultraviolet irradiation and was prehybridized for 3 h at 42 °C in Ultrahyb solution (Ambion). Double-stranded DNA oligonucleotide probes were created by random primer labeling with a High Prime DNA Labeling kit (Roche) and [α-32P]dCTP. Sμ RNA probes were created by cloning of the Sμ fragment into pGEM-T Easy (Promega) and were tested for orientation relative to SP6 RNA polymerase promoters. Plasmids containing Sμ in a defined orientation were then used for in vitro transcription assays with the MAXIscript SP6-T7 kit (Ambion) and [α-32P]UTP. Samples were hybridized for 24 h at 42 °C, followed by washing for 1 h at 55 °C in 0.2× SSC and 0.2% (vol/vol) SDS. Bands were then visualized with phosphor screens and a Typhoon scanner (GE Healthcare). Band intensity was analyzed with ImageQuant software (Molecular Dynamics).
Immunoblot analysis
For analysis of the intracellular abundance of AID, whole-cell extracts from 2 × 106 cells were resuspended in 20 μl PBS solution and were lysed by the addition of 30 μl SDS loading buffer. Samples were boiled and separated by electrophoresis through a 10.5–14% precast gel (Bio-Rad). Proteins were transferred onto a polyvinylidene difluoride membrane (Invitrogen) and visualized with antibody to AID27 or antibody to β-actin (AC15; Sigma), followed by detection with a West Dura Pico kit (Pierce).
Supplementary Material
Acknowledgments
We thank J. Stivers (John Hopkins University) for UNG; S. Wilson (National Institute of Environmental Health Sciences, National Institutes of Health) for polymerase β-lyase; R. Kohli (University of Pennsylvania), J. Buerstedde and H. Arakawa (Max Planck Institute of Biochemistry) for reagents and advice; S. Fugmann and R. Sen for discussions; T. Wolf, C. Nguyen and R. Wersto for assistance in flow cytometry; and the Comparative Medicine Section for mouse maintenance. Supported by the National Institute on Aging and the National Institute of Child Health and Human Development of the National Institutes of Health (Intramural Research Program) and Howard Hughes Medical Institute (D.G.S.).
Footnotes
Author Contributions
R.W.M., H.S., S.A.M. and P.J.G. designed the study; R.W.M., H.S., R.L.M., W.Y., A.V. and H.S.G. did experiments; D.G.S., R.W. and D.M.W. provided reagents and suggestions; and R.W.M. and P.J.G. wrote the manuscript.
Note: Supplementary information is available on the Nature Immunology website.
Additional methods. Information on primer-extension assays by DNA polymerase and kinetic analysis of replication products is available in the Supplementary Methods.
References
- 1.Muramatsu M, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 2.Honjo T, Nagaoka H, Shinkura R, Muramatsu M. AID to overcome the limitations of genomic information. Nat Immunol. 2005;6:655–661. doi: 10.1038/ni1218. [DOI] [PubMed] [Google Scholar]
- 3.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 4.Rada C, et al. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 5.Imai K, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- 6.Di Noia JM, Neuberger MS. Immunoglobulin gene conversion in chicken DT40 cells largely proceeds through an abasic site intermediate generated by excision of the uracil produced by AID-mediated deoxycytidine deamination. Eur J Immunol. 2004;34:504–508. doi: 10.1002/eji.200324631. [DOI] [PubMed] [Google Scholar]
- 7.Kavli B, et al. B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil. J Exp Med. 2005;201:2011–2021. doi: 10.1084/jem.20050042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saribasak H, et al. Uracil DNA glycosylase disruption blocks Ig gene conversion and induces transition mutations. J Immunol. 2006;176:365–371. doi: 10.4049/jimmunol.176.1.365. [DOI] [PubMed] [Google Scholar]
- 9.Begum NA, et al. Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch. Science. 2004;305:1160–1163. doi: 10.1126/science.1098444. [DOI] [PubMed] [Google Scholar]
- 10.Begum NA, et al. Further evidence for involvement of a noncanonical function of uracil DNA glycosylase in class switch recombination. Proc Natl Acad Sci USA. 2009;106:2752–2757. doi: 10.1073/pnas.0813252106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Begum NA, et al. Requirement of non-canonical activity of uracil DNA glycosylase for class switch recombination. J Biol Chem. 2007;282:731–742. doi: 10.1074/jbc.M607439200. [DOI] [PubMed] [Google Scholar]
- 12.Stivers JT. Comment on “Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch”. Science. 2004;306:2042. doi: 10.1126/science.1105225. [DOI] [PubMed] [Google Scholar]
- 13.Di Noia JM, et al. Dependence of antibody gene diversification on uracil excision. J Exp Med. 2007;204:3209–3219. doi: 10.1084/jem.20071768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guikema JE, et al. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204:3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabouri Z, et al. Apex2 is required for efficient somatic hypermutation but not for class switch recombination of immunoglobulin genes. Int Immunol. 2009;21:947–955. doi: 10.1093/intimm/dxp061. [DOI] [PubMed] [Google Scholar]
- 16.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 17.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10:1247–1253. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 20.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6−/−Ung −/− double-knockout mice. J Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- 22.Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med. 2006;203:2085–2094. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martomo SA, Fu D, Yang WW, Joshi NS, Gearhart PJ. Deoxyuridine is generated preferentially in the nontranscribed strand of DNA from cells expressing activation-induced cytidine deaminase. J Immunol. 2005;174:7787–7791. doi: 10.4049/jimmunol.174.12.7787. [DOI] [PubMed] [Google Scholar]
- 24.Slupphaug G, et al. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- 25.Buerstedde JM, et al. Light chain gene conversion continues at high rate in an ALV-induced cell line. EMBO J. 1990;9:921–927. doi: 10.1002/j.1460-2075.1990.tb08190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Humphries EH, Tjoelker L, Carlson L, Thompson CB. Ongoing diversification of the rearranged immunoglobulin light-chain gene in a bursal lymphoma cell line. Mol Cell Biol. 1990;10:3224–3231. doi: 10.1128/mcb.10.6.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohli RM, et al. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. [6 October 2010];J Biol Chem. doi: 10.1074/jbc.M110.177402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J Exp Med. 2005;202:561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajagopal D, et al. Immunoglobulin switch μ sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med. 2009;206:1237–1244. doi: 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson J, Coutinho A, Lernhardt W, Melchers F. Clonal growth and maturation to immunoglobulin secretion in vitro of every growth-inducible B lymphocyte. Cell. 1977;10:27–34. doi: 10.1016/0092-8674(77)90136-2. [DOI] [PubMed] [Google Scholar]
- 31.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 32.Bransteitter R, Pham P, Calabrese P, Goodman MF. Biochemical analysis of hypermutational targeting by wild type and mutant activation-induced cytidine deaminase. J Biol Chem. 2004;279:51612–51621. doi: 10.1074/jbc.M408135200. [DOI] [PubMed] [Google Scholar]
- 33.Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. J Biol Chem. 2004;279:6496–6500. doi: 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- 34.Larijani M, Frieder D, Basit W, Martin A. The mutation spectrum of purified AID is similar to the mutability index in Ramos cells and in ung−/−msh2−/− mice. Immunogenetics. 2005;56:840–845. doi: 10.1007/s00251-004-0748-0. [DOI] [PubMed] [Google Scholar]
- 35.Rada C, Ehrenstein MR, Neuberger MS, Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9:135–141. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- 36.Ehrenstein MR, Neuberger MS. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. EMBO J. 1999;18:3484–3490. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martomo SA, Yang WW, Gearhart PJ. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J Exp Med. 2004;200:61–68. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delbos F, Aoufouchi S, Faili A, Weill JC, Reynaud CA. DNA polymerase η is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J Exp Med. 2007;204:17–23. doi: 10.1084/jem.20062131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuberger MS, et al. Somatic hypermutation at A.T pairs: polymerase error versus dUTP incorporation. Nat Rev Immunol. 2005;5:171–178. doi: 10.1038/nri1553. [DOI] [PubMed] [Google Scholar]
- 40.Roche B, Claes A, Rougeon F. Deoxyuridine triphosphate incorporation during somatic hypermutation of mouse VkOx genes after immunization with phenyloxazolone. J Immunol. 2010;185:4777–4782. doi: 10.4049/jimmunol.1001459. [DOI] [PubMed] [Google Scholar]
- 41.Sharbeen G, et al. Incorporation of dUTP does not mediate mutation of A:T base pairs in Ig genes in vivo. Nucleic Acids Res. 2010 Aug 12; doi: 10.1093/nar/gkq682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petersen S, et al. AID is required to initiate Nbs1/γ-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reina-San-Martin B, et al. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nilsen H, et al. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell. 2000;5:1059–1065. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- 45.Chaudhuri J, et al. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 46.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 47.Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Res. 2003;31:2990–2994. doi: 10.1093/nar/gkg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008;18:521–527. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc Natl Acad Sci USA. 2004;101:12997–13002. doi: 10.1073/pnas.0404974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang FT, et al. Sequence dependence of chromosomal R-loops at the immunoglobulin heavy-chain Sμ class switch region. Mol Cell Biol. 2007;27:5921–5932. doi: 10.1128/MCB.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohli RM, et al. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J Biol Chem. 2009;284:22898–22904. doi: 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M, Rada C, Neuberger MS. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J Exp Med. 2010;207:141–153. S141–146. doi: 10.1084/jem.20092238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu M, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 54.Arakawa H, Saribasak H, Buerstedde JM. Activation-induced cytidine deaminase initiates immunoglobulin gene conversion and hypermutation by a common intermediate. PLoS Biol. 2004;2:E179. doi: 10.1371/journal.pbio.0020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taneyhill LA, Adams MS. Investigating regulatory factors and their DNA binding affinities through real time quantitative PCR (RT-QPCR) and chromatin immunoprecipitation (ChIP) assays. Methods Cell Biol. 2008;87:367–389. doi: 10.1016/S0091-679X(08)00219-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.