

Abstract
A vicinal disulfide ring (VDR) results from disulfide bond formation between two adjacent cysteine residues. This 8-membered ring is a rare motif in protein structures and is functionally important to those few proteins that posses it. This article focuses on the construction of strained and unstrained VDR mimics, discernment of the preferred conformation of these mimics, and the determination of their respective disulfide redox potentials.
Keywords: cyclocystine, dithiocine, dithiazacanones, eight-membered ring, vicinal cysteines, thiol disulfide redox
1.0 Introduction
1.1 VDR biological function
A vicinal disulfide ring (VDR; 1) is an eight-membered ring structure that occurs as a result of disulfide bond formation between vicinal cysteines and is a very rare occurrence in protein structures (Figure 1). In the most current search for this structure in the Brookhaven Protein Data Bank (PDB), Perczel and coworkers found 31 occurrences out of ca. ~28,000 deposited protein structures.1 Given its rare occurrence in the PDB, this ring system must have a unique structural/functional assignment where it is found. In the Janus-faced atracotoxins mutation of this motif to a vicinal serine pair resulted in no change in tertiary geometry of the protein, however the mutant was devoid of toxicity.2 In the case of methanol dehydrogenase, reduction of the VDR is accompanied by complete loss of enzymatic activity. Here it has been proposed that the ring aids in either electron transfer or conformational rigidity of the protein during substrate binding.3,4 We believe one very important role for this ring system is to act as a conformational switch and it is able to do so in two ways. First it may act as a conformational switch by lowering the trans/cis isomerization barrier of the peptide backbone. Several NMR studies of model peptides containing this ring have demonstrated that an 8-membered ring formed by a vicinal disulfide is in dynamic equilibrium between trans and cis conformations about the amide bond of the ring and that conversion from the trans form to the cis form would dramatically alter protein topology.5,6 Such a process is proposed to occur in the nicotinic acetylcholine receptor’s (nAChR) binding site. Upon binding with acetylcholine, or other agonists, the ring undergoes a conformational change that makes it two orders of magnitude more resistant to reduction.7 This type of conformational regulation has also been proposed for the vicinal disulfide ring of cytochrome C from Utricularia.8 Second, it may act as a redox conformational switch. Examples of this type of switch have been shown in human RNase H1,9 and the hSH3N domain of the adhesion and degranulating promoting adapter protein (ADAP).10 The mechanism by which the switch acts is through the change in peptide backbone conformation that is observed on going from the reduced state to the oxidized state. Upon oxidation of vicinal cysteines, formation of the 8-membered ring has the effect of constraining backbone torsion angles ϕ, ψ, and ω, similar to the effect that proline (a 5-membered ring) has on the backbone.11,12 This change in backbone conformation is rather dramatic as has been observed in the case of the hSH3N domain of ADAP. In both RNase H1 and the hSH3N domain of ADAP, it has been proposed that this motif acts as a cellular redox state sensor, as oxidation of the vicinal cysteine pair in RNase H1 leads to inactivation of the enzyme.9 The redox potential of the vicinal cysteine pair for the hSH3N domain has been determined to be −228 mV.10 This value is within the range of disulfide redox potentials found in proteins, but is near the oxidizing end of the scale. We posit that the relative instability of this particular disulfide bond is by design and is due to the strained nature of the ring. The strain in the ring is due to the distorted trans conformation that the ring adopts as determined by NMR spectroscopy. This next point is addressed in the following section.
Figure 1.

VDR mimics
1.2 VDR conformation
When the field of peptide structure was relatively new, it was predicted by Chandrasekaran and Balasubramanian that a VDR dipeptide should have a nonplanar cis geometry with a dihedral angle (ω) of −12°.13 The dihedral angle for the cyclic disulfide L-cysteinyl-Lcysteine was later experimentally determined to be −7° by X-ray crystallography.14,15 Similar cis ω-dihedral angles were also observed in the bicyclic diketopiperazine ring system of cyclo-L-cystine.16,17 A NMR study of VDR 6 in which R2 is replaced with OtBu and the acetyl group replaced with Boc (Figure 1) found evidence for two conformers in chloroform, both with cis amide geometry, in agreement with the earlier crystallographic and NMR studies.18 However, if the amino-terminus in 6 is replaced with gem-dimethyl substituents, two conformations are observed both with trans amide geometries by NMR and X-ray crystallography.19 Another solid state and solution conformational study revealed that a trans conformer was observed if the stereochemistry of the carbon α to the ring amide nitrogen is inverted, as is in the case of phenylacetyl-L-cysteinyl-D-penicillamine.20 When VDR 6 was placed in the context of a heptamer peptide, the conformer population that was cis was found to be 70 ± 5% as determined by NMR spectroscopy.21 However, when VDR 6 was part of the pentameric peptide TCCPD, found in the nAChR, both cis and trans conformers of the VDR were found to exist in solution and these conformers were found to interconvert, lending support for the idea that the ring motif could act as a protein conformational regulatory switch.7 A NMR study of the isolated dipeptide 6 done in aqueous solution revealed the presence of two trans conformers (designated as T- and T′-) and two cis conformers (designated as C+ and C−), with trans and cis isomers being in equilibrium with a ratio of ~60:40, with trans being slightly favored.5
In contrast to the model studies of small peptides above, a complete analysis of the PDB performed by Perczel and coworkers of the 31 deposited structures containing a VDR showed that all of the ring structures were in a strained, distorted trans conformation with average values of ω lying between 161° and −172° with deviations from 180° as large as 44° being observed.1 This discrepancy may be explained by the dynamic nature of the VDR not properly being accounted for in X-ray protein structures in particular. In a NMR study of the bass hepcidin structure it was noted that the most flexible region of the protein was the area containing the VDR.22 Peptide bonds prefer a trans conformation, with a torsion angle of 180°, so that the nitrogen lone-pair can have maximal delocalization into the π-system. This also minimizes steric repulsions from large peptidyl side-chains. However, small rings that have a central bond with restricted rotation will prefer to adopt a cis conformation to minimize ring strain. These disparate facts raise the question of what the preferred amide geometry of a VDR will be. This point was first raised by North, whose inspection of molecular models of smaller amide rings (less than 8 atoms) closed by a disulfide were found to have exclusively cis amide geometry (analogous to cycloalkenes), while larger rings (more than 8 atoms) had trans amide geometry.23 The transition point between amide-disulfide ring systems with all cis geometry and all trans geometry was 8 atoms. These 8-membered ring systems (VDRs) seem to be a special case as they can adopt either geometry and can oscillate between the two types in solution. This ability to easily switch between cis and trans amide geometries may make a VDR a good “molecular switch”,24 regulating the conformation of proteins as has been proposed for the nAChR.8,25
Multiple amide (trans/cis) and disulfide (+/−90°) conformations are energetically feasible in the unique case of a VDR. An all carbon analogue of a VDR that could be used as a point of comparison is cyclooctene. Both are 8-membered ring systems with a central bond that has some type of unsaturation; the element of unsaturation in cyclooctene is a carbon-carbon double bond, while a VDR contains an amide bond with only 40% double bond character. While both a VDR and cyclooctene are similar in having restricted rotation about a central bond and ring size, there are significant conformational differences. For cyclooctene, the cis isomer is more energetically stable than the trans as a result of the ring strain required to incorporate a trans double bond. This ring strain is demonstrated by the higher ΔHH2 of trans-cyclooctene (34.4 kcal/mol) compared to the cis isomer (23.0 kcal/mol).26 By contrast and as noted above, a VDR has a fluxional nature that permits both cis and trans conformers due to: (i) the longer carbon-sulfur and sulfur-sulfur bonds, which makes this 8-membered ring larger and more flexible than its all carbon analogue (ii) the amide bond having only partial double bond character allows for rotation about the C-N axis.
Since a VDR is a potential regulatory conformational switch in proteins, it would be useful to understand the factors that potentiate amide geometry in this system. Studies by North and coworkers found that cyclo-[(R)-cysteinyl-(R)-penicillamine] adopts a cis conformation, while a trans conformation is observed upon changing the penicillamine stereochemistry from R to S.23,27 We have confirmed this observation by constructing cyclo-[L-Cys-D-Cys]. Similar conformational studies showed that switching the stereochemistry of the Cα on the N-terminal Cys results in a strained trans conformation of the molecule with no cis conformers present (data not shown). Thus both stereochemistry and substituents about the ring effect amide geometry of a VDR.
As we were interested in VDRs in the context of a redox regulatory switch, we wanted to determine the redox potential of a cis VDR and a trans VDR. We wanted to mimic the situation in proteins, thus we avoided changing the stereochemistry of the α-carbon and focused instead on changing the properties of the amide bond by N-methylation. It has been previously established that N-methylation of the amide bond increases the population of the cis conformer in peptides.28 Construction of cis and trans VDRs would then allow us to assess how VDR conformation affects the redox potential of the disulfide-bond, and explore our VDR hypothesis that a VDR with a cisoid peptide bond will be a weaker oxidant than a VDR with a transoid peptide bond. Correspondingly, this means that the dithiol with a cisoid peptide bond will be the stronger reductant.
2.0 Results and Discussion
2.1 Determination of redox potentials
In order to test our hypothesis, variants of the VDR are needed with different elements of rigidity (2–7, Figure 1). Fully cis or trans dithiocines 2 and 3 mimic opposite ends of the amide rotational spectrum. Dithiazacanones 4 and 5 are not as rigid as compared to 2 and 3 due to the replacement of the central alkene with an amide bond. North has previously reported that amide 4 exists exclusively in a trans conformation.29 While N-Me amide 5 is unknown in the literature it is predisposed for a cis conformation as a result of steric interactions.28 Dithiazacanones 4 and 5 are the “parent” compounds for VDR mimics as they contain no appendages that would affect the conformation of the ring. Disulfides 6 and 7 however are dipeptides that have substituents on Cα and represent a minimalist VDR. Compound 6 was constructed by Leo and coworkers and displayed 4 major conformations, two being cis and two trans, with a trans/cis ratio of 60:40.5 Compound 7 has not been reported in the literature, but N-methylation should favor a cis amide orientation as is the case in 5.28 The redox potentials are determined by equilibrium thiol-disulfide exchange, with the varying concentrations of reduced and oxidized forms monitored by 1H-NMR (Figure 2).30,31 Employment of oxidized or reduced butane dithiol (BDTox and BDTred, respectively), a species of known redox potential E0(BDT), allows for the redox potential of the VDR mimics to be determined by equations 1 and 2.
| (1) |
| (2) |
Figure 2.

1H-NMR thiol-disulfide equilibrium redox experiment for cis-dithiocine 2
2.2 Synthesis, conformation, and redox potential of dithiocines 2 and 3
Our earlier communication involves the synthesis and redox properties of dithiocines 2 and 3.31 The dithiocine sulfhydryl precursors were readily available from diol intermediates 11 (Scheme 1) and 15 upon Mitsunubo thioesterification and hydrolysis (Scheme 1).32,33 Dithiocine 2 was isolated in good yield upon CsF-celite mediated oxidation.34 However, dithiocine 3 was never isolated even under dilute conditions with the cyclic dimer 18 predominating. The redox data (Table 1) supports our hypothesis by displaying a monomeric redox potential for cis-olefin 2 (−318 mV) and a dimeric redox potential for the trans-olefin 3 (−329 mV).31 The inability to observe a monomeric potential for the trans mimic and its propensity to dimerize suggests a strong oxidative potential. The ring-strain necessary to incorporate the trans-olefin is represented by a fragile disulfide-bond and results in an inability to form the cyclic monomer. Thus the cis-dithiocine is more stable than trans, making 3 a much stronger oxidant in comparison to 2. The dithiol of 2 is thus a very strong reductant. These initial results supported our VDR hypothesis.
Scheme 1.

Reagents and conditions for dithiocine synthesis: (a) mCPBA, K2HPO4, DCM:H2O; (b) 10% H2SO4, THF; (c) NaIO4, THF; (d) NaBH4, H2O, THF; (e) DEAD, Ph3P, AcSH, Et2O; (f) K2CO3, MeOH; (g) CsF-celite, air, ACN 30 mM; (h) H2SO4, MeOH (97%), (i) LAH, Et2O.
Table 1.
VDR redox potentials
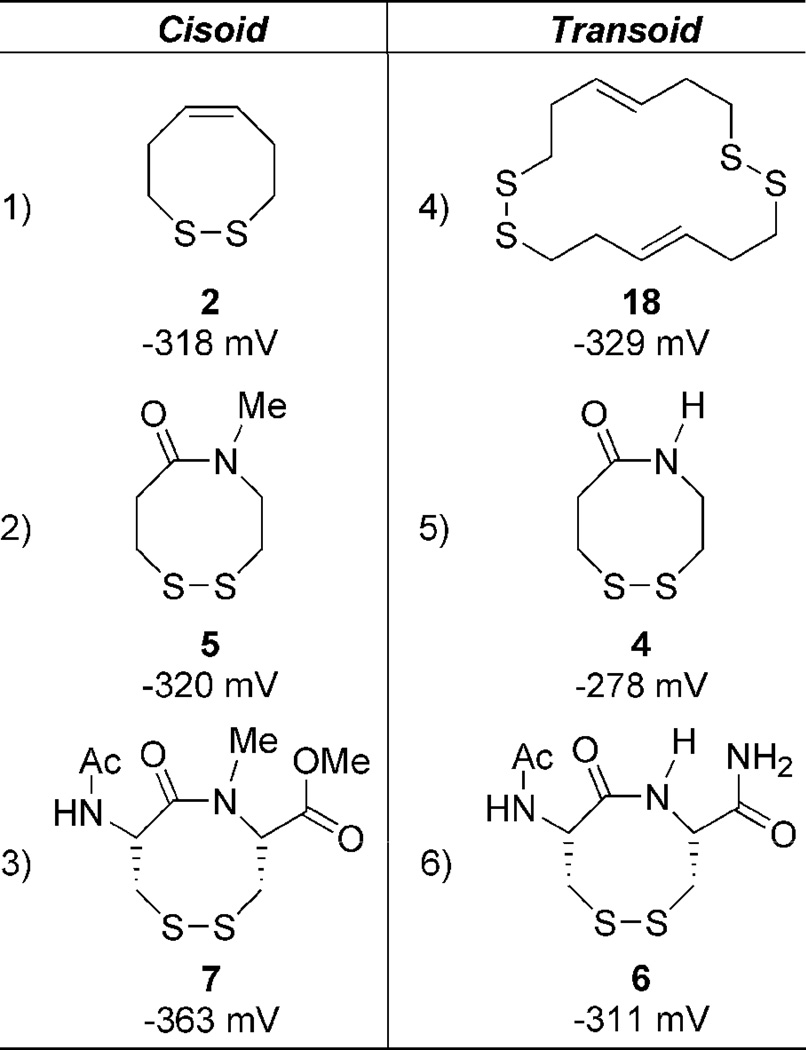 |
2.3 Synthesis, conformation, and redox potential of dithiazacanones 4 and 5
Construction of dithiazacanones 4 and 5 begins with sulfhydryl trityl protection of both acid 19 and amine 20 (Scheme 2).29,35 Standard amide coupling provides common intermediate 23.29,36 Dithiol deprotection,37 followed by oxidation provides 4 in low yield along with two unidentified oligiomeric products.34 Amide N-methylation of 23 generates 24 in mediocre yield,38 after which analogous sulfhydryl deprotection and oxidation affords 5.34,37 Despite the fact that the dithiazacanones are similar in structure they differ in conformation (Figure 3). Prior to this report, North observed that disulfide 4 existed in one trans-conformation.29 However here we have observed that VDR 4 populates two trans-conformations with a ratio of 1.7:1.0. In support of our result, after determining the separate spin-systems in VDR 4 by COSY NMR, ROESY experiments showed magnetization transfer between the two species specifically between the conformers CαN protons and CβC=O protons. This result supports the existence of two conformers (Supporting Information). The trans amide bond geometry was determined at 5 °C by ROESY signals between the amide proton and CαC=O, CβC=O, and CβN protons. As a result of the strong ROESY signals between the amide proton and axial CαC=O and CβN protons, a cis-amide conformation can be ruled out. Disulfide 5 exists in one predominating conformation, however the amide geometry is now cis. Here, TOCSY NMR determined the different spin-systems within VDR 5 and ROESY signals showed a strong through-space interaction between the CαN and CβC=O protons as well as a weaker CαN and CαC=O contact. These through space interactions would not be possible if the amide geometry was trans. A much weaker contact between the N-Me and CβN protons was also observed. Additional support of a cis-amide geometry is demonstrated by this species flexibility at room temperature, reminiscent of dithiocine 2.31 This flexibility was demonstrated by 1H-NMR coalescence experiments (Supporting Information). The observed redox potentials (Table 1), determined by equations 1 and 2, for both 4 (−278 mV) and 5 (−320 mV) are in support of the VDR hypothesis. Amide 4 exists in a trans conformation and as a result of the ring strain needed to accommodate this geometry, it is the stronger oxidant when compared to 5. The trans geometry of the precursor dithiol of 4 disfavors disulfide bond formation since the sulfur atoms are distant, e.g., not close to each other in space. Amide 5 is the weaker oxidant as a result of the cis amide bond relieving ring strain and thus stabilizing the disulfide. In this case, the precursor dithiol of 5 most likely has a significant population of the cis conformer, which brings the sulfur atoms closer in space and favors intramolecular disulfide bond formation. This fact makes the dithiol form of 5 a good reductant. These redox data for the dithiazacanone VDR mimics supports our VDR hypothesis since the more strained system is the stronger oxidant.
Scheme 2.

Reagents and conditions for dithiazacanone synthesis: (a) 4-DMAP, TrtCl, Et3N, DMF; (b) 21, 22, DCC, HOBt, Et3N, DCM:DMF; (c) TES, TFA, DCM; (d) CsF-celite, air, ACN, 1 mM; (e) NaH, MeI, DMF.
Figure 3.

Dithiazacanone VDR conformations
The redox potential of disulfide 4 (−278 mV) is particular interesting in light of the determined redox potential of the single vicinal disulfide bond of the hSH3N domain in ADAP being −228 mV.10 Disulfide 4 has the highest redox potential (strongest oxidant) of all the mimics in Table 1. Since both North and ourselves have demonstrated that the amide bond geometry is in an all trans configuration, it is expected to be the most strained of the amide-containing disulfides that are in this study. While it is not a perfect mimic of the strained disulfide bond within the hSH3N domain of ADAP, the results in Table 1 clearly demonstrate how straining the amide bond within a VDR affects the redox potential of the disulfide bond. Clearly a disulfide bond can be more strained in the context of a folded domain such as that found in the hSH3N domain of ADAP compared to when the disulfide bond is isolated and brought out of its protein context, as is disulfide 4. This is the first study that we are aware of that quantitates redox potentials of vicinal disulfide bonds and makes a comparison to strain in the peptide torsion angle.
2.4 Synthesis, conformation, and redox potential of dipeptides 6 and 7
Construction of the VDR dipeptides begins with fashioning 6.5 Amide coupling of 27 and 28 provides 29 in good yield (Scheme 3).36 Sulfhydryl deprotection and oxidation affords 6 in modest isolated yield.34,37 Construction of 7 was much more labor intensive. Amine methylation proved difficult under a variety of conditions. Fortunately N-methylation was achieved via oxazolidinone intermediate 32.39,40 The oxazolidinone ring opens upon treatment with TES/TFA to provide N-Me cysteine 33, whose sulfhydryl is StBu protected.41 Methyl esterification and Fmoc deprotection ensues,42,43 followed by amide coupling with acid 27 to generate dipeptide 35.44 The StBu-group is removed,45 and after treatment with DTNP cyclizes spontaneously under acidic conditions to produce N-Me VDR 7.46 While the true pathway for this cyclization is currently under investigation, a possible mechanism employs an addition-elimination sequence (Scheme 4). First the trityl-protected sulfur atom is deprotected which generates a free sulfhydryl. The unmasked thiol now attacks the vicinal cysteinyl sulfur atom, whose electrophilicity is enhanced by the Npys group, expelling pyridinyl-thione. The attacking sulfhydyl’s proton is subsequently scavenged. Problematic to this step was also the production of diNpys-protected dipeptide 37, resultant from DTNP protection of both sulfur atoms. Compound 37 can also be transformed into VDR 7 upon treatment with dithiothreitol in similar yield.45 As with the dithiazacanones, VDR 6 and 7 are similar in structure but not in conformation (Figure 4). VDR 6 exists as a mixture of four detectable interconverting cisoid (C+:C− 36:5) and transoid (T-:T’-42:17) amide conformers with transoid being preferred in accordance with what was observed prior by Leo and coworkers (Figure 4, trans/cis 59:41).5 VDR 7 exists in one major conformation that is cisoid in nature (Figure 4). These conformations were determined in a similar manner as dithiazacanone VDRs 4 and 5. The observed redox potentials, determined by equations 1 and 2, for both 6 (−311 mV) and 7 (−363 mV) are in support of our hypothesis (Table 1). While both VDR dipeptides are flexible the N-Me VDR 7 exists preferentially in a cisoid confirmation while 6 is primarily transoid. In accord with prior results VDR 6 is a stronger oxidant, when compared to 7, as a result of a weak disulfide-bond that is a consequence of incorporating a strained transoid amide geometry within the eight-membered ring. Conversely VDR 7 is a weaker oxidant, when compared to 6, due to a stronger disulfide bond that is a result of a less strained cisoid ring system. These redox data for the dipeptide VDR mimics supports our hypothesis that dithiols that form cisoid 8-membered rings will be stronger reductants in comparison to dithiols that form transoid 8-membered rings. This means that a transoid VDR will be much more reactive towards thiol/disulfide exchange reactions and this enhanced reactivity could be harnessed by proteins for either catalytic or regulatory mechanisms. As noted above, all of the vicinal disulfide bonds in proteins so far have been found to be in such a strained state.
Scheme 3.

Reagents and conditions for dipeptide synthesis: (a) DCC, HOBt, Et3N, DCM:DMF; (b) TES, TFA, DCM; (c) CsF-celite, air, ACN, 1 mM; (d) CSA, (CH2O)n, PhH, reflux; (e) TFA, TES, DCM; (f) K2CO3, MeI, DMF; (g) Et2NH, DCM; (h) 27, HATU, Hünigs, DCM; i) DTT, NMM, DMF; (j) 1) DTNP, 2) TFA, 3) TES, DCM; (k) DTT, NMM, DCM.
Scheme 4.

Proposed Mechanism for Formation of VDR 7
Figure 4.

Dipeptide VDR conformations
3.0 Summary
It is interesting to compare trans-dithiol 17 to that of a VDR found in proteins. When the central torsional angle is constrained to 180°, as is the case for 17, intramolecular disulfide-bond formation is impossible. A disulfide-bond can form between nearest neighbors in peptidyl systems because the peptide bond is not as rigid as an olefin. This allows the central peptide bond of the VDR to adopt a strained trans geometry with the lone-pair of electrons on the nitrogen atom out of phase with the π system to varying degrees depending on the strain in the system. This amide bond strain allows disulfide bond formation to occur. Here we have demonstrated a correlation between amide strain and redox potential in a VDR. We also have demonstrated that this strain can be relieved, by forcing the VDR to adopt the cis conformation via N-methylation. A VDR with cis geometry is less strained as evidenced by the lower redox potentials of all of the VDR mimics in this study. While there are currently no known examples of a vicinal disulfide bond with cis amide geometry in the PDB, several redox enzymes contain a vicinal disulfide bond as part of their redox cycle and could use cis amide geometry to influence the redox potential of the VDR. Of particular interest to us are eukaryotic thioredoxin reductases that contain a C-terminal vicinal disulfide bond as part of their active site. The redox potential of the disulfide bond of the protein substrate (thioredoxin) is −270 mV.47 Thus for efficient catalysis to occur, the redox potential of the vicinal disulfide bond of thioredoxin reductase should be more negative (more reducing) than this value. One way to achieve a lower redox potential would be to have the VDR of thioredoxin reductase to adopt a cis amide geometry as shown here. A problem of having the VDR of a protein adopt a cis conformation is that it forces the peptide main chain to make a sharp turn, greatly altering protein topology and affecting protein function. This is most likely the cause of enzyme inactivation in RNase H1 upon oxidation of the vicinal cysteine pair. This problem is largely avoided in the cases of VDRs found in thioredoxin reductase and mercuric ion reductase since the vicinal cysteine pair occurs at the C-terminus of these enzymes, which would minimize the effect of the conformation on the rest of peptide chain. The conformation of the VDR of thioredoxin reductase is an area of intense research in our laboratory.
4.0 Experimental Details
4.1 Materials and methods
Reactions employed oven-dried glassware under argon unless otherwise noted. Argon was passed through a column of anhydrous CaSO4 before use. Amino acids were purchased from Synpep Corp (Dublin, CA). All other chemicals were purchased from either Sigma-Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA) and were used as received or purified by standard procedures.48 Reactions were monitored by thin layer chromatography (TLC) using glass 0.25 mm silica gel plates with UV indicator. Flash chromatography was performed using columns packed with 230–400 mesh silica gel as a slurry in the elution solvent, unless otherwise noted. Gradient flash chromatography was conducted by adsorption of product mixtures on silica gel, packing onto fresh silica bed as a slurry in minimal hexanes, and eluting with a continuous gradient as noted in parenthesizes. Reverse-phase high performance liquid chromatography (RP-HPLC) was executed using a Shimadzu VP system with a Symmetry© C18-5 µm column from Waters (4.6 × 150 mm) for analytical analysis and a SymmetryPrep™ C18-7 µm column from Waters (19 × 150 mm) for prepatory separation. Melting points were determined on a Meltemp apparatus and are uncorrected. Proton and carbon NMR data were obtained with a Varian or Bruker ARX 500 spectrometer at 20 °C unless otherwise noted. Chemical shifts for 1H NMR and 13C NMR are reported in parts per million (ppm) relative to tetramethylsilane (δ = 7.24 ppm for 1H NMR) or chloroform-d (δ = 77.0 ppm for 13C NMR) respectively. Infrared spectra were recorded with a Perkin-Elmer 2000 FT-IR spectrophotometer. Low resolution mass spectra were obtained with a Hewlett Packard 5988 GCMS. High resolution mass spectra were performed by the University of South Carolina Mass Spectrometry Laboratory. A Voyager-DE™ PRO Workstation (Applied Biosystems) was used for mass spectral analysis of peptide samples.
4.2 Synthesis of dithiocine 2
4.2.1 7-Oxa-bicyclo[4.1.0]hept-3-ene (10)
In an air-dried flask 10.0 mL (106 mmol) 1,4-cyclohexadiene was added quickly to a stirring room temperature biphasic solution of 24.38 g (108 mmol) m-CPBA and 18.8 g (108 mmol) K2HPO4 in 762 mL DCM/H2O (151:1). The reaction was stirred at room temperature for 18 hr. Quenched reaction with 200 mL aqueous NaHCO3(sat) and layers were separated. DCM layer was washed once with 150 mL 5% aqueous Na2SO3 and once with 150 mL aqueous NaHCO3(sat). The combined aqueous washes were extracted two sequential times with 100 mL DCM. The DCM extractions were combined, dried over MgSO4, filtered and concentrated to afford 9.56 g (94%) of 10 as a clear oil. Rf = 0.63 (1:2 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.42 (s, 2 H), 3.22 (s, 2 H), 2.56 (d, J = 14.0 Hz, 2 H), 2.43 (d, J = 13.5 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 121.5 (CH), 50.8 (CH), 24.9 (CH2); Ir (neat) (neat) 1736 (m), 1422 (m), 1214 (m), 1021 (m) cm−1; LRMS (EI) m/z 96.0 [(M+), calcd. for C6H8O: 96.0].49
4.2.2 Hex-3-ene-1,6-diol (11)
In an air-dried flask 12 mL H2SO4 was added to a stirring room temperature solution of 6.17 g (64.2 mmol) epoxide 10 in 200 mL THF/H2O (1:1). Upon complete addition the reaction was refluxed (oil-bath=85 °C) for 1 hr and then cooled to 0 °C. Over 30 min 14.4 g (67.4 mmol) NaIO4 was added in three portions after which the reaction was stirred for 1.5 hr. A solution of 3.66 g (96.7 mmol) NaBH4 in 15 mL H2O was then added dropwise via addition funnel to the cold reaction. Upon complete addition the reaction was allowed to warm to room temperature and subsequently stirred for 30 min. After quenching with 100 mL aqueous NH4Cl(sat), the layers were separated and the aqueous was extracted three consecutive times with 100 mL DCM. The DCM extractions were combined, dried over MgSO4, filtered and concentrated. The diol was purified after impregnation onto flash silica gel via gradient flash-chromatography (2:1 hexanes/EtOAc to 1:2 to EtOAc) to provide 3.55 g (48%) of 11 as a light yellow oil. Rf = 0.28 (1:2 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.50 (m, 2 H), 3.59 (t, J = 6.0 Hz, 4 H), 3.52 (br s, 2 H), 2.29 (q, J = 6.2 Hz, 4 H); 13C NMR (125 MHz, CDCl3) δ 128.7 (CH), 61.5 (CH2), 30.4 (CH2); Ir (neat) (neat) 3376 (m), 3336 (m), 1740 (m), 1462 (m), 1350 (m), 1259 (m), 1042 (m) cm−1; LRMS (EI) m/z 116.0 [(M+), calcd. for C6H12O2: 116.0].50
4.2.3 Thioacetic acid S-(6-acetylsulfanyl-(Z)-hex-3-enyl) ester (12)
To a chilled (0 °C) stirring solution of 13.5 g (51.4 mmol) Ph3P in 500 mL anhydrous Et2O, 8.11 mL (51.4 mmol) DEAD was added. After 1 hr a solution of 3.80 mL (53.4 mmol) AcSH and 2.30 g (19.8 mmol) diol 11 in 165 mL anhydrous Et2O was added dropwise via addition funnel over 30 min. Upon complete addition the reaction was kept at 0 °C for 1 hr and then warmed naturally to room temperature. Stirred for 10 hr. The reaction was filtered and triturated with cold anhydrous Et2O. The filtrate was concentrated. The oil was purified via flash-chromatography using 10:1 hexanes/EtOAc eluent to provide 3.49 g (76%) of 12 as a clear oil. Rf = 0.46 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.37 (t, J = 4.8 Hz, 2 H), 2.82 (t, J = 6.8 Hz, 4 H), 2.26 (s, 6 H), 2.24 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 195.1 (C), 128.8 (CH), 30.3 (CH3), 28.5 (CH2), 27.0 (CH2); Ir (neat) (neat) 3038 (w), 1682 (s), 1355 (m), 1130 (s), 1100 (s) cm−1; HRMS (EI) m/z 233.0672 [(M+), calcd. for C10H16O2S2: 233.0670].
4.2.4 Hex-3-ene-1,6-dithiol (8)
In an air-dried flask 0.872 g (6.31 mmol) K2CO3 was added to a stirring solution of 1.00 g (4.30 mmol) 12 in MeOH at room temperature. The reaction was filtered, concentrated, and taken up in 100 mL DCM:H2O (1:1). After separation the DCM layer was washed once with 50 mL aqueous NH4Cl(aq) and two sequential times with 50 mL H2O. The aqueous layers were combined and extracted two consecutive times with 50 mL DCM. The DCM extractions were combined, dried over MgSO4, filtered and concentrated to afford 0.59 g (94%) of 8 as a foul-smelling oil. Rf = 0.11 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.49 (t, J = 4.7 Hz, 2 H), 2.72 (t, J = 7.0 Hz, 4 H), 2.47 (q, J = 7.0 Hz, 4 H), 1.25 (br s, 2 H); 13C NMR (125 MHz, CDCl3) δ 128.9 (CH), 38.4 (CH2), 27.2 (CH2); Ir (neat) 3010 (vw), 1690 (m), 1640 (s), 1415 (m), 1360 (m), 1235 (w), 1199 (s), 1100 (w) cm−1; HRMS (EI) m/z 148.0382 [(M+) calcd. for C6H12S2: 148.0380].
4.2.5 3,4,7,8-Tetrahydro-[1,2]dithiocine (2)
In an air-dried flask 0.853 g (4.02 mmol) CsF-celite was added to a stirring solution of 0.390 g (2.66 mmol) 8 in 89 mL ACN (30 mM) at room temperature. The reaction was vigorously stirred at room temperature for 5 d. After filtration the solid was triturated with ACN and the filtrate concentrated to provide an oil which was purified via alumina (basic; activity 1) chromatography using hexanes eluent to provide 0.249 g (65%) of 2 as a clear oil with a distinct disulfide odor. Rf = 0.87 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.67 (m, 2 H), 2.87 (m, 2 H), 2.64 (m, 4 H), 2.50 (2 H); 13C NMR (125 MHz, CDCl3) δ 129.7 (CH), 39.4 (CH2), 27.2 (CH2); Ir (neat) 3010 (w), 1645 (w), 1448 (m), 1407 (m), 1279 (m), 1249 (w), 966 (w), 886 (w), 726 (s) cm−1; HRMS (EI) m/z 146.0225 [(M+), calcd. for C6H10S2: 146.0224].
4.3 Synthesis of dithiocine dimer 18
4.3.1 (E)-Hex-3-enedioic acid dimethyl ester (14)
In an air-dried flask 1 drop H2SO4 was added to a stirring solution of 2.01 g (13.9 mmol) trans-mucionic acid in 50 mL MeOH at room temperature. The reaction was heated to reflux (oil-bath=90 °C) and stirred for 16 hrs. Cooled to room temperature and partitioned with 40 mL DCM:H2O (1:1). After separation the aqueous layer was extracted two consecutive times with 20 mL DCM The DCM extractions were combined, dried over MgSO4, filtered and concentrated to afford 2.32 g (97%) of 14 as a sweet-smelling oil. Rf = 0.41 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.61 (m, 2 H), 3.59 (s, 6 H), 3.01 (d, J = 1.5 Hz, 4 H); 13C NMR (125 MHz, CDCl3) δ 171.5 (C), 125.7 (CH), 51.5 (CH2), 37.3 (CH3); Ir (neat) 1729 (s), 1453 (m), 1360 (w), 1251 (m), 1154 (s) cm−1; LRMS (EI) m/z 172.0 [(M+) calcd. for C8H12O4: 172.0].51
4.3.2 (E)-Hex-3-ene-1,6-diol (15)
To a stirring solution of 0.919 g (24.2 mmol) LAH in 16 mL anhydrous THF previous cooled to 0 °C, a solution of 2.08 g (12.1 mmol) 14 in 4 mL anhydrous THF was added dropwise via syringe. Upon compete addition the reaction was stirred at 0 °C for 10 min then heated to reflux (oil-bath=40 °C) and stirred for 4 hrs. Cooled to room temperature and quenched cautiously with H2O. After filtration the filtrate layers were separated. The aqueous layer was extracted two consecutive times with 50 mL Et2O. The ethereal extractions were combined, dried over MgSO4, filtered and concentrated to afford 1.23 g (88%) of 15 as a clear oil. Rf = 0.17 (1:2 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.49 (t, J = 1.5 Hz, 2 H), 3.59 (t, J = 6.0 Hz, 4 H), 3.57 (br s, 2 H), 2.24 (q, J = 2.0 Hz, 4 H); 13C NMR (125 MHz, CDCl3) δ 129.2 (CH), 61.4 (CH2), 35.7 (CH2); Ir (neat) 3386 (s), 3336 (s), 1741 (m), 1435 (m), 1365 (m), 1051 (s) cm−1; LRMS (EI) m/z 116.0 [(M+) calcd. for C6H12O2: 116.0].51
4.3.3 Thioacetic acid S-(6-acetylsulfanyl-(E)-hex-3-enyl) ester (16)
Procedure and workup analogous to 12 using 2.32 g (19.9 mmol) 15. After workup and concentration the oil obtained was purified via flash-chromatography using 10:1 hexanes/EtOAc eluent to provide 3.61 g (78%) of 16 as a clear oil. Rf = 0.42 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.44 (m, 2 H), 2.88 (t, J = 7.3 Hz, 4 H), 2.30 (s, 6 H), 2.23 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 195.6 (C), 129.7 (CH), 32.3 (CH2), 30.5 (CH3), 28.7 (CH2); Ir (neat) 3015 (w), 1686 (s), 1429 (m), 1353 (ms), 1131 (s) cm−1; HRMS (EI) m/z 233.0662 [(M+H), calcd. for C10H16O2S2: 233.0670].
4.3.4 (E)-Hex-3-ene-1,6-dithiol (17)
Procedure and workup analogous to 8 using 0.718 g (3.09 mmol) 16 as substrate. After workup and concentration the oil obtained was purified without delay, to avoid oligomer formation, after impregnation onto flash silica gel via gradient flash-chromatography (hexanes to 10:1 to 4:1 to 1:1 to EtOAc) to provide 0.421 g (92%) of 17 as a clear foul-smelling oil. Rf = 0.55 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.47 (m, 2 H), 2.57 (q, J = 7.7 Hz, 4 H), 2.33 (m, 4H), 1.42 (t, J = 7.7 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 129.6 (CH), 38.7 (CH2), 25.6 (CH2); Ir (neat) 3015 (w), 1739 (ms), 1429 (s), 1278 (ms), 1227 (ms) cm−1; HRMS (EI) m/z 148.0379 [(M+) calcd. for C6H12S2: 148.0380].
4.3.5 1,2,9,10-tetrathia-Cyclohexadeca-5,13-diene (18)
Procedure and workup analogous to 2 using 1.85 g (12.7 mmol) 17 in ACN (30.0 µM). After workup and concentration the oil obtained was purified via gradient alumina (basic; activity 1) chromatography (hexanes to 50:1 to 25:1 to 10:1 to 4:1 to EtOAc) to provide 0.192 g (21%) of 18 as a distinctly disulfide-smelling oil. Rf = 0.65 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.62 (m, 2 H), 2.79 (t, J = 7.1 Hz, 4 H), 2.40 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 129.8 (CH), 39.7 (CH2), 31.8 (CH2); Ir (neat) 3010 (w), 1450 (m), 1410 (m), 1268 (m) cm−1; HRMS (EI) m/z 292.0446 [(M+), calcd. for C12H20S4: 292.0448].
4.4 Synthesis of dithiazacanone 4
4.4.1 3-Tritylsulfanyl-propionic acid (21)
In an air-dried flask 10.5 g (37.6 mmol) TrtCl was added to a stirring solution of 3.30 mL (37.6 mmol) 19 in 754 mL (0.05 M) DCM at room temperature. The reaction was stirred at room temperature for 14 hrs and then concentrated. The solid was purified upon recrystallization from MeOH/H2O to provide 10.7 g (82%) 21 as a white solid.39 Rf = 0.60 (4:1 hexanes/EtOAc); mp = 202–204 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.2 (br s, 1 H), 7.33 (s, 12 H), 7.24 (m, 3 H), 2.29 (t, J = 7.3 Hz, 2 H), 2.17 (t, J = 7.3 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 178.6 (C), 144.5 (C), 129.4 (CH), 127.7 (CH), 126.4 (CH), 66.6 (C), 33.3 (CH2), 26.7 (CH2); Ir (neat) 3463 (w), 3010 (s), 2748 (m), 2650 (m), 2569 (m), 1700 (vs), 1429 (s) cm−1; 1231 (s); HRMS (EI) m/z 347.1110 [(M-H), calcd. for C22H20O2S: 347.1106].29,35
4.4.2 2-Tritylsulfanyl-ethylamine (22)
In an air-dried flask 4.90 g (17.6 mmol) TrtCl was added to a stirring solution of 2.00 g (17.6 mmol) 20 in TFA at room temperature where upon dissolution resulted in a deep red color. The reaction was set-aside for 1 hr and subsequently poured into 200 mL water. The solution was made basic by the addition of concentrated KOHaq and vacuum-filtered. The solid was dried over CaSo4 and high-vacuum. The mono/di-protected mixture was dissolved in 1:1 HCl/ACN and poured into ether (10:1 ether/HCl:ACN). The solution was filtered and the solid triturated with ether. Concentrated KOHaq was added to the ethereal solution (1:1) and stirred vigorously mixed for 45 min. After partitioning the ethereal was dried over MgSO4, filtered, and concentrated to afford a 5.62 g (100%) of 22 as a white solid. Rf = 0.17 (4:1 hexanes/EtOAc); mp = 146–148 °C; 1H NMR (500 MHz, CDCl3) 7.41 (m, 6 H), 7.27 (m, 6 H), 7.20 (m, 3 H), 2.68 (br s, 2 H), 2.52 (t, J = 6.4 Hz, 2 H), 2.36 (t, J = 6.4 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 144.6 (C), 129.5 (CH), 127.9 (CH), 126.7 (CH), 66.6 (C), 40.4 (CH2), 34.7 (CH2); Ir (neat) 3379 (br m), 3057 (w), 2932 (w), 1680 (m), 1484 (m), 1439 (m), 1206 (m), 1130 (m), 698 (s) cm−1; HRMS (EI) m/z 320.1489 [(M+H), calcd. for C21H21NS: 320.1473].29,35
4.4.3 3-Tritylsulfanyl-N-(2-tritylsulfanyl-ethyl)-propionamide (23)
To a stirring solution of 1.39 g (3.99 mmol) acid 21 and 1.26 g (3.95 mmol) amine 22 in 40 mL DCM was added 0.69 mL (5.01 mmol) Et3N followed by 1.06 g (7.84 mmol) HOBt. For complete dissolution 2 mL DMF was added. The reaction was stirred at room temperature for 18 hrs and then vacuum-filtered. The filtrate was partitioned with the addition of 50 mL 10% aq, NaHCO3. After separation the DCM layer was washed once with 50 mL 10% aq, NaHCO3. The DCM layer was dried over MgSO4, filtered and concentrated. After impregnation onto flash silica the acid was purified via gradient flash-chromatography (hexanes to 10:1 to 4:1 to 1:1 to EtOAc) to afford 1.72 g (67%) 23 as a white solid. Rf = 0.07 (4:1 hexanes/EtOAc); mp = 138–141 °C; 1H NMR (500 MHz, CDCl3) δ 7.39 (m, 12 H), 7.25 (m, 12 H), 7.19 (m, 6 H), 5.29 (t, J = 6.0 Hz, 1 H), 2.99 (q, J = 6.2 Hz, 2 H), 2.44 (t, J = 7.3 Hz, 2 H), 2.34 (t, J = 6.2 Hz, 2 H), 1.92 (t, J = 7.5 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 170.5 (C), 144.6 (C), 144.5 (C), 129.5 (CH), 129.4 (CH), 127.9 (CH), 127.8 (CH), 129.7 (CH), 126.6 (CH), 66.8 (C), 66.7 (C), 38.0 (CH2), 35.5 (CH2), 31.8 (CH2), 27.6 (CH2); Ir (neat) 3469 (m), 3058 (m), 2933 (m), 1703 (s), 1488 (s), 1444 (s), 1252 (m), 1155 (m), 1074 (m), 1009 (m), 695 (s) cm−1; HRMS (EI) m/z 688.2094 [(M+K), calcd. for C43H39NOS2: 688.2110].29
4.4.4 3-Mercapto-N-(2-mercapto-ethyl)-propionamide (25)
In an air-dried flask 256 mL TFA was added to a stirring solution of 5.00 g (7.69 mmol) 23 in 512 mL (15 mM) DCM at room temperature. The yellow color produced was then quenched by the addition of 4.96 mL (30.7 mmol) TES. Upon complete addition the reaction was stirred at room temperature for 1 hr and then concentrated. The oil was redissolved in 500 mL DCM and concentrated. This was repeated two additional times. After impregnation onto flash silica the dithiol was purified via flash-chromatography (4:1hexanes/EtOAc) to provide 1.16 g (92%) 25 as a foul-smelling oil. Rf = 0.72 (1:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.98 (br s, 1 H), 3.46 (q, J = 6.3 Hz, 2 H), 2.82 (q, J = 8.7 Hz, 2 H), 2.68 (q, J = 8.7 Hz, 2 H), 2.52 (t, J = 6.3 Hz, 2 H), 1.63 (t, J = 8.7 Hz, 1 H), 1.39 (t, J = 8.7 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 170.7 (C), 42.3 (CH2), 40.3 (CH2), 24.6 (CH2), 20.4 (CH2); Ir (neat) 3283 (m), 3076 (w), 2933 (w), 1640 (s), 1539 (s), 1420 (m), 1355 (m), 258 (m), 1198 (m) cm−1; HRMS (EI) m/z 243.4738 [(M+2K), calcd. for C5H11NOS2: 243.4735].
4.4.5 [1,2,5]-Dithiazocan-6-one (4)
Procedure and workup analogous to 2 using 0.39 g (2.35 mmol) 25 as substrate in ACN (1 mM). After workup the oil was triturated with hexanes overnight. The white solid obtained after filtration with triturated with excess DCM to provide 70.2 mg (18%) 4 in two conformations (1.7:1.0) as a white solid. Rf = 0.65 (1:1 hexanes/EtOAc); mp = 178–181 °C; 1H NMR (500 MHz, DMSO-d6) δ Major conformer: 8.15 (m, 1 H), 3.32 (m, 2 H), 2.90 (m, 2 H), 2.79 (m, 2 H), 2.47 (m, 2 H); Minor conformer: 8.22 (m, 1 H), 3.31 (m, 2 H), 2.89 (m, 2 H), 2.84 (m, 2 H), 2.52 (m, 2 H); 13C NMR (125 MHz, DMSO-d6) δ Major conformer: 170.1 (C), 37.6 (CH2), 37.0 (CH2), 34.4 (CH2), 33.9 (CH2); Minor conformer: 173.3 (C), 39.0 (CH2), 38.9 (CH2), 37.5 (CH2), 34.9 (CH2); Ir (neat) 3287 (m), 3068 (w), 2925 (w), 1633 (s), 1542 (s), 1416 (m), 1358 (m), 1258 (m), 1201 (m), 710 (w) cm−1; HRMS (EI) m/z 163.0120 [(M+), calcd. for C5H9NOS2: 163.0126].29
4.5 Synthesis of N-Me dithiazacanone 5
4.5.1 N-Methyl-3-tritylsulfanyl-N-(2-tritylsulfanyl-ethyl)-propionamide (24)
A solution of 1.55 g (2.38 mmol) 23 in 5 mL anhydrous DMF was added dropwise to a solution of 0.22 g (5.72 mmol) NaH in 20 mL anhydrous DMF previously chilled to 0 °C. After complete addition the reaction was stirred at 0 °C for 10 min, after which 1.18 mL (19.0 mmol) MeI was added rapidly dropwise. The reaction was stirred at 0 °C for 30 min and then warmed naturally to room temperature. The reaction was stirred at room temperature for 14 hrs and then quenched with excess phosphate buffer (pH=7). The solution was then poured into 100 mL EtOAc and the biphasic solution separated. The EtOAc layer was washed once with 100 mL brine, dried over MgSO4, filtered and concentrated. After impregnation onto flash silica the N-methyl amide was purified via gradient flash-chromatography (hexanes to 10:1 to 4:1 to 1:1 to EtOAc to DCM) to provide 1.37 g (87%) 24 in two indistinguishable conformations (~1.0:1.0) as a white solid. Rf = 0.45 (4:1 hexanes/EtOAc); mp = 146–148; 1H NMR (500 MHz, CDCl3) δ Conformer mixture: 7.45 (dd, J = 16.1, 7.3 Hz, 24 H), 7.28 (m, 24 H), 7.22 (m, 12 H), 3.06 (t, J = 7.3 Hz, 2 H), 2.80 (t, J = 7.3 Hz, 2 H), 2.56 (s, 3 H), 2.49 (m, 4 H), 2.47 (s, 3 H), 2.34 (t, J = 7.3 Hz, 2 H), 2.29 (t, J = 7.3 Hz, 2 H), 2.02 (t, J = 7.3 Hz, 2 H), 1.93 (t, J = 7.3 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ Conformer mixture: 170.6 (C), 170.5 (C), 144.8 (C), 144.7 (C), 144.6 (C), 144.4 (C), 129.6 (CH), 129.5 (CH), 129.5 (CH), 129.4 (CH), 127.9 (CH), 127.8 (CH), 127.7 (CH), 127.7 (CH), 126.8 (CH), 126.6 (CH), 126.5 (CH), 126.5 (CH), 67.2 (C), 66.7 (C), 66.7 (C), 66.6 (C), 48.8 (CH2), 47.2 (CH2), 35.6 (CH3), 33.1 (CH3), 32.7 (CH2), 32.1 (CH2), 29.8 (CH2), 29.5 (CH2), 27.2 (CH2), 27.0 (CH2); Ir (neat) 3464 (m), 3062 (m), 2924 (m), 1694 (m), 1489 (m), 1444 (s), 1148 (s), 1009 (s), 755 (s), 695 (s) cm−1; HRMS (EI) m/z 664.2714 [(M+K), calcd. for C44H41NOS2: 664.2708].
4.5.2 3-Mercapto-N-(2-mercapto-ethyl)-N-methyl-propionamide (26)
Procedure and workup analogous to 25 using 1.37 g (2.06 mmol) 24 as substrate. After impregnation onto flash silica the disulfide was purified via gradient flash-chromatography (hexanes to 10:1 to 4:1 to EtOAc to DCM) to provide 0.33 g (92%) 25 in two indistinguishable conformations (~1.0:1.0) as a foul-smelling oil. Rf = 0.32 (1:2 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ Conformer mixture: 3.65 (br m, 2 H), 3.56 (br m, 1 H), 3.36 (br m, 1 H), 3.07 (br m, 4 H), 2.95 (br s, 6 H), 2.86 (br m, 4 H), 2.76 (br m, 3 H), 2.62 (br m, 1 H), 1.72 (br m, 2 H), 1.62 (br m, 2 H); 13C NMR (125 MHz, CDCl3) δ Conformer mixture: 176.0 (C), 175.6 (C), 49.8 (CH2), 49.5 (CH2), 33.5 (CH3), 33.4 (CH3), 29.7 (CH2), 29.6 (CH2), 25.4 (CH2), 25.3 (CH2), 24.7 (CH2), 24.7 (CH2); Ir (neat) 3301 (w), 2927 (m), 1671 (s), 1628 (s), 1548 (m), 1414 (m), 1135 (s), 1046 (m), 704 (m) cm−1; HRMS (EI) m/z 179.0437 [(M+), calcd. for C6H13NOS2: 179.0439].
4.5.3 5-Methyl-[1,2,5]dithiazocan-6-one (5)
Procedure and workup analogous to 2 using 96.1 mg (0.53 mmol) 26 in ACN (1 mM). After impregnation onto flash silica the disulfide was purified via gradient flash-chromatography (hexanes to 10:1 to 4:1 to 2:1 to EtOAc to DCM to 10%MeOH/DCM) to provide 33.8 mg (36%) 5 in indistinguishable interconverting conformations as a water-white oil. Rf = 0.09 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ Conformer mixture: 4.25 (m, 2 H), 3.51 (m, 2 H), 3.16 (m, 2 H), 2.93 (m, 8 H), 2.88 (m, 6 H), 2.76 (m, 2 H); 13C NMR at −18 °C (125 MHz, CDCl3) δ Major: 174.2 (C), 54.3 (CH2), 37.8 (CH2), 36.8 (CH2), 34.1 (CH3), 30.7 (CH2); Minor: 176.4 (C), 47.2 (CH2), 37.9 (CH2), 37.3 (CH2), 36.6 (CH2), 32.2 (CH3); Ir (neat) 3464 (m), 2921 (m), 1632 (s), 1462 (m), 1397 (m), 1284 (w), 1172 (w) cm−1; LRMS (EI) m/z 2921 (m), 1632 (s), 1462 (m), 1397 (m), 1284 (w), 1172 (w); HRMS (EI) m/z 177.0283 [(M+), calcd. for C6H11NOS2: 177.0282].
4.6 Synthesis of dipeptide 6
4.6.1 2R-(2R-Acetylamino-3-tritylsulfanyl-propionylamino)-3-tritylsulfanyl-propionamide (29)
Peptide coupling of 0.50 g (1.23 mmol) 27 and 0.45 g (1.25 mmol) 28 was analogous to the production of compound 23 in DCM:DMF (0.1 M DCM). After impregnation onto flash silica the protected dipeptide was purified via gradient flash-chromatography (10:1 hexanes/EtOAc to 4:1 to 1:1 to EtOAc to DCM to 2.5%MeOH/DCM to 10%MeOH/DCM) to provide 0.72 g (79%) 29 as a white-solid. Rf=0.56 (5%MeOH/DCM); (c 0.09, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.47 (m, 12 H), 7.32 (m, 12 H), 7.26 (m, 6 H), 6.45 (d, J = 7.8 Hz, 2 H), 6.16 (d, J = 6.8 Hz, 1 H), 5.67 (s, 1 H), 4.21 (dd, J = 12.6, 6.8 Hz, 1 H), 4.09 (dd, J = 13.1, 6.3 Hz, 1 H), 2.90 (dd, J = 12.6, 6.3 Hz, 1 H), 2.73 (dd, J = 13.1, 6.3 Hz, 1 H), 2.66 (dd, J = 13.1, 6.3 Hz, 1 H), 2.56 (dd, J = 13.1, 5.3 Hz, 1 H), 1.92 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 171.6 (C), 170.3 (C), 169.7 (C), 144.2 (CH), 144.0 (CH), 129.4 (CH), 129.2 (CH), 128.0 (CH), 127.9 (CH), 129.9 (C), 126.8 (C), 67.1 (C), 67.0 (C), 52.2 (CH), 51.8 (CH), 33.2 (CH2), 32.9 (CH2), 22.8 (CH3); Ir (neat) 3255 (m), 3056 (m), 2930 (w), 1744 (w), 1650 (s), 1488 (s), 1442 (s), 1032 (w), 697 (s) cm−1; HRMS (EI) m/z 772.2637 [(M+K), calcd. for C46H43N3O3S2: 772.2644].
4.6.2 2R-(2R-Acetylamino-3-mercapto-propionylamino)-3-mercapto-propionamide (30)
Procedure and workup analogous to 25 using 0.20 g (0.26 mmol) 29 as substrate in DCM (2 mM). After impregnation onto flash silica the dithiol was purified via gradient flash-chromatography (hexanes to DCM to EtOAc) to provide 64.5 mg (91%) of 30 as a foul-smelling oil. Rf = 0.1 (10%MeOH/DCM); (c 0.22, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.99 (br m, 2 H), 6.39 (br m, 1 H), 5.59 (br m, 1 H) 4.60 (dd, J = 9.6, 6.4 Hz, 1 H), 4.51 (dd, J = 13.3, 6.2 Hz, 1 H), 3.08 (dd, J = 7.9 Hz, 6.2 Hz, 1 H), 3.00 (dd, J = 13.3, 6.6 Hz, 1 H), 2.90 (dd, J = 12.4, 7.3 Hz, 1 H), 2.81 (dd, J = 13.1, 6.6 Hz, 1 H), 2.08 (s, 3 H), 1.70 (m, 1 H), 1.60 (m, 1 H); 13C NMR (125 MHz, CDCl3) δ 178.2 (C), 175.4 (C), 173.9 (C), 67.1 (CH), 65.0 (CH), 32.3 (CH2), 31.9 (CH2), 23.7 (CH3); Ir (neat) 3282 (m), 3021 (m), 1741 (m), 1633 (s), 1535 (s), 1444 (s), 1211 (m), 1171 (s), 699 (s) cm−1; HRMS (EI) m/z 304.0186 [(M+K), calcd. for C8H15N3O3S2: 304.0192].
4.6.3 7R-Acetylamino-6-oxo-[1,2,5]dithiazocane-4R-carboxylic acid amide (6)
Procedure and workup analogous to 2 using 70.9 mg (0.26 mmol) 30 as substrate in ACN (1 mM). After impregnation onto flash silica the disulfide was purified via gradient flash-chromatography (DCM to 2.5%MeOH/DCM to 5%MeOH/DCM to 10%MeOH/DCM). Rf=0.27 (10%MeOH/DCM); (c 0.08, D2O); 1H NMR (500 MHz, DMSO-d6/D2O) δ T- conformer: 8.09 (s, 1 H), 7.73 (s, 1 H), 7.39 (s, 1 H), 7.28 (s, 1 H), 4.96 (m, 1 H), 4.29 (m, 1 H), 3.68 (m, 1 H), 3.42 (m, 1 H), 3.39 (m, 1 H), 3.28 (m, 1 H), 2.11 (s, 3 H); T’- conformer: 8.62 (d, J = 5.0 Hz, 1 H), 8.03 (d, J = 5.1 Hz, 1 H), 7.53 (m, 1 H), 7.09 (s, 1 H), 4.95 (m, 1 H), 3.98 (m, 1 H), 3.63 (m, 1 H), 3.59 (m, 1 H), 3.44 (m, 1 H), 3.43 (m, 1 H), 2.07 (s, 3 H); C- conformer: 8.54 (m, 1 H), 8.22 (d, J = 7.2 Hz, 1 H), 8.15 (d, J = 7.2 Hz, 1 H), 6.92 (m, 1 H), 5.10 (m, 1 H), 4.98 (m, 1 H), 3.53 (m, 1 H), 3.05 (m, 1 H), 3.02 (m, 1 H), 2.74 (m, 1 H), 2.05 (s, 3 H); C+ conformer: 8.29 (s, 1 H), 7.58 (s, 1 H), 7.42 (s, 1 H), 7.37 (s, 1 H), 5.23 (m, 1 H), 5.11 (m, 1 H), 3.41 (m, 1 H), 3.20 (m, 1 H), 3.15 (m, 1 H), 2.74 (m, 1 H), 2.02 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ T- conformer: 180.3 (C), 176.4 (C), 175.3 (C), 59.1 (CH), 56.7 (CH), 49.8 (CH2), 48.6 (CH2), 24.4 (CH3); T’- conformer: 176.1 (C), 175.6 (C), 174.9 (C), 57.4 (CH), 54.4 (CH), 50.9 (CH2), 43.3 (CH2), 24.4 (CH3); C− conformer: 176.5 (C), 174.0 (C), 173.8 (C), 55.6 (CH), 55.2 (CH), 45.0 (CH2), 43.3 (CH2), 24.4 (CH3); C+ conformer: 176.1 (C), 174.7 (C), 173.6 (C), 61.7 (CH), 52.2 (CH), 44.5 (CH2), 41.2 (CH2), 24.3 (CH3); Ir (neat) 3265 (br s), 1642 (s), 1516 (m), 1403 (m), 1179 (w) cm−1; HRMS (EI) m/z 264.0466 [(M+), calcd. for C8H13N3O3S2: 264.0476].5
4.7 Synthesis of N-Me dipeptide 7
4.7.1 4R-tert-Butyldisulfanylmethyl-5-oxo-oxazolidine-3-carboxylic acid 9H-fluoren-9-ylmethyl ester (32)
To a room temperature solution of 10.0 g (23.1 mmol) Fmoc-Cys(StBu)-OH in 230 mL PhH under natural atmosphere was added 3.59 g (155.1 g:1 mmol) p-formaldehyde and 0.37 g (7 mol%) camphorsulfonic acid. The reaction was heated to reflux (oil-bath=100 °C) in a Dean-Stark apparatus and stirred for 14 h. The reaction was concentrated to provide a clear oil. After impregnation onto flash silica the oxazolidinone was purified via gradient flash-chromatography (hexanes to 10:1 to 4:1 to 1:1 to EtOAc) to provide 9.63 g (94%) 32 as a white viscous gum. Rf = 0.71 (EtOAc); (c 0.23, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.6 Hz, 2 H), 7.55 (d, J = 7.6 Hz, 2 H), 7.40 (t, J = 7.6 Hz, 2 H), 7.31 (m, 2 H), 5.5−5.2 (br m's, 2 H), 4.75−4.35 (br m’s, 2 H), 4.24 (br s, 1 H), 4.01 (br s, 1 H), 3.53 (br s, 0.5 H), 3.20 (br s, 0.5 H), 2.97 (br s, 0.5 H), 2.66 (br s, 0.5 H), 1.26 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ 170.8 (C), 152.2 (C), 143.4 (C), 141.4 (C), 127.9 (CH), 127.2 (CH), 124.6 (CH), 120.0 (CH), 78.4 (CH2), 73.9 (CH2), 67.6 (CH), 55.3 (C), 48.2 (CH), 47.2 (CH2), 29.6 (CH3); IR (neat) 1800 (s), 1714 (s), 1417 (s), 1288 (m), 1129 (m), 1052 (m), 910 (w), 737 (m) cm−1; HRMS (EI) m/z 444.1308 [(M+), calcd. for C23H25NO4S2: 444.1303].39
4.7.2 3-tert-Butyldisulfanyl-2R-[(9H-fluoren-9-ylmethoxycarbonyl)-methyl-amino]-propionic acid (33)
To a room temperature stirring solution of 9.03 g (20.3 mmol) 32 in 101 mL CHCl3 under natural atmosphere was added 32.8 mL (203 mmol) TES followed by the rapid addition of 50 mL TFA. The reaction was stirred at room temperature for 16 h and then concentrated. The oil was dissolved in 150 mL DCM and concentrated. This was repeated three consecutive times. After impregnation onto flash silica, MeCys 33 was purified via gradient flash-chromatography (hexanes to 10:1 to 4:1 to 2:1; 1:1 to EtOAc) to provide 8.05 g (89%) 33 as a white solid in a 1.5:1.0 conformer ratio. Rf=0.32 (EtOAc); (c 0.33, CHCl3); 1H NMR (500 MHz, CDCl3) δ Major conformer: 10.36 (br s, 1 H), 7.82 (m, 2 H), 7.69 (m, 2 H), 7.46 (m, 2 H), 7.39 (m, 2 H), 4.91 (dd, J = 10.5, 4.3 Hz, 1 H), 4.52 (m, 2 H), 4.36 (t, J = 7.1 Hz, 1 H), 3.47 (dd, J = 13.9, 4.3 Hz, 1 H), 3.25 (dd, J = 13.9, 10.7 Hz, 1 H), 3.13 (s, 3 H), 1.43 (s, 9 H); Minor conformer: 10.36 (br s, 1 H), 7.81 (m, 2 H), 7.68 (m, 2 H), 7.46 (m, 2 H), 7.39 (m, 2 H), 4.86 (m, 1 H), 4.63 (dd, J = 10.5, 6.4 Hz, 1 H), 4.57 (m, 1 H), 4.32 (t, J = 6.0 Hz, 1 H), 3.33 (dd, J = 12.2, 5.5 Hz, 0.5 H), 3.23 (dd, J = 13.9, 4.7 Hz, 1 H), 3.06 (s, 3 H), 2.84 (dd, J = 13.7, 10.5 Hz, 0.5 H), 1.41 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ Major conformer: 174.4 (C), 156.5 (C), 143.5 (C), 141.0 (C), 127.4 (CH), 126.8 (CH), 124.8 (CH), 124.6 (CH), 67.9 (CH2), 59.5 (CH), 48.0 (C), 46.8 (CH), 38.8 (CH2), 33.4 (CH3), 29.7 (CH3); Minor conformer: 174.3 (C), 156.1 (C), 143.6 (C), 141.0 (C), 127.2 (CH), 126.9 (CH), 124.9 (CH), 124.7 (CH), 67.8 (CH2), 58.5 (CH), 47.9 (C), 46.7 (CH), 39.0 (CH2), 32.7 (CH3), 29.7 (CH3); IR (neat) 3353 (w), 1702 (s), 1449 (s), 1399 (m), 1317 (s), 1165 (s), 1129 (m), 976 (w), 738 (s) cm−1; HRMS (EI) m/z 446.1457 [(M+), calcd. for C23H27NO4S2: 446.1460].39
4.7.3 3-tert-Butyldisulfanyl-2R-[(9H-fluoren-9-ylmethoxycarbonyl)-methyl-amino]-propionic acid methyl ester (34)
In an air-dried flask 0.08 g (0.59 mmol) K2CO3 was added to a stirring solution of 0.26 g (0.54 mmol) 33 in 0.5 mL DMF previously chilled to 0 °C. Upon complete addition 0.06 mL (1.08 mmol) MeI was added and the reaction was stirred at 0 °C for 30 min. The ice-bath was removed and the reaction was stirred for 1 hr. The reaction was then filtered and concentrated. After impregnation onto flash silica the N-Me amino ester was purified via gradient flash-chromatography (hexanes to 10:1 to 4:1 to 1:1 to EtOAc to DCM) to provide (95%) 34 as a water-white oil in a 2.0:1.0 conformer ratio. Rf=0.86 (EtOAc); (c 0.35, CHCl3); 1H NMR (500 MHz, CDCl3) δ Major conformer: 7.76 (d, J = 7.5 Hz, 2 H), 7.60 (d, J = 7.5 Hz, 2 H), 7.40 (t, J = 7.5 Hz, 2 H), 7.31 (dt, J = 7.5, 7.3 Hz, 2 H), 4.79 (dd, J = 10.3, 4.5 Hz, 1 H), 4.41 (dd, J = 13.9, 7.0 Hz, 2 H), 4.30 (t, J = 7.0 Hz, 1 H), 3.74 (s, 3 H), 3.33 (dd, J = 13.9, 4.7 Hz, 1 H), 3.11 (dd, J = 13.9, 10.5 Hz, 1 H), 3.01 (s, 3 H), 1.34 (s, 9 H); Minor conformer: 7.76 (d, J = 7.5 Hz, 2 H), 7.58 (d, J = 7.3 Hz, 2 H), 7.40 (t, J = 7.5 Hz, 2 H), 7.31 (dt, J = 7.5, 7.3 Hz, 2 H), 4.72 (dd, J = 9.0, 4.5 Hz, 1 H), 4.52 (dd, J = 10.1, 5.5 Hz, 1 H), 4.46 (dd, J = 9.6, 5.5 Hz, 1 H), 4.2.5 (t, J = 5.5 Hz, 1 H), 3.61 (s, 3 H), 3.11 (dd, J = 13.9, 10.5 Hz, 1 H), 2.93 (s, 3 H), 2.73 (dd, J = 13.9, 9.8 Hz, 1 H), 1.32 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ Major conformer: 169.5 (C), 150.5 (C), 143.8 (C), 141.3 (C), 127.6 (CH), 127.0 (CH), 125.1 (CH), 124.8 (CH), 73.1 (CH2), 59.6 (CH), 48.1 (CH3), 47.1 (CH2), 42.7 (C), 39.6 (CH2), 33.2 (CH3), 29.9 (CH3); Minor conformer: 170.6 (C), 153.2 (C), 144.5 (C), 139.9 (C), 127.4 (CH), 126.8 (CH), 125.0 (CH), 124.8 (CH), 67.8 (CH2), 62.0 (CH), 52.5 (CH3), 46.5 (CH2), 42.8 (C),39.4 (CH2), 33.1 (CH3), 29.8 (CH3); Ir (neat) 3010 (w), 1743 (s), 1700 (s), 1449 (m), 1312 (m), 1164 (s), 1000 (m), 737 (s) cm−1; HRMS (EI) m/z 459.1539 [(M+), calcd. for C24H29NO4S2: 459.1538].
4.7.4 2R-[(2R-Acetylamino-3-tritylsulfanyl-propionyl)-methyl-amino]-3-tert-butyldisulfanyl-propionic acid methyl ester (35)
To a room temperature solution of 0.89 g (1.95 mmol) 34 in 19.5 mL DCM was added 2.04 mL (19.5 mmol) Et2NH. Stirred for 1.5 h and then concentrated. After dissolution in 21 mL DMF, 0.86 g (2.14 mmol) 27 and 0.81 g (2.14 mmol) HATU was added. Upon complete dissolution 0.48 mL (2.92 mmol) Hünigs base was added and the reaction was stirred at room temperature for 3 h. Reaction was poured into 100 mL EtOAc. The EtOAc layer was washed two consecutive times with 100 mL 10% aqueous NaHCO3, once with 100 mL H2O, and once with 100 mL brine. The EtOAc layer was dried over MgSO4, filtered and concentrated. After impregnation onto flash silica the dipeptide was purified via gradient flash-chromatography (hexanes to 10:1 to 5:1 to 2:1 to 1:1 to EtOAc to 10%MeOH/DCM) to provide (68%) 35 as a white solid in a 1.0:1.0 conformer ratio. Rf=0.45 (EtOAc); (c 0.06, CHCl3); 1H NMR (500 MHz, CDCl3) δ Conformer mixture: 7.39 (m, 12 H), 7.25 (m, 12 H), 7.18 (m, 6 H), 6.46 (br t, J = 7.8 Hz, 2 H), 4.93-4.82 (series of m's, 3 H), 4.64 (dd, J = 9.7, 4.8 Hz, 1 H), 3.61 (s, 3 H), 3.60 (s, 3 H), 3.29 (t, J = 4.3 Hz, 1 H), 3.26 (t, J = 4.3 Hz, 1 H), 3.05 (dd, J = 13.6, 10.2 Hz, 1 H), 2.93 (dd, J = 14.1, 10.2 Hz, 1 H), 2.87 (s, 3 H), 2.79 (s, 3 H), 2.51 (m, 4 H), 1.91 (s, 3 H), 1.90 (s, 3H), 1.29 (s, 9 H), 1.28 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ Conformer mixture: 175.1 (C), 174.6 (C), 171.2 (C), 170.9 (C), 169.8 (C), 169.7 (C), 144.4 (C), 144.4 (C), 129.5 (CH), 129.5 (CH), 127.9 (CH), 127.8 (CH), 126.7 (CH), 126.7 (CH), 66.9 (CH), 66.9 (CH), 61.0 (C), 59.4 (C), 58.0 (CH), 57.8 (CH), 52.4 (CH3), 52.4 (CH3), 48.4 (C), 48.1 (C), 38.7 (CH2), 38.4 (CH2), 34.4 (CH2), 34.3 (CH2), 29.9 (CH3), 29.8 (CH3), 29.7 (CH3), 29.7 (CH3), 23.1 (CH3), 23.1 (CH3); Ir (neat) 3327 (m), 2926 (s), 1734 (m), 1623 (m), 1567 (m), 1242 (m) cm−1; HRMS (EI) m/z 624.2140 [(M+H), calcd. for C33H40N2O4S3: 624.2150].
4.7.5 2R-[(2R-Acetylamino-3-tritylsulfanyl-propionyl)-methyl-amino]-3-mercapto-propionic acid methyl ester (36)
To a room temperature solution of 32.8 mg (52.4 µmol) 35 in 0.5 mL DMF was added 80.9 g (0.52 mmol) dithiothreitol and 5 µL (52.4 mol) N-methyl morpholine. Stirred for 24 h and then concentrated. Reaction was transferred into 50 mL EtOAc and the EtOAc layer was washed two consecutive times with 50 mL H2O and once with 50 mL brine. The EtOAc layer was dried over MgSO4, filtered and concentrated. The solid was purified via reverse-phase HPLC to provide 23.6 mg (84%) 36 as a white solid in a 1.0:1.0 conformer ratio. Rf=0.38 (EtOAc); (c 0.07, CHCl3); 1H NMR (500 MHz, CDCl3) δ Conformer mixture: 7.37 (m, 12 H), 7.29 (m, 12 H), 7.22 (m, 6 H), 6.59 (m, 2 H), 4.86 (m, 3 H), 4.64 (m, 1 H), 3.67 (s, 3 H), 3.66 (s, 3 H), 3.07 (m, 2 H), 2.91 (s, 3 H), 2.83 (s, 3 H), 2.75 (m, 1 H), 2.70 (m, 1 H), 2.56 (m, 2 H), 2.54 (m, 2 H), 2.03 (s, 3 H), 2.00 (s, 3 H), 1.59 (t, J = 8.3 Hz, 1 H), 1.51 (t, J = 8.7 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ Conformer mixture: 173.3 (C), 172.3 (C), 171.4 (C),170.7 (C),170.2 (C), 169.7 (C), 144.5 (C), 144.4 (C), 129.5 (CH), 129.5 (CH), 127.9 (CH), 127.9 (CH), 126.8 (CH), 126.7 (CH), 67.1 (CH), 67.0 (CH), 59.4 (C), 58.9 (C), 54.0 (CH), 53.2 (CH), 52.6 (CH3), 52.5 (CH3), 34.4 (CH2), 34.1 (CH2), 33.8 (CH2), 31.7 (CH2), 30.0 (CH3), 29.6 (CH3), 23.1 (CH3), 23.0 (CH3); Ir (neat) 3288 (w), 3058 (w), 1740 (s), 1637 (s), 1487 (s), 1443 (s), 1241 (s), 1154 (s), 1097 (m), 1033 (m), 742 (s), 699 (s) cm−1; HRMS (EI) m/z 575.1443 [(M+K), calcd. for C29H32N2O4S2: 575.1441].
4.7.6 7R-Acetylamino-5-methyl-6-oxo-[1,2,5]dithiazocane-4R-carboxylic acid methyl ester (7)
In an air-dried flask 21.0 mg (67.9 µmol) 2,2′-Dithiobis(5-nitropyridine) was added to a stirring solution of 36.1 mg (67.2 µmol) 36 in 25 mL DCM at room temperature producing a yellow color over time. After 14 h, 10 mL TFA was added and the reaction an even brighter yellow color being produced over time. After 2 h the reaction was scavenged by the addition of 0.05 mL (0.33 mmol) TES creating a water-white reaction. Upon complete addition the reaction was stirred at room temperature for 8 h, once again with slow evolution of yellow color, and concentrated. The yellow solid was purified via reverse-phase HPLC to provide 7.0 mg (36%) 7 as a white solid in a single conformation and 12.5 mg (31%) 37 as a white solid in a single conformation. Rf=0.14 (EtOAc); (c 0.02, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.85 (s, 1 H), 5.31 (dd, J = 11.9, 3.6 Hz, 1 H), 5.13 (t, J = 7.6 Hz, 1 H), 3.81 (s, 3 H), 3.38 (dd, J = 13.7, 3.9 Hz, 1 H), 3.04 (d, J = 14.0 Hz, 1 H), 2.96 (d, J = 13.7 Hz, 1 H), 2.89 (dd, J = 11.3, 2.4 Hz, 1 H), 2.86 (s, 3 H), 2.01 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 171.7 (C), 169.3 (C), 169.0 (C), 57.5 (CH), 52.1 (CH), 53.1 (CH3), 42.4 (CH2), 38.9 (CH2), 30.3 (CH3), 22.7 (CH3); Ir (neat) 3332 (m), 1741 (s), 1637 (s), 1436 (w), 1407 (w), 1240 (w) cm−1; HRMS (EI) m/z 293.0623 [(M+), calcd. for C10H16N2O4S2: 293.0630].
4.7.7 2R-{[2R-Acetylamino-3-(5-nitro-pyridin-2-yldisulfanyl)-propionyl]-methyl-amino}-3-(5-nitro-pyridin-2-yldisulfanyl)-propionic acid methyl ester (37)
Compound 37 was obtained as a single conformer during the 36 to 7 transformation (see section 4.7.6). Rf=0.34 (EtOAc); (c 0.06, CHCl3); 1H NMR (500 MHz, CDCl3) δ 9.31 (s, 2 H), 8.43 (d, J = 8.7 Hz, 2 H), 7.82 (t, J = 8.7 Hz, 2 H), 6.65 (d, J = 8.7 Hz, 1 H), 5.39 (m, 1 H), 5.05 (m, 1 H), 3.75 (s, 3 H), 3.56 (dd, J = 14.1, 5.3 Hz, 1 H), 3.36 (dd, J = 13.6, 4.8 Hz, 1 H), 3.26 (dd, J = 13.6, 9.7 Hz, 1 H), 3.18 (s, 3 H), 3.12 (dd, J = 13.6, 7.8 Hz, 1 H), 2.09 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 191.2 (C), 191.0 (C), 165.9 (C), 165.3 (C), 164.3 (C), 145.7 (CH), 145.2 (CH), 141.2 (C), 139.8 (C), 131.9 (CH), 131.7 (CH), 119.9 (CH), 119.9 (CH), 63.8 (CH), 52.9 (CH), 48.4 (CH3), 37.6 (CH2), 29.8 (CH2), 23.4 (CH3), 19.6 (CH3); Ir (neat) 3291 (br w), 3060 (w), 2925 (w), 1739 (m), 1645 (s), 1587 (s), 1563 (s), 1514 (s), 1430 (vs), 1096 (s), 1007 (w) cm−1; HRMS (EI) m/z 603.0459 [(M+), calcd. for C20H22N6O8S4: 603.0460].
Compound 37 is a byproduct of the 36 to 7 transformation, but can be treated with DTT to generate 7 as follows. To a room temperature stirring solution of 13.1 mg (22.0 µmol) 37 in 20 mL DCM was added 3.4 g (22.2 µmol) dithiothreitol. After 14 h the reaction was concentrated and purified via prep-HPLC to provide 2.0 mg (32%) 7 as a white solid in a single conformer.
Supplementary Material
Acknowledgements
This study was supported by NIH grant GM070742 to RJH. ELR was supported by NIH grant PHS T32 HL07594.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Hudáky I, Gáspári Z, Carugo O, Cemzar M, Pongor S, Perczel A. Proteins: Struct., Func. Bioinf. 2004;55:152–168. doi: 10.1002/prot.10581. [DOI] [PubMed] [Google Scholar]
- 2.Wang X-H, Connor M, Smith R, Maciejewski MW, Howden MEH, Nicholson GM, Christie MJ, King GF. Nat. Struct. Biol. 2000;7:505–513. doi: 10.1038/75921. [DOI] [PubMed] [Google Scholar]
- 3.Blake CCF, Ghosh M, Harlos K, Avezoux A, Anthony C. Struct. Biol. 1994;1:102–105. doi: 10.1038/nsb0294-102. [DOI] [PubMed] [Google Scholar]
- 4.Oubrie A, Rozeboom HJ, Kalk KH, Huizinga EG, Dijkstra BW. J. Biol. Chem. 2002;277:3727–3732. doi: 10.1074/jbc.M109403200. [DOI] [PubMed] [Google Scholar]
- 5.Creighton CJ, Reynolds CH, Lee DHS, Leo GC, Reitz AB. J. Am. Chem. Soc. 2001;123:12664–12669. doi: 10.1021/ja016505m. [DOI] [PubMed] [Google Scholar]
- 6.Avizonis DZ, Farr-Jones S, Kosen PA, Basus VJ. J. Am. Chem. Soc. 1996;118:13031–13039. [Google Scholar]
- 7.Kao PN, Karlin A. J. Biol. Chem. 1986;261:8085–8088. [PubMed] [Google Scholar]
- 8.Laakkonen L, Jobson RW, Albert VA. Plant Biol. (Stuttg) 2006;8:758–764. doi: 10.1055/s-2006-924459. [DOI] [PubMed] [Google Scholar]
- 9.Lima WF, Wu H, Nichols JG, Manalili SM, Drader JJ, Hofstadler SA, Crooke ST. J. Biol. Chem. 2003;278:14906–14912. doi: 10.1074/jbc.M211279200. [DOI] [PubMed] [Google Scholar]
- 10.Zimmermann J, Kühne R, Sylvester M, Freund C. Biochemistry. 2007;46:6971–6977. doi: 10.1021/bi700437r. [DOI] [PubMed] [Google Scholar]
- 11.Esposito L, De Simone A, Zagari A, Vitagliano L. J. Mol. Biol. 2005;347:483–487. doi: 10.1016/j.jmb.2005.01.065. [DOI] [PubMed] [Google Scholar]
- 12.Che Y, Marshall GR. Biopolymers. 2006;81:392–406. doi: 10.1002/bip.20431. [DOI] [PubMed] [Google Scholar]
- 13.Chandrasekaran R, Balasubramanian R. Biochim. Biophys. Acta. 1969;188:1–9. doi: 10.1016/0005-2795(69)90039-7. [DOI] [PubMed] [Google Scholar]
- 14.Capasso S, Mattia C, Mazzarella L, Pulit R. Acta Cryst. 1977;B33:2080–2083. [Google Scholar]
- 15.Tanaka N, Ashida T, Kakudo M. Acta Cryst. 1977;B33:3561–3564. [Google Scholar]
- 16.Mez HC. Cryst. Struct. Commun. 1974;3:657–660. [Google Scholar]
- 17.Varughese KI, Lu CT, Kartha G. Int. J. Peptide Protein Res. 1981;18:88–102. doi: 10.1111/j.1399-3011.1981.tb02043.x. [DOI] [PubMed] [Google Scholar]
- 18.Horne A, North M, Parkinson JA, Sadler IH. Tetrahedron. 1993;49:5891–5904. [Google Scholar]
- 19.Fujimura K, Ito S, Suhara H, Kawashima Y. J. Chem. Res. (S) 1992:88–89. [Google Scholar]
- 20.Baxter RL, Glover SSB, Gordon EM, Gould RO, McKie MC, Scott AI, Walkinshaw MD. J. Chem. Soc. Perkin Tran.s 1. 1988:365–371. [Google Scholar]
- 21.Sukumaran DK, Prorok M, Lawrence DS. J. Am. Chem. Soc. 1991;113:706–707. [Google Scholar]
- 22.Lauth X, Babon JJ, Stannard JA, Singh S, Nizet V, Carlberg JM, Ostland VE, Pennington MW, Norton RS, Westerman ME. J. Biol. Chem. 2005;280:9272–9282. doi: 10.1074/jbc.M411154200. [DOI] [PubMed] [Google Scholar]
- 23.Cumberbatch S, North M, Zagotto G. Tetrahedron. 1993;49:9049–9066. [Google Scholar]
- 24.Lu KP, Finn G, Lee TH, Nicholson LK. Nat. Chem. Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 25.Moise L, Piserchio A, Basus VJ, Hawrot E. J Biol. Chem. 2002;277:12406–12417. doi: 10.1074/jbc.M110320200. [DOI] [PubMed] [Google Scholar]
- 26.Rogers DW, von Voithenberg H, Allinger NL. J. Org. Chem. 1978;43:360–361. [Google Scholar]
- 27.Cumberbatch S, North M, Zagotto G. J. Chem. Soc. Chem. Commun. 1993:641–642. [Google Scholar]
- 28.Vitoux B, Aubry A, Cung MT, Marraud M. Int. J. Pept. Protein Res. 1986;27:617–632. doi: 10.1111/j.1399-3011.1981.tb02016.x. [DOI] [PubMed] [Google Scholar]
- 29.Cepas SC, North M. Tetrahedron. 1997;53:16859–16866. [Google Scholar]
- 30.Houk J, Whitesides GM. J. Am. Chem. Soc. 1987;109:6825–6836. [Google Scholar]
- 31.Ruggles EL, Hondal RJ. Tetrahedron Lett. 2006;47:4281–4284. doi: 10.1016/j.tetlet.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobbs AP, Guesné SJJ, Martinovic S, Coles SJ, Hursthouse MB. J. Org. Chem. 2003;68:7880–7883. doi: 10.1021/jo034981k. [DOI] [PubMed] [Google Scholar]
- 33.Redman JE, Sanders JKM. Org. Lett. 2000;2:4141–4144. doi: 10.1021/ol006615i. [DOI] [PubMed] [Google Scholar]
- 34.Shah STA, Khan KM, Fecker M, Voelter W. Tetrahedron Lett. 2003;44:6789–6791. [Google Scholar]
- 35.Rao PV, Bhaduri S, Jiang J, Holm RH. Inorg. Chem. 2004;43:5833–5849. doi: 10.1021/ic040055s. Acid 21. [DOI] [PubMed] [Google Scholar]; Maltese M. J. Org. Chem. 2001;66:7615–7625. doi: 10.1021/jo0156971. Amine 22. [DOI] [PubMed] [Google Scholar]
- 36.Montalbetti CAGN, Falque V. Tetrahedron. 2005;61:10827–10852. [Google Scholar]
- 37.Pearson DA, Blanchette M, Baker ML, Guindon CA. Tetrahedron Lett. 1989;30:2739–2742. [Google Scholar]
- 38.Patel HM, Tao J, Walsh CT. Biochemistry. 2003;42:10514–10527. doi: 10.1021/bi034840c. [DOI] [PubMed] [Google Scholar]
- 39.Ruggles EL, Flemer SF, Jr, Hondal RJ. Biopolymers. 2008;90:61–68. doi: 10.1002/bip.20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aurelio L, Box JS, Brownlee RTC, Hughes AB, Sleebs MM. J. Org. Chem. 2003;68:2652–2667. doi: 10.1021/jo026722l. [DOI] [PubMed] [Google Scholar]
- 41.Freidinger RM, Hinkle JS, Perlow DS, Arison BH. J. Org. Chem. 1983;48:77–81. [Google Scholar]
- 42.Garner P, Park JM. Organic Syntheses, Coll. Vol. 1998;9:300–307. [Google Scholar]
- 43.Greene TW, Wuts PGM. Protective Groups in Organic Synthesis 4th Edition. New York: John Wiley & Sons; 1999. pp. 711–713. [Google Scholar]
- 44.Han S-Y, Kim Y-A. Tetrahedron. 2004;60:2447–2467. [Google Scholar]
- 45.Greene TW, Wuts PGM. Protective Groups in Organic Synthesis 4th Edition. New York: John Wiley & Sons; 1999. pp. 688–689. [Google Scholar]
- 46.Flemer SF, Jr, Lacey BM, Hondal RJ. J. Pept. Sci. 2008;14:637–647. doi: 10.1002/psc.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mossner E, Huber-Wunderluch M, Glockshuber R. Prot. Sci. 1998;7:1233–1244. doi: 10.1002/pro.5560070519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. 4th ed. Woburn, MA: Butterworth-Heinemann; 1998. [Google Scholar]
- 49.Perlman N, Albeck A. Syn. Comm. 2000;30:4443–4449. [Google Scholar]
- 50.Eya BK, Otsuka T, Kubo I, Wood DL. Tetrahedron. 1990;46:2695–2706. [Google Scholar]
- 51.Gassman PG, Bonser SM, Mlinaric-Majerski K. J. Am. Chem. Soc. 1989;111:2652–2662. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.