

Abstract
Earlier studies indicated a role for reactive oxygen species (ROS) in host defense against Pseudomonas aeruginosa infection. However, the role of nicotinamide adenine dinucleotide phosphate–reduced (NADPH) oxidase (NOX) proteins and the mechanism of activation for NADPH oxidase in P. aeruginosa infection are not well-defined. Here, we investigated the role of NOX2 and NOX4 proteins in P. aeruginosa infection, ROS generation, and endothelial barrier function in murine lungs and in human lung microvascular endothelial cells (HLMVECs). Airway instillation of P. aeruginosa strain 103 (PA103) significantly increased ROS concentrations in bronchial alveolar lavage (BAL) fluid, along with the expression of NOX2 and NOX4, but not NOX1 and NOX3, in lung tissue. In addition, PA103-infected HLMVECs revealed elevated concentrations of ROS, NOX2, and NOX4. In murine lungs and HLMVECs, PA103 induced the NF-κB pathway, and its inhibition blocked PA103-dependent NOX2 and NOX4 expression. Barrier function analysis showed that heat-killed PA103 induced endothelial permeability in a dose-dependent manner, which was attenuated by treatment with small interfering (si)RNA specific for NOX4, but not NOX2. Furthermore, the knockdown of NOX4, but not NOX2, with siRNA reduced PA103-mediated apoptosis in HLMVECs. In vivo, the down-regulation of NOX4 with NOX4 siRNA attenuated PA103-induced lung vascular permeability. The deletion of NOX2 in mice exerted no effect on permeability, but offered significant resistance to P. aeruginosa–induced lung inflammation. These data show that P. aeruginosa lung infection up-regulates NOX2 and NOX4 expression and ROS generation, which play distinct roles in regulating lung inflammation, apoptosis, and permeability.
Keywords: Pseudomonas aeruginosa, NADPH oxidase, reactive oxygen species, apoptosis, vascular permeability
Clinical Relevance
Using in vivo and in vitro Pseudomonas aeruginosa infection models, we show that nicotinamide adenine dinucleotide phosphate–reduced oxidase–2 (NOX2) and NOX4 are up-regulated in response to P. aeruginosa in murine lung tissue and human lung microvascular endothelial cells. The inhibition of the NF-κB pathway attenuates the effects of P. aeruginosa on NOX2 and NOX4 expression. However, blocking NOX4, but not NOX2, attenuated P. aeruginosa–mediated apoptosis and lung permeability, whereas NOX2 deficiency offered resistance to lung inflammation. These results suggest differential roles for NOX4 and NOX2 in P. aeruginosa–mediated lung inflammation, apoptosis, and permeability.
Reactive oxygen species (ROS) have long been recognized as pivotal contributors to inflammatory injury caused by a variety of pathogens (1–3). The nicotinamide adenine dinucleotide phosphate–reduced (NADPH) oxidase–2 (NOX2) protein plays an important role in a variety of signaling pathways, and offers protection against bacterial invasion. To date, seven members have been identified, including NOX1–5, dual oxidase (DUOX) 1 and DUOX2 (4). The NOX2 NADPH oxidase system consists of six subunits, namely, Nox2, p22 phagocyte oxidase (p22phox), p40phox, p47phox, p67phox, and Ras-related C3 botulinum toxin substrate 1 (Rac1). The activation of NOX2 requires the sequential phosphorylation and membrane translocation of cytosolic cofactors, p40phox, p47phox, p67phox, and Rac1 (4). NADPH oxidase activation by bacterial infection results in increased and bactericidal ROS production (5).
NOX4 was originally identified as an NADPH oxidase homologue highly expressed in kidneys (6). It is also found in endothelial cell (7), epithelial cells (8), smooth muscle cells (9), and fibroblasts (10). Unlike NOX2, NOX4 produces ROS at low concentrations, which is believed to play a role in signal transduction rather than in killing bacteria. Furthermore, NOX4 activity is independent of the translocation of key cofactors to the membrane-anchored NOX4 that are usually critical in the activation of other NOX proteins. Various stimuli, such as endoplasmic reticulum stress (11), shear stress (12), hypoxia (13), hyperoxia (14), ischemia (15), transforming growth factor–β1 (16), and TNF-α (17), have all been shown to induce the expression of NOX4. A recent study showed that NOX4 accounts for approximately 75% of vascular endothelial ROS (18).
Pseudomonas aeruginosa is a common pathogen associated with respiratory-tract infections in diverse clinical settings. Studies have shown that the oxidative stress induced by P. aeruginosa infection accentuates lung injury. Its infection induces an activation of phagocytes to produce a burst of ROS, an innate immune defense mechanism. However, an uncontrolled response can aggravate tissue damage. In addition, P. aeruginosa causes both endothelial and epithelial barrier dysfunction by direct contact with the endothelium and epithelium (19, 20). The mechanisms of NOX activation and involvement in P. aeruginosa infection remain largely unknown. Here, we investigated the effects of P. aeruginosa infection on the expression of NOX proteins, ROS generation, and lung permeability in a murine model. We found that P. aeruginosa induced NOX4 and NOX2 expression in the lung by activation of the NF-κB signaling pathway. The down-regulation of NOX4 with small interfering (si)RNA attenuated P. aeruginosa–mediated murine lung permeability and endothelial barrier dysfunction, without affecting inflammatory responses and ROS in bronchoalveolar lavage (BAL) fluid. However, the genetic knockdown of NOX2 in mice inhibited P. aeruginosa–induced cytokines and ROS concentrations in BAL fluid, without altering total cell counts and lung permeability. These studies are the first to demonstrate a differential role for NOX4 and NOX2 in P. aeruginosa–induced lung inflammation and endothelial barrier dysfunction.
Materials and Methods
Reagents and Cells
Details regarding reagents and cells are provided in the online supplement.
Preparation of P. aeruginosa
The parent strain P. aeruginosa 103 (PA103) was selected for most experiments because it is a well-characterized and highly toxic strain. The PA103 strain expresses Type III proteins and secretes exoenzyme U (Exo U) and exoenzyme T (Exo T). Strain PA103 stock was prepared as described previously (21). Briefly, bacteria from frozen stocks were streaked onto trypticase soy agar plates and grown in a deferrated dialysate of trypticase soy broth supplemented with 10 mM nitrilotriacetic acid (Sigma Chemical Co., St. Louis, MO), 1% glycerol, and 100 mM monosodium glutamate at 33°C for 13 hours in a shaking incubator. Cultures were centrifuged at 8,500 × g for 5 minutes, and the bacterial pellet was washed twice in Ringer lactate and diluted into the appropriate number of colony-forming units (CFU) per milliliter in Ringer lactate solution, as determined by a spectrophotometer. The bacterial concentration was confirmed by diluting all samples and plating out the known dilution on sheep blood agar plates.
Murine Model
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago. Adult C57BL/6J mice, weighing approximately 20–25 g, were purchased from Jackson Laboratories (Bar Harbor, ME). Male gp91phox−/− mice on a C57BL/6 background were maintained as a homozygous colony, and female mice were used as wild-type controls. In vivo NOX4 knockdown was performed by intratracheal NOX4 siRNA transfection, using jetPEI (Polyplus-Transfection Inc., New York, NY) as a transfecting agent (for details, please see the online supplement). For bacterial infection, mice received PBS or PA103 intratracheally at a dose of 1 × 106 CFU/mouse under anesthesia. Twenty-four or 48 hours after treatment, BAL was obtained using 1 ml of sterile Hanks’ Balanced Salt Buffer for the measurement of cell count, protein concentration, H2O2, and cytokines. Left lungs were removed and fixed for hematoxylin–eosin staining or immunohistochemistry with NOX antibodies, and right lungs were snap-frozen in liquid nitrogen and stored at −80°C for Western blotting.
In Vivo Inhibition of NF-κB Activity
Details are provided in the online supplement.
Detection of ROS in Live HLMVECs, and Measurement of H2O2
Details are provided in the online supplement.
Immunofluorescence Staining
Details are provided in the online supplement.
Permeability Assays
Endothelial permeability was monitored by a highly sensitive biophysical assay with an electrical cell-substrate impedance sensing system, as described previously (22), or by transwell tracer experiment with fluorescent dextran. Details are provided in the online supplement.
Analysis of Apoptosis
The apoptosis of HLMVECs exposed to P. aeruginosa was analyzed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and the Western blotting of cleaved caspase 3. Details are provided in the online supplement.
Real-Time and Quantitative PCR
Total RNA from control and experimental lung samples was isolated using the TRIzol solution (Invitrogen-GIBCO BRL, Life Technologies, Foster City, CA). Details are provided in the online supplement.
Western Blot Analysis
Protein extracts from murine lungs or HLMVEC lysates were separated by SDS-PAGE and transferred onto nitrocellulose membranes, followed by incubation with specific antibodies of interest. Equal protein loading was verified by reprobing with anti–β-actin antibody.
Statistical Analysis
Results are presented as the means ± SDs of 3 to 10 independent experiments. Stimulated samples were compared with control samples by unpaired Student t test. For multiple-group comparisons, ANOVA, followed by the post hoc Fisher test, was used. P < 0.05 was considered statistically significant.
Results
P. aeruginosa Induces Acute Lung Injury with Increased Production of Hydrogen Peroxide in the Lungs
We first determined the extent of inflammatory response and ROS generation induced by P. aeruginosa (1 × 106 CFU/mouse) in vivo. After 24-hour and 48-hour infections of mice with P. aeruginosa strain PA103, no significant mortality was observed at either time point. The infection of mice with P. aeruginosa led to a significant increase in inflammatory cells in the lungs, compared with PBS control samples (12.6 ± 2.81 × 105/ml versus 1.18 ± 0.09 × 105/ml, respectively; P < 0.01) at 24 hours and 48 hours of infection (14.5 ± 2.36 × 105/ml versus 1.18 ± 0.09 × 105/ml, respectively; P < 0.01) (Figure 1A). Likewise, a significant increase in BAL protein content was observed at 24 hours and 48 hours after infection (0.43 ± 0.03 mg/ml versus 0.15 ± 0.05 mg/ml, and 0.48 ± 0.08 mg/ml versus 0.15 ± 0.05 mg/ml, respectively; both P < 0.01) (Figure 1B). In addition, P. aeruginosa induced a significant increase in BAL H2O2 (2.06 ± 0.15 μM versus 1.31 ± 0.15 μM, respectively, P < 0.05, at 24 hours; 2.19 ± 0.18 μM versus 1.31 ± 0.15 μM, respectively, P < 0.01, at 48 hours) (Figure 1C). These data suggest that P. aeruginosa induced an inflammatory response with a time-dependent increase in ROS.
Figure 1.

Pseudomonas aeruginosa (PA) induces lung inflammation and oxidative stress. Bronchoalveolar lavage (BAL) was collected at 24 and 48 hours after bacterial treatment. (A) A cell pellet was resuspended in 200 μl PBS for cell counting. (B) BAL protein and (C) H2O2 concentrations are shown. P. aeruginosa induced significant lung injury, as evidenced by increased BAL cell count, protein content, and reactive oxygen species (ROS) production. *P < 0.01, compared with PBS control samples (n = 4). (D) Human lung microvascular endothelial cells (HLMVECs), cultured in 35-mm dishes to approximately 90% confluence, were exposed to Pseudomonas aeruginosa strain 103 (PA103) at multiplicities of infections (MOIs) 1, 5, and 10 for 9 hours. Cells were then loaded with 5-(and-6)-carboxy-2',7'-dichlorodihydro-fluorescein diacetate (carboxy-H2DCFDA) (5 μM) for 30 minutes, and the oxidation of DCFDA was monitored under fluorescent microscopy (×200). Scale bars, 30 μm. (E) ROS production in each group was quantified. (F) H2O2 concentrations in culture media were assayed. PA103 induced significant ROS production in HLMVECs in a dose-dependent manner. *P < 0.05 and **P < 0.01, versus PBS control samples (n = 6). con, control.
P. aeruginosa Exerts Oxidative Stress In HLMVECs
Next, we determined whether the P. aeruginosa infection of endothelial cells leads to the generation of ROS in vitro. P. aeruginosa induced ROS production in HLMVECs in a dose-dependent manner (Figures 1D and 1E). The release of H2O2 into the culture media of infected HLMVECs was also dose-dependent on P. aeruginosa infection (Figure 1F). Combined with the in vivo murine lung data, our in vitro results clearly demonstrate that P. aeruginosa infection is associated with an increase in ROS production.
P. aeruginosa Lung Infection Induces NOX2 and NOX4 Expression, but Not NOX1 and NOX3
We next investigated whether NOX activation is the source of elevated ROS underlying the oxidative stress in P. aeruginosa–infected lungs. The expressions of NOX2 and NOX4 proteins were significantly up-regulated at 24 hours after P. aeruginosa treatment, and this up-regulation persisted up to 48 hours, as determined by Western blotting (Figure 2A). Interestingly, no increase in NOX1 and NOX3 expression in lung tissue was evident at either time point. Densitometry analysis revealed that the expression of NOX4 increased approximately 3.2-fold, and that of NOX2 increased approximately 2.3-fold (Figure 2B). The immunohistochemical staining of lung tissue for NOX proteins confirmed an increase in the expression of NOX2 and NOX4 proteins, but not NOX1 or NOX3 (Figure 2C). The expression of NOX2 was detectable in neutrophils, lung endothelial cells, and lung epithelial cells, whereas the expression of NOX4 was seen mainly in lung endothelial and epithelial cells (Figure 2C). Real-time PCR showed an increase only in NOX2 mRNA (∼ 4.5-fold) and NOX4 mRNA (∼ 3.8-fold) (Figure 2D). These data suggest that P. aeruginosa infection selectively induces the expression of NOX2 and NOX4 in the lung.
Figure 2.


P. aeruginosa infection induces nicotinamide adenine dinucleotide phosphate–reduced oxidase–2 (NOX2) and NOX4 expression in murine lung tissue and HLMVECs. (A) Lungs were harvested at 24 hours and 48 hours for analyses of NOX proteins by Western blotting, or of mRNA by real-time PCR (D) (n = 4/group). Western blots were examined using an image analyzer, and NOX protein expression was quantified and normalized to total actin. (B) Approximately 2.5-fold and approximately 3.5-fold increases in NOX2 and NOX4 protein expression, respectively, were evident. *P < 0.01, compared with PBS control samples. #P < 0.01, compared with PBS control samples. (C) Immunohistochemical staining of NOX proteins. Scale bar, 200 μm. P. aeruginosa induced significant NOX2 and NOX4 protein expression in lung tissue, 48 hours after infection. However, it did not induce the expression of NOX1 or NOX3. (D) Real-time PCR quantification of NOX mRNA. **P < 0.01, compared with PBS control samples. ##P < 0.01, compared with PBS control samples. (E) HLMVECs were exposed to PA103 at MOI = 1, 5, and 10 for the indicated periods of time. Total cell lysates were subjected to SDS-PAGE and Western blotting for NOX protein expression. Actin was blotted to monitor protein loading. PA103 induced NOX2 expression in HLMVECs as early as 16 hours and 24 hours. MOI = 1 and 5 did not show significant NOX2 expression at 16 hours. PA also induced NOX4 expression at 16 hours and 24 hours after infection. However, PA103 did not induce NOX1 and NOX3 expression at any of the time points tested. The blot shown is representative of four independent experiments. (F) Statistical analysis for NOX proteins expression normalized to actin at various time points.
P. aeruginosa Induces NOX2 and NOX4 Expression in HLMVECs
Consistent with the in vivo data, only NOX2 and NOX4 were up-regulated in HLMVECs in response to P. aeruginosa treatment, whereas NOX1 and NOX3 expression levels remained unaltered (Figures 2E and 2F). Of the three doses tested, P. aeruginosa induced NOX2 expression as early as 9 hours at the maximum dose of 10 multiplicity of infection (MOI). However, NOX4 expression was induced at 5 MOI and at later time points (16 hours and 24 hours). All three doses of P. aeruginosa elevated NOX2 and NOX4 expression. These results show the increased expression of NOX2 and NOX4, but not NOX1 and NOX3, by P. aeruginosa in HLMVECs.
P. aeruginosa–Induced NOX2 and NOX4 Expression Is Mediated by NF-κB Activation
The immunomodulation of the NF-κB pathway seems to affect host defense against infection by P. aeruginosa in murine airway epithelia (23). We therefore wanted to determine whether the induction of NOX2 and NOX4 by P. aeruginosa in HLMVECs and lungs is mediated by NF-κB signaling. As shown in Figure 3A, after 24 hours of the infection of murine lungs with P. aeruginosa, inhibitor of kappa B (IκB) phosphorylation was observed, and this phosphorylation was further enhanced at 48 hours after P. aeruginosa instillation. Consequently, IκB degradation was noticeable at 24 hours, and IκB had reached nondetectable concentrations by 48 hours after infection. The inhibition of NF-κB in vivo by the intravenous injection of 1 mg/kg NF-κB inhibitor attenuated the induction of NOX2 and NOX4 in lung tissue by P. aeruginosa (Figure 3B). Next, we investigated the effects of blocking NF-κB activation on P. aeruginosa–induced NOX2 and NOX4 expression in HLMVECs. As shown in Figure 3C, P. aeruginosa activated NF-κB, as evidenced by the enhanced translocation of p65/RelA from the cytosol to the nucleus, and by the time-dependent phosphorylation of IκB in HLMVECs (Figure 3D). Compared with scrambled siRNA (scRNA) control samples, the knockdown of p65 with p65 siRNA in HLMVECs inhibited the induction of NOX2 and NOX4 by P. aeruginosa (Figure 3E). These data show that P. aeruginosa infection activates the NF-κB pathway both in vivo in murine lungs and in vitro in endothelial cells. Further, these results show for the first time that the P. aeruginosa–induced expression of NOX2 and NOX4 proteins in endothelial cells or in the lungs is mediated by the NF-κB signaling pathway.
Figure 3.
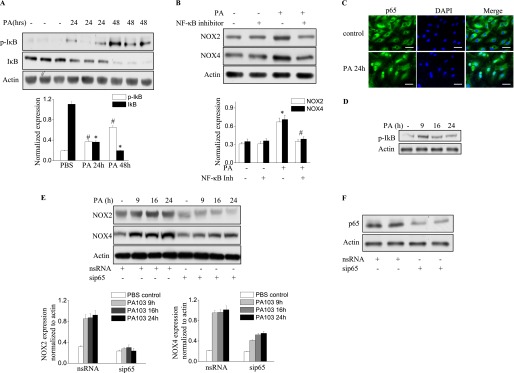
P. aeruginosa induces NOX2 and NOX4 expression via the NF-κB pathway. (A) Lung tissue from mice exposed to PA103 was used for the immunoblot analysis of IκB and phosphorylated IκB. A significant phosphorylation of IκB (#P < 0.01, compared with PBS control samples) and degradation of IκB occurred in the lungs (*P < 0.01, compared with PBS control samples). (B) The inhibition of NF-κB activity in the lung by an intravenous injection of NF-κB inhibitor at a dose of 1 mg/kg before PA103 stimulation blocked NOX2 and NOX4 induction (*P < 0.05, compared with PBS control samples; #P < 0.05, compared with the PA103 treatment group). (C) Immunofluorescence staining of NF-κB subunit p65 was performed after P. aeruginosa treatment for 6 hours. Images were captured using a fluorescent microscope (×200), and the images shown are representative of three independent experiments. Scale bars, 30 μm. (D) The activation of NF-κB was verified by immunoblotting. HLMVECs were exposed to PA103 for the indicated times. The phosphorylation of IκB was analyzed by Western blotting. *P < 0.01, compared with vehicle control samples. (E) HLMVECs were transfected with scRNA or NF-κB subunit p65 small interfering (si)RNA (sip65) for 72 hours, followed by PA103 challenge for the indicated times. Western blotting was performed to detect NOX4 and NOX2 expression. *P < 0.01, compared with scRNA PA MOI = 10 (n = 3). #P < 0.01, compared with scRNA PA MOI = 10 at 9 hours, 16 hours, and 24 hours (n = 3). (F) The knockdown of p65 was confirmed by Western blotting.
NOX4, but Not NOX2, Mediates P. aeruginosa–Induced Endothelial Barrier Dysfunction via Vascular Endothelial Cadherin
Next, we assessed the role of NOX2 and NOX4 on P. aeruginosa–mediated endothelial barrier function. As shown in Figure 4A, P. aeruginosa significantly increased endothelial permeability in a dose-dependent and time-dependent manner, compared with vehicle control samples. The knockdown of the NOX4 gene in HLMVECs by siRNA (as evidenced by Western blotting, approximately 80% of NOX4 protein was down-regulated), restored endothelial permeability (Figure 4B). Interestingly, unlike NOX4, the knockdown of NOX2 exerted no effect on P. aeruginosa–induced endothelial cell (EC) barrier disruption (Figure 4C). To verify these results, an FITC-dextran transwell assay was performed. The down-regulation of NOX4 attenuated P. aeruginosa–induced permeability, whereas the knockdown of NOX2 exerted no effect on FITC-dextran permeability (Figure 4D). These results indicate an important role for NOX4, but not for NOX2, in mediating the loss of endothelial barrier integrity attributable to P. aeruginosa infection.
Figure 4.



P. aeruginosa–induced barrier dysfunction in HLMVECs is blocked by NOX4 siRNA. HLMVECs were grown to confluence on gold electrodes and exposed to heat-killed PA103 at MOI = 1, 5, and 10. Transendothelial electrical resistance (TER) was monitored by electrical cell-substrate impedance sensor (ECIS). The basal monolayer TER was between 1,600 and 1,800 mΩ, and was normalized to 1 before initiating measurements. (A) P. aeruginosa significantly induced endothelial barrier permeability in a dose-dependent manner. *P < 0.01, versus PA MOI = 10. (B) HLMEVCs were transfected with scrambled siRNA or NOX4 siRNA (50 nM) for 72 hours before TER measurement. The P. aeruginosa–induced decrease in TER was partly blocked by NOX4 siRNA, compared with scrambled control siRNA–treated cells. **P < 0.01, versus scRNA PA MOI = 10. The knockdown of NOX4 was confirmed by Western blot, as shown below. (C) HLMEVCs were transfected with scrambled siRNA or NOX2 siRNA (50 nM) for 72 hours before TER measurements. The P. aeruginosa–induced decrease in TER was not blocked by NOX2 siRNA, compared with control siRNA–treated cells. The knockdown of NOX2 was confirmed by Western blotting, as shown below. (D) HLMVECs were first transfected with scrambled RNA, NOX2 siRNA, or NOX4 siRNA. Twenty-four hours later, cells were reseeded onto transwells, followed by P. aeruginosa challenge (MOI = 10). Permeability was indexed by the FITC-dextran leaked into the outside medium from the inner medium, 48 hours after reseeding. Only the knockdown of NOX4 inhibited the FITC-dextran leakage induced by P. aeruginosa. #P > 0.05, versus scRNA PA. ##P < 0.05, versus siNOX4. (E) NOX4 siRNA attenuated P. aeruginosa–induced VE-cadherin phosphorylation and decrease in expression, whereas NOX2 siRNA exerted no effect on VE-cadherin phosphorylation and expression. (F) Immunofluorescent staining of VE-cadherin, ZO-1, and actin was performed 24 hours after P. aeruginosa challenge (MOI = 10). For scRNA-transfected and siNOX2-transfected cells, P. aeruginosa induced significant stress fiber formation, and disruption of VE-Cadherin and ZO-1 from the cell adhesion junction, which was attenuated by NOX4 siRNA. Images are representative of the entire cell monolayer in two independent experiments. Scale bars, 10 μm. p-, phosphorylated; Veh Con, vehicle control.
The effects of NOX4 and NOX2 on permeability were further studied by analyzing the expression and phosphorylation of VE-cadherin in response to P. aeruginosa infection. As shown in Figure 4E, the P. aeruginosa infection of scrambled siRNA–transfected HLMVECs caused significant phosphorylation of VE-cadherin, which is necessary for the disassembly of endothelial junctions (at 6 and 24 h). The down-regulation of NOX4, but not NOX2, with siRNA attenuated the phosphorylation of VE-cadherin at both time points. Furthermore, P. aeruginosa infection caused a significant decrease in VE-cadherin expression in both scrambled siRNA–transfected and NOX2 siRNA–transfected cells. In contrast, the decrease in VE-cadherin expression by P. aeruginosa was significantly attenuated by the down-regulation of NOX4 (Figure 4E). Similarly, VE-cadherin immunofluorescent staining showed that HLMVECs exposed to P. aeruginosa showed significant stress fiber formation and disruption of VE-cadherin at adherens junctions in both scrambled siRNA–transfected and NOX2 siRNA–transfected cells (Figure 4F). In contrast, NOX4 siRNA–transfected cells showed a restoration of VE-cadherin at adherens junctions and fewer stress fibers. In addition, P. aeruginosa caused significant dissociation of Zonula Occluden-1 (ZO-1) from cell–cell junctions in both scrambled siRNA–transfected and NOX2 siRNA–transfected cells, but not in NOX4 siRNA–transfected cells. These results suggest a major role for NOX4 and not NOX2 in P. aeruginosa–induced VE-cadherin phosphorylation and endothelial permeability.
Knockdown of NOX4 Blocks P. aeruginosa–Induced Apoptosis in HLMVECs
Because apoptosis may be involved in barrier dysfunction (24), we sought to learn whether NOX4 is involved in P. aeruginosa–mediated apoptosis. The apoptosis of endothelial cells was induced by P. aeruginosa and analyzed by TUNEL staining and Western blotting of cleaved caspase 3. As shown in Figure 5A, PA103 (MOI = 10; 6 h) induced significant endothelial apoptosis (TUNEL staining) in both scrambled RNA–treated and NOX2 siRNA–treated cells. However, the knockdown of NOX4 attenuated the apoptosis caused by PA103. Further, the exposure of scrambled or NOX2 siRNA–treated HLMVECs to PA103 activated the caspase 3 pathway, as evidenced by increased cleaved caspase 3, and this activation was inhibited by the knockdown of NOX4 (Figure 5B). These results suggest a role for NOX4, but not for NOX2, in the PA103-induced apoptosis of HLMVECs.
Figure 5.

The knockdown of NOX4 attenuates P. aeruginosa–induced apoptosis in HLMVECs. HLMVECs were transfected with scrambled NOX2 or NOX4 siRNA (50 nM) for 72 hours and treated with PA103 (MOI = 10) for 6 hours. (A) Endothelial cell apoptosis was assessed by TUNEL staining. (B) Apoptosis was quantified by the number of TUNEL-positive cells/field. (C) Cell lysates were Western blotted for cleaved caspase 3, an indicator of apoptosis activation. Images are representative of two independent experiments. *P < 0.05, scRNA + PA or siNOX2 + PA versus siNOX4 + PA. Scale bars, 20 μm. Relative intensity versus actin is depicted. All values represent means ± SDs. *P < 0.05, scRNA + PA or siNOX2 + PA versus siNOX4 + PA.
Genetic Knockdown of NOX2 in Mice Attenuates the Inflammatory Response to P. aeruginosa, but Not the Permeability
Next, P. aeruginosa–mediated lung injury was assessed by determining the concentrations of select inflammatory factors in a wild-type C57BL/6J murine model, and the results were compared with those obtained using gp91phox−/− (NOX2) mice in the same background. In wild-type, PBS-instilled control mice, P. aeruginosa induced a significant increase in IL-6, TNF-α, and H2O2 in BAL fluid (Figures 6A–6C)/ However, in gp91phox−/− mice, the concentrations of IL-6, TNF-α, and H2O2 in BAL fluid, in response to P. aeruginosa challenge, were significantly lower. Moreover, P. aeruginosa infection induced a significant infiltration of cells into the alveolar space (Figure 6D), and increased protein concentrations in BAL fluid (Figure 6E), indices of pulmonary leakage. Interestingly, no significant change was evident in the BAL fluid cell count and protein content in gp91phox−/− mice compared with wild-type mice infected with P. aeruginosa. Histological analysis showed that P. aeruginosa induced significant lung inflammation and injury, including neutrophil infiltration into the parenchyma and alveolar space, edema of the alveolar wall, and hemorrhages in wild-type mice (Figure 6F). On the other hand, gp91phox−/− mice showed less injury and inflammation compared with wild-type mice. These results confirm that NOX2 is essential for the P. aeruginosa–mediated release of proinflammatory cytokines in murine lungs, but may not be involved in pulmonary leakage.
Figure 6.

The knockdown of NOX2 expression attenuates P. aeruginosa–induced lung inflammation, but with no loss of endothelial permeability. (A–E) gp91phox−/− knockout mice were exposed to P. aeruginosa for 24 hours. BAL was collected, and its cell count (D), protein content (E), and IL-6 (A), TNF-α(Β), and H2O2 (C) concentrations were measured as described in Materials and Methods. *P < 0.01, compared with wild-type (WT) control samples. **P < 0.01, compared with gp91phox−/− control samples (n = 5). #P < 0.01, compared with wild-type control samples. ##P < 0.05, compared with the wild-type PA treatment group (n = 5). (F) C57BL/6J and gp91phox−/− were exposed to P. aeruginosa for 24 hours C57BL/6J had significant evidence of lung injury and inflammation, whereas gp91phox−/− mice exhibited significantly fewer instances of alveolar damage and inflammatory response. Images are representative of four mice for each group. Original magnification, ×200.
Down-Regulation of NOX4 in Lung Tissue by siRNA Attenuates P. aeruginosa–Induced Pulmonary Permeability
To compare the effects of NOX4 with those of NOX2 in terms of pulmonary permeability and inflammation, we used NOX4-specific siRNA to down-regulate NOX4 expression in murine lung tissue. The instillation of NOX4 siRNA significantly reduced the expression of NOX4 without affecting NOX2 expression (Figure 7F). Unlike gp91phox−/− mice, the knockdown of NOX4 inhibited P. aeruginosa–induced pulmonary permeability, as evidenced by BAL fluid cell counts and protein concentrations (Figures 7A and 7B). On the other hand, the knockdown of NOX4 exerted no effect on IL-6, TNF-α, and H2O2 in BAL fluid (Figures 7C–7E). Furthermore, histological analysis showed that the down-regulation of NOX4 inhibited P. aeruginosa–induced lung tissue injury (Figure 7F). These results suggest that NOX2 and NOX4 play distinct roles in regulating P. aeruginosa–mediated permeability and inflammatory responses in murine lungs.
Figure 7.

The knockdown of NOX4 expression attenuates P. aeruginosa–induced pulmonary permeability but not inflammatory reactions. siRNA NOX4–transfected C57BL/6J mice were exposed to P. aeruginosa for 24 hours. (A–E) BAL was collected, and its cell count (A), protein content (B), and IL-6 (C), TNF-α (D), and H2O2 (E) concentrations were measured as described in Materials and Methods. *P < 0.01, compared with wild-type control samples. **P < 0.01, compared with RNA PA treatment (n = 5). #P < 0.01, compared with scRNA control samples. (F) NOX4 siRNA–treated mice demonstrated less cell accumulation in lung tissue and less lung injury by P. aeruginosa, compared with nsRNA-treated mice. Images are representative of four mice for each group. Original magnification, ×200. Below: Western blotting of cell lysates from scrambled or NOX4 siRNA–treated murine lungs.
Discussion
Here, using murine and HLMVEC models, we studied the mechanism underlying the inflammatory response and lung injury triggered by P. aeruginosa infection. In the mouse, P. aeruginosa infection of the lungs resulted in an increased leakage of proteins and cells into the BAL fluid, along with elevated concentrations of inflammatory cytokines such as IL-6 and TNF-α and oxidative stress, as evidenced by elevated concentrations of H2O2 compared with uninfected, wild-type control samples. In HLMVECs exposed to P. aeruginosa, a clear loss in endothelial barrier integrity was evident. Interestingly, under both in vitro and in vivo infection conditions, a preferential increase in concentrations of the NADPH oxidase proteins NOX2 and NOX4 was noted. Based on in vitro experiments using HLMVECs, P. aeruginosa infection triggered the activation of NF-κB, which was responsible for the observed increase in NOX2 and NOX4 expression levels. Moreover, NOX4, but not NOX2, appeared to be involved in the P. aeruginosa–induced loss of EC barrier integrity. Finally, using NOX2 knockout mice and the siRNA-mediated knockdown of NOX4 in the murine lung, we showed a significant attenuation of inflammatory response and oxidative stress, with no change in permeability after P. aeruginosa challenge in NOX2 knockout mice. Unlike NOX2, the knockdown of NOX4 in the murine lung significantly inhibited pulmonary permeability and lung injury, but exerted no effect on the production of proinflammatory cytokines TNF-α and IL-6 in BAL fluid. Of note, NOX2 knockout mice and the siRNA-mediated knockdown of NOX4 showed an amelioration of lung injury in response to P. aeruginosa. Apparently the inhibitory effects on lung injury by NOX2 and NOX4 down-regulation were achieved through different mechanisms. In gp91phox−/− mice, the improved oxidative stress attributable to the lack of NOX2 expression accounts for the inhibitory effects against P. aeruginosa–induced lung injury. Unlike gp91phox−/− mice, the mechanism underlying the inhibitory effects against P. aeruginosa–induced lung injury in NOX4 knockdown mice with siRNA occurs through improvements in pulmonary vascular permeability, because we did not see improved oxidative stress in BAL fluid from NOX4 siRNA–treated murine lungs.
The NOX family comprises seven members, NOX 1–5, DUOX1, and DUOX2. Of these, in vascular endothelial cells, NOX2, NOX4, and to a lesser extent NOX1 are known to be expressed (25). In agreement with previous reports, we observed a significant expression of NOX 1–4 proteins in HLMVECs. However, only the expression of NOX2 and NOX4 increased in response to P. aeruginosa infection (Figure 2), suggesting that these two NOX proteins could be the principal players in P. aeruginosa–induced lung pathology. The expression level of DUOX1 and DUOX2 in control and infected lung tissue was not determined. However, DUOX1 and DUOX2 are the major NADPH oxidase proteins expressed in airway epithelial cells (26). A recent study showed that flagellin isolated from P. aeruginosa (Type A flagellin, ATCC 27,853; American Type Culture Collection, Manassas, VA) up-regulated DUOX2 expression in human bronchial epithelial cells, whereas the down-regulation of DUOX2 by siRNA up-regulated NOX4 expression, suggesting the existence of a potential compensation mechanism between DUOX2 and NOX4 (27).
The transcription factor NF-κB is known to be activated by various stimuli, including bacterial pathogens, and triggers the expression of inflammatory genes. An airway instillation of P. aeruginosa was shown to activate NF-κB in epithelial cells as early as 4 hours after treatment, followed by a much later activation of NF-κB in a variety of other cell types (23). Moreover, both LPS-induced and TNF-α–induced NOX2 expression is dependent on NF-κB activation (28, 29), and the expression of other NADPH oxidase components, such as p47phox and p67phox, is also regulated by NF-κB (29). Our experiments were based on IκB phosphorylation/degradation and the nuclear translocation of p65/RelA, and clearly demonstrated that P. aeruginosa infection also triggers NF-κB activation, which then induces the expression of NOX 2 and NOX4 proteins. In cell culture experiments, we consistently found a significant phosphorylation of IκB by 9 hours upon exposure to P. aeruginosa, followed by consistent but less amounts of phosphorylation (Figure 5B). In parallel, we also showed that the kinetics of induction for NOX4 and NOX2 occurred as early as 9 hours after infection, and mirrored NF-κB activation. Moreover, the induction of NOX4 and NOX2 was inhibited either by the siRNA-mediated knockdown of p65 or by the treatment of animals with an NF-κB inhibitor (Figures 3A and 3E). Our results are in agreement with an earlier study in which NF-κB–mediated NOX4 expression was also observed in human pulmonary artery smooth muscle cells exposed to hypoxia (30).
In addition to NF-κB, other transcription factors such as E2 transcription factor (E2F) (31), mothers against decapentaplegic homolog (Smad)-binding elements (32), hypoxia-inducible factor 1α (HIF-1α) (33), polymerase (DNA-directed) δ–interacting protein–2 (34), and nuclear factor–erythroid factor 2 (Nrf2) (14) are known to regulate NOX4 in mammalian cells. Interestingly, although hyperoxia increased the expression of both NOX2 and NOX4 in human lung endothelial cells (35), only NOX4, but not NOX2, expression was transcriptionally regulated by Nrf2 (14). Our present findings on distinct cellular responses to NOX2 and NOX4 suggest a differential regulation of NOX isoforms in response to P. aeruginosa. The P. aeruginosa–mediated up-regulation of NOX4 may involve Nrf2 in addition to NF-κB, and this potential pathway is under investigation.
Among the many arsenals used by P. aeruginosa to establish infection in the host, it encodes a Type III secretion system (T3SS) that is a major determinant of virulence, and that allows the bacterium to inject toxins into the host cell (36, 37). The Type III secretion system is associated with acute invasive infections, and requires pilin-mediated bacterial–epithelial contact (35, 36). This system is activated on contact with eukaryotic cell membranes and interferes with signal transduction, resulting in cell death or alterations in host immune responses. The Type III secretion system can be functionally divided into five components, namely, the needle complex, the translocation apparatus, the regulatory proteins, the effecter proteins, and the chaperones. These five parts work together to inject effecter proteins into host cells in a highly regulated manner (37). Thus far, four P. aeruginosa effector proteins have been identified, namely, exoenzyme S (ExoS), ExoT, ExoU (also called peptide A [Pep A]), and exoenzyme Y (ExoY) (38).
In this study, we found that P. aeruginosa increased permeability both in vitro and in vivo. Our experiments identified a positive role for NOX4, but not NOX2, in the loss of EC barrier integrity upon P. aeruginosa infection. However, the exact role of NOX4 in barrier dysfunction remains unclear. Evidence indicates that NOX4 is colocalized with vinculin in focal adhesions (39). This suggests that NOX4 may produce ROS locally in focal adhesions, which may then activate focal adhesion–mediated stress fiber formation, triggering an increase in permeability. Our in vivo experiments using NOX2 knockout mice revealed a lack of significant effect on P. aeruginosa–induced permeability. However, in some studies, NOX2-dependent ROS has been shown to induce vascular permeability in response to hyperoxia (35). ROS-induced signaling is recognized to be highly dependent on the compartmentalization of oxidases (40). We therefore believe that the differences in the function of NOX4 and NOX2 proteins may be attributed to their different tissue distributions and cellular localizations. In other words, the results from the present study support distinct functional roles for NOX4 and NOX2 in lung inflammation.
The present study also led to the interesting observation that the down-regulation of NOX4, but not NOX2, attenuated P. aeruginosa–mediated apoptosis in HLMVECs (Figure 5). Cell–cell detachment, a salient feature of endothelial apoptosis, requires the coordinated participation of a variety of signal transduction pathways, resulting in changes in cell morphology, cell matrix interactions, and adherens junction interactions. Earlier studies indicated that the activation of apoptotic signaling pathways (24, 41), and the dissociation of catenins and VE-cadherins from the adherens junction (42, 43), may be linked to changes in vascular permeability. In fact, our data show that the knockdown of NOX4, but not NOX2, with siRNA partly restores VE-cadherin distribution at the adherens junction in P. aeruginosa–challenged HLMVECs (Figure 4F). However, apoptosis may not be the only mechanism involved in P. aeruginosa–induced endothelial barrier function.
In addition to apoptosis, the direct regulation of cytoskeletal remodeling by ROS and ExoT may also contribute to changes in endothelial permeability by P. aeruginosa. In the present experiments, we used the PA103 strain, which expresses ExoU and ExoT proteins. ExoU is cytotoxic, and the disruption of this gene results in decreased virulence in animal models of acute pneumonia (44). ExoU also demonstrates potent phospholipase activity, and has been shown to be a cofactor of superoxide dismutase in vitro, causing cell rounding and the rapid lysis of host cells (45, 46). ExoT demonstrates GTPase-activating protein and ADP ribosyltransferase activities, and has been implicated in the apoptosis of cells (37, 47). We speculate that ExoT may plausibly contribute to the organization of the actin and stress fibers in vitro in endothelial cells. Thus the Type III secretion proteins ExoU and Exo T may be directly involved in the cell rounding and structural changes seen in our in vitro experiments with HLMVECs. However, whether T3SS proteins that include ExoS, ExoT, ExoU, and ExoY interact with NOX proteins to induce structural and functional changes remains unknown, and calls for further investigation.
In conclusion, P. aeruginosa–induced pneumonia appears to be the result of NOX2-mediated and NOX4-mediated effects on lung vasculature. The involvement of the canonical NF-κB pathway is clear, both in the enhanced expression of NOX2 and NOX4 proteins and in the increased expression of inflammatory cytokines. In addition, our experiments clearly delineate the functions of NOX2 and NOX4 proteins. NOX2 proteins are critical in promoting inflammation and an oxidative response to P. aeruginosa infection, and NOX4 proteins are involved in apoptosis and the disruption of endothelial barrier integrity. Inhibitors that specifically target NOX2 and NOX4 are likely to be useful in the treatment of the highly recalcitrant P. aeruginosa infections most commonly seen in patients with cystic fibrosis and nosocomial infections, and in immunocompromised individuals.
Footnotes
This work was supported by National Institutes of Health grants RO1 HL085553 and PO1 HL58064 (V.N.) and by Merit Review Funding through the Department of Veterans Affairs (R.T.S.).
Author Contributions: P.F. performed in vivo and in vitro experiments and data analysis and interpretation, and drafted the manuscript. V.M. performed nicotinamide adenine dinucleotide phosphate–reduced oxidase–4 siRNA in vivo transfection, lung infection experiments, and data analysis. S.M. performed the in vivo experiments and bacterial preparations. C.T. performed the in vivo experiments involving NF-κB inhibition. R.T.S. designed the experiment with PA103 and performed data analysis and interpretation, and prepared the manuscript. V.N. designed the experiments, interpreted the data, prepared the manuscript, and gave final approval of the article.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0242OC on January 10, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal 2007;9:1991–2002 [DOI] [PubMed] [Google Scholar]
- 2.Suntres ZE, Omri A, Shek PN. Pseudomonas aeruginosa–induced lung injury: role of oxidative stress. Microb Pathog 2002;32:27–34 [DOI] [PubMed] [Google Scholar]
- 3.Ward PA. Oxidative stress: acute and progressive lung injury. Ann N Y Acad Sci 2010;1203:53–59 [DOI] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313 [DOI] [PubMed] [Google Scholar]
- 5.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol 2008;30:279–289 [DOI] [PubMed] [Google Scholar]
- 6.Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of RENOX, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA 2000;97:8010–8014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, Iida M. NOX4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004;109:227–233 [DOI] [PubMed] [Google Scholar]
- 8.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 2010;15:607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of NOX4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res 2005;65:495–504 [DOI] [PubMed] [Google Scholar]
- 10.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor–beta1–induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 2005;97:900–907 [DOI] [PubMed] [Google Scholar]
- 11.Amanso AM, Debbas V, Laurindo FR. Proteasome inhibition represses unfolded protein response and NOX4, sensitizing vascular cells to endoplasmic reticulum stress-induced death. PLoS ONE 2011;6:e14591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, Guo X, Wassall CD, Kemple MD, Unthank JL, Kassab GS. Reactive oxygen species cause endothelial dysfunction in chronic flow overload. J Appl Physiol 2011;110:520–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad M, Kelly MR, Zhao X, Kandhi S, Wolin MS. Roles for NOX4 in the contractile response of bovine pulmonary arteries to hypoxia. Am J Physiol Heart Circ Physiol 2010;298:H1879–H1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pendyala S, Moitra J, Kalari S, Kleeberger SR, Zhao Y, Reddy SP, Garcia JG, Natarajan V. Nrf2 regulates hyperoxia-induced NOX4 expression in human lung endothelium: identification of functional antioxidant response elements on the NOX4 promoter. Free Radic Biol Med 2011;50:1749–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben Mkaddem S, Pedruzzi E, Werts C, Coant N, Bens M, Cluzeaud F, Goujon JM, Ogier-Denis E, Vandewalle A. Heat shock protein GP96 and NAD(P)H oxidase 4 play key roles in Toll-like receptor 4–activated apoptosis during renal ischemia/reperfusion injury. Cell Death Differ 2010;17:1474–1485 [DOI] [PubMed] [Google Scholar]
- 16.Tong X, Hou X, Jourd'heuil D, Weisbrod RM, Cohen RA. Upregulation of NOX4 by TGF{beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res 2010;107:975–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.St Hilaire C, Koupenova M, Carroll SH, Smith BD, Ravid K. TNF-alpha upregulates the A2B adenosine receptor gene: the role of NAD(P)H oxidase 4. Biochem Biophys Res Commun 2008;375:292–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, et al. NOX4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 2012;110:1217–1225 [DOI] [PubMed] [Google Scholar]
- 19.Chun J, Prince A. TLR2-induced calpain cleavage of epithelial junctional proteins facilitates leukocyte transmigration. Cell Host Microbe 2009;5:47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song Y, Pittet JF, Huang X, He H, Lynch SV, Violette SM, Weinreb PH, Horan GS, Carmago A, Sawa Y, et al. Role of integrin alphaV beta6 in acute lung injury induced by Pseudomonas aeruginosa. Infect Immun 2008;76:2325–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadikot RT, Zeng H, Azim AC, Joo M, Dey SK, Breyer RM, Peebles RS, Blackwell TS, Christman JW. Bacterial clearance of Pseudomonas aeruginosa is enhanced by the inhibition of COX-2. Eur J Immunol 2007;37:1001–1009 [DOI] [PubMed] [Google Scholar]
- 22.Usatyuk PV, Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal–induced actin remodeling and barrier function in endothelial cells. J Biol Chem 2004;279:11789–11797 [DOI] [PubMed] [Google Scholar]
- 23.Sadikot RT, Zeng H, Joo M, Everhart MB, Sherrill TP, Li B, Cheng DS, Yull FE, Christman JW, Blackwell TS. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J Immunol 2006;176:4923–4930 [DOI] [PubMed] [Google Scholar]
- 24.Petrache I, Verin AD, Crow MT, Birukova A, Liu F, Garcia JG. Differential effect of MLC kinase in TNF-alpha–induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol 2001;280:L1168–L1178 [DOI] [PubMed] [Google Scholar]
- 25.Brown DI, Griendling KK. NOX proteins in signal transduction. Free Radic Biol Med 2009;47:1239–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid Redox Signal 2009;11:2453–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gattas MV, Forteza R, Fragoso MA, Fregien N, Salas P, Salathe M, Conner GE. Oxidative epithelial host defense is regulated by infectious and inflammatory stimuli. Free Radic Biol Med 2009;47:1450–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of GP91phox. J Biol Chem 2006;281:5657–5667 [DOI] [PubMed] [Google Scholar]
- 29.Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, DeLeo FR, Quinn MT. Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor–alpha. J Leukoc Biol 2007;82:729–741 [DOI] [PubMed] [Google Scholar]
- 30.Lu X, Murphy TC, Nanes MS, Hart CM. PPAR{gamma} regulates hypoxia-induced NOX4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B. Am J Physiol 2010;299:L559–L566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Sheppard OR, Shah AM, Brewer AC. Positive regulation of the NADPH oxidase NOX4 promoter in vascular smooth muscle cells by E2F. Free Radic Biol Med 2008;45:679–685 [DOI] [PubMed] [Google Scholar]
- 32.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, et al. Transforming growth factor–beta1 induces NOX4 NAD(P)H oxidase and reactive oxygen species–dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol 2006;290:L661–L673 [DOI] [PubMed] [Google Scholar]
- 33.Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Gorlach A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 2007;27:755–761 [DOI] [PubMed] [Google Scholar]
- 34.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. POLDIP2, a novel regulator of NOX4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 2009;105:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pendyala S, Gorshkova IA, Usatyuk PV, He D, Pennathur A, Lambeth JD, Thannickal VJ, Natarajan V. Role of NOX4 and NOX2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal 2009;11:747–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by Type III secretion machines. Nature 2006;444:567–573 [DOI] [PubMed] [Google Scholar]
- 37.Hauser AR. The Type III secretion system of Pseudomonas aeruginosa: infection by injection. Natl Rev 2009;7:654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kipnis E, Sawa T, Wiener-Kronish J. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Med Mal Infect 2006;36:78–91 [DOI] [PubMed] [Google Scholar]
- 39.Heeb S, Valverde C, Gigot-Bonnefoy C, Haas D. Role of the stress sigma factor RPOS in GACA/RSMA-controlled secondary metabolism and resistance to oxidative stress in Pseudomonas fluorescens cha0. FEMS Microbiol Lett 2005;243:251–258 [DOI] [PubMed] [Google Scholar]
- 40.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase–derived ROS. Antioxid Redox Signal 2009;11:1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Childs EW, Tharakan B, Hunter FA, Tinsley JH, Cao X. Apoptotic signaling induces hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol 2007;292:H3179–H3189 [DOI] [PubMed] [Google Scholar]
- 42.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol 1996;135:497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong RK, Baldwin AL, Heimark RL. Cadherin-5 redistribution at sites of TNF-alpha and IFN-gamma–induced permeability in mesenteric venules. Am J Physiol 1999;276:H736–H748 [DOI] [PubMed] [Google Scholar]
- 44.Finck-Barbancon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SM, Wu C, Mende-Mueller L, Frank DW. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol 1997;25:547–557 [DOI] [PubMed] [Google Scholar]
- 45.Sato H, Feix JB, Frank DW. Identification of superoxide dismutase as a cofactor for the Pseudomonas Type III toxin, ExoU. Biochemistry 2006;45:10368–10375 [DOI] [PubMed] [Google Scholar]
- 46.Sato H, Frank DW. ExoU is a potent intracellular phospholipase. Mol Microbiol 2004;53:1279–1290 [DOI] [PubMed] [Google Scholar]
- 47.Shafikhani SH, Morales C, Engel J. The Pseudomonas aeruginosa Type III secreted toxin ExoT is necessary and sufficient to induce apoptosis in epithelial cells. Cell Microbiol 2008;10:994–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]