

Abstract
Lung epithelial cell death is a prominent feature of hyperoxic lung injury, and has been considered a very important underlying mechanism of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Here we report on a novel mechanism involved in epithelial cytoprotection and homeostasis after oxidative stress. p62 (sequestosome 1; SQSTM1) is a ubiquitously expressed cellular protein. It interacts with ubiquitinated proteins and autophagic marker light chain 3b (LC3b), thus mediating the degradation of selective targets. In this study, we explored the role of p62 in mitochondria-mediated cell death after hyperoxia. Lung alveolar epithelial cells demonstrate abundant p62 expression, and p62 concentrations are up-regulated by oxidative stress at both the protein and mRNA levels. The p62/LC3b complex interacts with Fas and truncated BID (tBID) physically. These interactions abruptly diminish after hyperoxia. The deletion of p62 robustly increases tBID and cleaved caspase-3, implying an antiapoptotic effect. This antiapoptotic effect of p62 is further confirmed by measuring caspase activities, cleaved poly ADP ribose polymerase, and cell viability. The deletion of the p62 PBI domain or the ubiquitin-associated domain both lead to elevated tBID, cleaved caspase-3, and significantly more cell death after hyperoxia. Moreover, p62 traffics in an opposite direction with LC3b after hyperoxia, leading to the dissociation of the p62/Cav-1/LC3b/BID complex. Subsequently, the LC3b-mediated lysosomal degradation of tBID is eliminated. Taken together, our data suggest that the p62/LC3b complex regulates lung alveolar epithelial cell homeostasis and cytoprotection after hyperoxia.
Keywords: p62/SQSTM1, hyperoxia, tBid, LC3b, apoptosis
Clinical Relevance
Cellular homeostasis is crucial in cytoprotection after oxidative stress. A better understanding of the detailed mechanisms by which the p62/light chain 3b complex regulates cellular homeostasis may provide additional therapeutic targets in the management of epithelial cell death during acute lung injury.
Acute lung injury (ALI) and its severe form, acute respiratory distress syndrome (ARDS), are devastating syndromes responsible for significant morbidity and mortality (1–3). Lung alveolar epithelial cell death is a prominent feature of ALI, and plays an important functional role in its pathogenesis (4–6). Hyperoxia-induced ALI and lung epithelial cell death comprise an established model used widely by investigators to understand the mechanisms involved in ALI/ARDS (4–6). A broad spectrum of signaling molecules regulates alveolar epithelial cell death, including a variety of apoptosis-related proteins such as Fas, Bcl-2 homology domain 3 (B3)-interacting domain death agonist (BID), and autophagic marker proteins, such as light chain 3b (LC3b) (6–9). Fas/BID mediates mitochondria-associated apoptosis (intrinsic pathway) in lung alveolar epithelial cells (7–9). After hyperoxia, Fas polymerizes and forms the death-inducing signaling complex (DISC) with the Fas-associated protein with death domain (7–9). Proapoptotic protein BID is brought adjacently to the DISC and cleaved into an active form, namely, truncated BID (tBID). tBID translocates to mitochondria and regulates the release of cytochrome C, resulting in apoptosis (7–9). tBid has been shown to be regulated by proteasomes, and is a substrate of the ubiquitin ligase (10). After BID cleavage, the N-terminal fragment (tBid-N) is ubiquitinated and subsequently degraded. The BH3 domain in the remaining C-terminal fragment (tBID-C) is free and thus mediates the proapoptotic activity (11).
Recent studies reveal that p62 sequestosome 1 (SQSTM1) regulates the degradation of some ubiquitinated proteins, particularly via lysosomal pathways through LC3b (12). The p62 protein contains several domains that mediate protein–protein interactions and the formation of signaling protein complexes. p62 is able to polymerize via its N-terminal Phox and Bem1p (PB1) domain (13–15). p62 interacts physically with ubiquitin and polyubiquitin via its C-terminal ubiquitin-associated (UBA) domain (13–15). Furthermore, p62 binds directly to LC3 (a and b) via the LC3-interacting region (LIR) domain, which is a 22 amino-acid region located N-terminally to the UBA domain in p62 (13–15). Currently, p62 is believed to be crucial in the regulation of cellular homeostasis, along with autophagic proteins. However, how exactly p62 regulates cell fate upon stressful stimulation, such as oxidative stress, remains unclear. Our previous work focused on hyperoxia-induced epithelial cell apoptosis, particularly the Fas/DISC/BID–mediated pathways (9, 16). We found that autophagic marker protein LC3b migrates toward lipid rafts, interacts with caveolin-1 (Cav-1), and exerts protective functions in the presence of hyperoxia (16). p62 and LC3b are both required for lysosome-mediated degradation of selective targets (13–16). In the present study, we further delineated the function of p62 in the regulation of Fas/tBID pathway–mediated apoptosis in hyperoxia-induced apoptosis. Our data, for the first time, uncovered the detailed functions and mechanisms of p62 involved in intrinsic apoptotic pathways after oxidative stress in alveolar epithelial cells. These data provide novel insights into the regulation of multiple important cellular events after hyperoxia, and suggest potential novel therapeutic targets.
Materials and Methods
Reagents and Chemicals
p62 and LC3b antibodies were purchased from Sigma Chemical Company (St. Louis, MO). All other antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) or Cell Signaling (Danvers, MA). p62 small interfering RNAs (siRNAs) and overexpression clones were from Santa Cruz Biotechnology and Genecopoeia (Rockville, MD), respectively. Immunohistochemistry reagents and kit were purchased from Vectors Laboratories (Burlingame, CA). The TaqMan PCR kit was purchased from Applied Biosystems (Foster City, CA). All other reagents and chemicals were from Sigma Chemical Company.
P62 wild-type and mutants were kindly provided by Dr. Maria Diaz-Meco (Sanford-Burnham Medical Research Institute, La Jolla, CA).
Cell Culture
Primary lung alveolar epithelial cell isolation.
Primary alveolar epithelial cells were isolated from C57BL/6J mice (aged 6–8 weeks). Mice were killed in a carbon dioxide chamber. An intratracheal catheter was cannulated into the trachea after the thoracic cavity was opened. Lungs were perfused with 20 ml PBS, followed by 2 ml of dispase and 0.5 ml agarose. Lungs were dissociated with forceps in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 25 mM HEPES and 200 U/ml DNase. The dissociated lungs were successively filtered through 100-μM and 40-μM cell strainers. The suspension was centrifuged, and the cell pellets were resuspended in DMEM containing 25 mM HEPES and 10% FBS. Cells were seeded on CD45-coated and CD16/32-coated dishes. After 1 hour of incubation in a CO2 incubator, the supernatants were transferred into new dishes for another 4 hours. After centrifuging, the pellets were resuspended in DMEM containing 10% FBS. Cells were then seeded onto dishes or transwell plates coated with fibronectin.
Air–liquid interface cell culture.
Air–liquid interface cell culture was established using transwell plates. DMEM (2.6 ml) with 10% FBS was added to the lower section of each well. Cells diluted into 1 ml DMEM containing 10% FBS were added to the apical side of the transwell inserts. The air–liquid interface cell culture is illustrated in Figure E1 in the online supplement.
Beas2B cells were obtained from the American Type Culture Collection (Manassas, VA). Beas2B cells were cultured in DMEM medium with 10% FBS (Gibco, Grand Island, NY). All cells were grown at 37°C in a humidified atmosphere of 5% CO2/95% air. For hyperoxic treatment, cells were exposed to hyperoxia (95% oxygen with 5% CO2) in modular exposure chambers.
Cell Viability Assay and Caspase 3/7 Activity Assay
A CellTiter-Glo Luminescent Cell Viability Kit and Caspase-Glo 3/7 Assay Kit were purchased from Promega (Madison, WI). Briefly, equal numbers of cells were seeded in each well, using a 96-well plate. Transfection was performed, and then cells were treated with hyperoxia (95% oxygen and 5% carbon dioxide) for designated times. Before measurements, cells were washed twice with PBS. One hundred microliters of CellTiter-Glo reagent or Caspase-Glo 3/7 reagent and 100 μl PBS were added to each well and then incubated for 30 minutes at room temperature. The luminescent signal was measured using a FLx800 Fluorescence Microplate Reader (BioTeck, Winooski, VT).
Western Blot Analysis
Cells were harvested after two washes in cold PBS, and were then resuspended in lysis buffer (radioimmunoprecipitation assay buffer) with protease inhibitors (Roche, Indianapolis, IN). Total protein samples were resolved by 4–12% NuPAGE gel (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were blocked in 5% nonfat milk in PBS with Tween-20 for 1 hour at room temperature and then blocked with primary antibodies at 4°C overnight. Membranes were washed and incubated with appropriate secondary antibodies (Santa Cruz Biotechnology). Detection was performed using the SuperSignal West Pico system (Pierce, Rockford, IL), with exposure to x-ray film (Fujifilm, Tokyo, Japan).
Confocal Microscopy
Briefly, cells were collected and fixed with 4% paraformaldehyde for 30 minutes and then permeabilized by 0.5% Triton X-100 for 1 hour at room temperature. Cells were blocked with 5% BSA in PBS for 2 hours at room temperature before incubating with primary antibodies at 4°C overnight. Cells were then washed three times, and secondary fluorescein-conjugated antibodies (Santa. Cruz Biotechnology) were applied at a 1:200 dilution at room temperature for 1 hour. Images were captured using an Olympus Fluoview BX 61 confocal microscope (Olympus, Center Valley, PA).
Immunohistochemistry
Immunohistochemistry was performed according to the manufacturer’s instructions, using a Vectastain Universal Elite ABC kit (Vector Laboratories). Briefly, Paraffin-embedded sections were incubated at 60°C for 1 hour and then dewaxed with xylene. Sections were rehydrated in serial ethanol solutions and then immersed in the reheated retrieval solution for 30 minutes at 95°C. Endogenous peroxidases were blocked by incubating sections in 0.3% H2O2 for 30 minutes. Sections were preblocked with normal donkey serum before being incubated with primary antibodies at 4°C overnight. The immunoreactions were visualized with peroxidase substrate. Nuclei were stained with Hematoxylin QS (Vector Labs, Burlingame, CA).
Coimmunoprecipitation
Cells were washed twice with PBS, and then resuspended in lysis buffer with protease inhibitor mixtures (Roche). The samples were centrifuged, and the supernatants were incubated with primary antibodies at 4°C overnight. Protein A/G Sepharoses (Santa Cruz Biotechnology) were incubated with samples for an additional 3 hours at 4°C. Immune complexes were precipitated by centrifugation.
Lipid Raft Isolation
Lipid raft fractions were isolated by sucrose gradient ultracentrifugation without detergent, as previously described (16). All procedures were performed on ice. Cell lysates were centrifuged at 39,000 × g for 18 hours before being separated into 12 subfractions. Western blotting was performed to analyze the fractions.
Animal Exposures
C57BL/6 mice (male) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice aged 8–12 weeks were maintained in a pathogen-free facility at Brigham and Women’s Hospital (Boston, MA). Mice were exposed to room air or hyperoxia (5% N2 balanced O2) for the designated times.
Statistical Analysis
The means of fold change were compared using two-way ANOVA to test the differences among independent samples. A difference was considered statistically significant at P < 0.05. Error bars indicate standard deviations.
Results
Hyperoxia Modulated p62 SQSTM1 Expression in Lung Alveolar Epithelial Cells in a Time-Dependent Manner
We initially evaluated the expression of p62 in murine lung tissue, and determined whether hyperoxia regulates its concentrations. As shown in Figure 1A, hyperoxia up-regulated the expression of p62 in lung parenchyma and alveolar epithelial cells (Figure 1A, red arrow). The peak level of p62 occurred between Day 1 and Day 2. Isolated primary lung alveolar Type II cells from C57/BJ6 mice also showed similar responses to hyperoxia (Figure 1B). We further characterized the expression of p62 in response to hyperoxia, using the human epithelial cell line Beas2B. p62 in Beas2B cells was up-regulated by hyperoxia in an identical pattern compared with p62 in primary murine alveolar Type II cells (Figure 1C). Initially, p62 expression increased after hyperoxia and subsided by 72 hours in both primary epithelial cells (Figure 1B) and Beas2B cells (Figure 1C). We next showed that p62 mRNA was also up-regulated by hyperoxia in Beas2B cells, using TaqMan real-time PCR (Figure 1D), as early as 4 hours after hyperoxia.
Figure 1.

Expression of p62 in lung epithelial cells. (A) p62 expression and distribution in lung parenchyma was determined by immunohistochemistry (IHC). C57/BJ6 mice were exposed to room air (RA) or hyperoxia (95%). After the indicated numbers of days, murine lung tissue was inflated, fixed, and dissected as previously described. IHC staining with a polyclonal p62 antibody was performed to determine the expression and localization of p62. Representative images from identical results are shown here. Red arrow indicates p62 in lung epithelial cells. (B) The expression of p62 after hyperoxia was determined by Western blot analysis in primary alveolar epithelial cells isolated from C57/BJ6 mice. Primary epithelial cells were isolated and cultured as described in Materials and Methods. After hyperoxia (95%), cells were collected at the indicated time points and subjected to Western blot analysis. p62 expression was determined using a polyclonal anti-p62 antibody. (C) Beas2B human lung epithelial cells. Beas2B cells were exposed to room air or hyperoxia (95%). After the indicated times of exposure, cell lysates were collected and fractioned, using SDS-PAGE. (D) Beas2B cells were exposed to room air or hyperoxia, as already described. At each indicated time point, mRNA was isolated and the p62 mRNA concentration was determined, using TaqMan real-time PCR. (B–D) All images represent three identical results. * P < 0.05. In B and C, the bar graphs indicate the relative folds of increase, as determined by densitometer. GAPDH, glyceraldehyde 3–phosphate dehydrogenase.
Hyperoxia and p62 Concentrations Affected LC3b in Epithelial Cells
Hyperoxia up-regulated LC3bII in a time-dependent manner, using both primary alveolar Type II cells (Figure 2A) and Beas2B cells (Figure 2B). However, hyperoxia-induced LC3bII became prominent after 48 hours of exposure to hyperoxia (i.e., later than p62 up-regulation). Again, a similar pattern of LC3bII induction was evident in both primary cells and Beas2B cells. We further found that the manipulation of p62 concentrations affected LC3b. As shown in Figure 2C, the deletion of p62 using siRNA resulted in an elevated concentration of LC3b (Figure 2C). In contrast, the overexpression of p62, using p62 cDNA clones, suppressed the amount of LC3b (Figure 2D). These results suggest that p62/LC3b is crucial in regulating epithelial cell homeostasis after hyperoxia, and prompted us to explore the cellular functions of p62 further.
Figure 2.

Effects of hyperoxia or p62 on the expression of light chain 3b (LC3b). (A) The expression of LC3b after hyperoxia was determined by Western blot analysis in primary alveolar epithelial cells isolated from C57/BJ6 mice. As described in Figure 1, primary epithelial cells were isolated and exposed to hyperoxia (95%). After the indicated times of exposure, cell lysates were collected and fractioned using SDS-PAGE. (B) Beas2B cells were exposed to room air or hyperoxia (95%). After the indicated times of exposure, cell lysates were collected and fractioned using SDS-PAGE. LC3b concentrations were determined using Western blot analysis. (C) Expression of LC3b after deletion of p62. Beas2B cells were transfected with control small interfering RNA (siRNA) or p62 siRNA. After 24 hours, cell lysates were collected and subjected to SDS-PAGE. p62, LC3b, and β-actin concentrations were determined by Western blot analysis. (D) Expression of LC3b after overexpression of p62. Beas2B cells were transfected with control clones or p62 overexpression clones. After 24 hours, p62, LC3b, and β-actin concentrations were determined by Western blot analysis. (A–D) All images represent three identical results. *P < 0.05. Bar graphs indicate the relative folds of increase, as determined by densitometer.
Deletion of p62 SQSTM1 Significantly Augmented BID Truncation and Caspase 3 Cleavage, Resulting in Elevated Apoptosis in the Presence and Absence of Hyperoxia
Our previous studies suggested that LC3b regulates lung epithelial cell apoptosis after hyperoxia (16). To evaluate whether p62 plays a role in epithelial cell apoptosis after hyperoxia, we deleted p62, using siRNA in Beas2B cells. As shown in Figure 3A, we found that the deletion of p62 resulted in robustly increased caspase-3 activity after hyperoxia (Figure 3A). Furthermore, the deletion of p62 increased tBID and cleaved caspase-3, as shown in Figure 3B. Our previous studies have shown that the deletion of LC3b also leads to elevated caspase-3 cleaved forms (16). Thus, we next investigated whether the deletion of both p62 and LC3b exerts a synergistic effect on hyperoxia-induced apoptosis. As shown in Figure 2C, the deletion of both p62 and LC3b (double “knockout”) led to markedly higher caspase-3 activities, compared with the deletion of p62 alone or LC3b alone (Figure 3C). Using Western blot analysis, the deletion of both p62 and LC3b (double “knockout”) consistently resulted in significantly elevated apoptosis marker proteins, compared with the deletion of p62 alone or LC3b alone, including cleaved poly ADP ribose polymerase (PARP), cleaved caspase-3, and tBID (Figure 3D). These results prompted us to investigate further the interactions between p62 and BID, Fas, and LC3b in lung epithelial cells after hyperoxia.
Figure 3.

p62 confers cytoprotection after hyperoxia. (A) Beas2B cells were transfected with control siRNA or p62 siRNA. After 24 hours, caspase-3 activities were determined, as described in Materials and Methods. Open bars: Cells were transfected with control siRNA. Striated bars: Cells were transfected with p62 siRNA. (B) Beas2B cells were transfected with control siRNA or p62 siRNA. After 24 hours, cell lysates were obtained and subjected to Western blot analysis. Truncated BID (tBID), cleaved caspase-3, p62, and β-actin concentrations were determined as already described. (C) Beas2B cells were grouped and transfected with several siRNAs: (1) control group, two parts of control siRNA; (2) one part of LC3b siRNA + one part of control siRNA; (3) one part of p62 siRNA + one part of control siRNA; and (4) one part of LC3b siRNA + one part of p62 siRNA. Cells were then incubated for 24 hours, with and without hyperoxia. Cell lysates were obtained and subjected to Western blot analysis. (A–D) All images represent three identical results. *Group 1, +Group 2; both P < 0.05. (C and D) Bar graphs indicate the relative folds of increase, as determined by densitometer. CTL, control; RFU, relative fluorescence units.
Hyperoxia Disrupted p62/Fas, p62/BID, and p62/LC3b Complexes
The effects of hyperoxia on p62/BID, Fas, and LC3b interactions were determined using coimmunoprecipitation (co-IP) assays. Hyperoxia rapidly dissociated the physical interactions between p62 and BID, and between p62 and Fas, as shown in Figures 4A and 4B, respectively. This effect was observed as early as 4 hours after hyperoxia. Similarly, p62 interacted and colocalized with LC3b in room air. Shortly after hyperoxia (4 hours), the interaction and colocalization between p62 and LC3b were significantly decreased, as determined by co-IP assays and confocal microscopy, respectively (Figures 4C and 4D). Interestingly, p62 did not precipitate Bim and Bax, suggesting a selective interaction after hyperoxia (data not shown).
Figure 4.
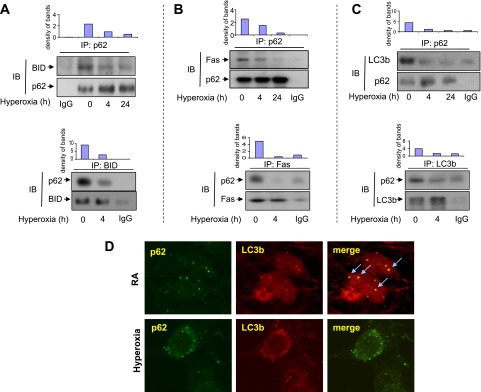
Effects of hyperoxia on p62/BID, Fas, and LC3b interactions. (A–C) Beas2B cells were exposed to hyperoxia. At indicated time points, coimmunoprecipitation (co-IP) assays were performed as described in Materials and Methods. Cell lysates were incubated with p62 polyclonal antibodies and A/G-coated beads (upper row). (A) After fractionation by SDS-PAGE, p62 and BID monoclonal antibodies (murine) were used to determine the input (p62) and the BID interacting with p62. Below: Reverse immunoprecipitation (IP). Cell lysates were incubated with anti-BID antibodies and blotted with anti-p62. (B) After fractionation by SDS-PAGE, p62 monoclonal antibody (murine) and Fas polyclonal antibody (goat) were used to determine the input (p62) and the Fas interacting with p62. Below: Reverse IP. Cell lysates were incubated with anti-Fas antibodies and blotted with anti-p62. (C) After fractionation by SDS-PAGE, p62 and LC3b monoclonal antibodies (murine) were used to determine the input (p62) and the LC3b interacting with p62. Below: Reverse IP. Cell lysates were incubated with anti-LC3b antibodies and blotted with anti-p62. Bar graphs indicate the relative folds of increase, as determined by densitometer. (D) Beas2B cells were exposed to room air or hyperoxia (4 hours). p62 and LC3b colocalization was determined using confocal microscopy. Green, p62; red, LC3b; yellow (arrow), merge. All images represent three identical results. *P < 0.05. (A–C) Bar graphs indicate the relative folds of increase, as determined by densitometer. IB, immunoblotting.
PB1 and UBA Domains Are Crucial for Epithelial Cell Survival after Hyperoxia
To illustrate further the detailed mechanisms by which p62 regulates Fas/BID-mediated apoptosis after hyperoxia, we transfected Beas2B cells with human influenza hemagglutinin–tagged p62 wild-type clones, p62 mutants of the PB1 domain, and p62 mutants of the LIR domain. After expressing these clones in Beas2B cells, we found that cells transfected with the wild-type p62 clone were resistant to hyperoxia-induced cell death. However, cells transfected with either the PB1 mutant clone or the UBA mutant clone showed significantly lower survival after hyperoxia (24 hours) (Figure 5A). Furthermore, cells transfected with mutated PB1 domain or UBA domain clones showed increased concentrations of tBID and cleaved caspase-3 (Figure 5B), implying that intact PB1 and UBA domains are required to eliminate proapoptotic proteins.
Figure 5.

Effects of Phox and Bem1p (PB1) or ubiquitin-associated (UBA) mutants on hyperoxia-induced cell death. (A) Beas2B cells were transfected with wild-type (WT) p62, p62 PB1 domain mutants, and p62 UBA domain mutants. Cells were then exposed to hyperoxia (95%). After 24 hours, cell viability was determined as described in Materials and Methods. Open bars: Cells were transfected with wild-type p62. Striated bars: Cells were transfected with PB1 domain mutant clones. Solid bars: Cells were transfected with UBA domain mutant clones. (B) Beas2B cells were transfected with wild-type p62, p62 PB1 domain mutants (ΔPB1), and p62 UBA domain mutants (ΔUBA). Cell lysates were collected and subjected to Western blot analysis. tBID, cleaved caspase-3, and β-actin concentrations were determined. Bar graphs indicate the relative folds of increase determined by densitometer. All figures represent three identical results. *Group 1, #Group 2; both P < 0.05.
After Hyperoxia, p62 Trafficked away from Lipid Rafts, whereas LC3b Trafficked toward Lipid Rafts
Thus far, our data indicate that p62 plays crucial roles in cytoprotection after hyperoxia, and this protective function may require physical interactions between p62 and BID, Fas, and LC3b.
To explore further the mechanism of hyperoxia-induced dissociation of p62/BID, Fas, and LC3b complexes, we studied hyperoxia-induced p62 and LC3b trafficking in Beas2B cells. As shown in Figures 6A and 6B, after hyperoxia, p62 trafficked away from lipid rafts, and the amount of p62 in lipid raft portion dramatically decreased. On the other hand, LC3b trafficked toward lipid rafts after hyperoxia (Figures 6A and 6B). Our previous studies suggested that LC3b/BID interacted with the lipid raft marker protein Cav-1 (9, 16). We next determined whether p62 interacts with Cav-1 in the presence or absence of hyperoxia. Consistent with our observations that p62 trafficked away from lipid rafts, hyperoxia rapidly disrupted p62 and Cav-1 interactions, using co-IP assays (Figure 6C). Thus, the discrepancy between p62 and LC3b trafficking after hyperoxia may result in the dissociation of the p62/LC3b/BID/Cav-1 complex.
Figure 6.

Effects of hyperoxia on p62 trafficking. Cells were exposed to normoxia or hyperoxia for 4 hours. p62 and LC3b concentrations in lipid rafts (LR) and non–lipid rafts (non-LR) were determined using Western blot analysis. LR and non-LR portions were determined based on caveolin-1 (Cav-1). (A) Beas2B cells. (B) Bar graphs indicate the relative folds of increase as determined by densitometer, correlated with the blots shown in A. (C) Beas2B cells were exposed to room air or hyperoxia (4 hours and 24 hours). Co-IP assays were performed to determine the interactions between p62 and Cav-1. Results are representative of three independent experiments. *P < 0.05. ns, no significance.
Discussion
Hyperoxia-induced alveolar epithelial cell death has been recognized as one of the most important features in acute lung injury (4–6). Apoptosis, necrosis, and autophagy have all been shown to be involved in epithelial cell death (4–6, 16). Upon stressful stimulations such as hyperoxia, all these cellular responses can occur in consequence or simultaneously. The initial cellular response involves attempts at self-salvage by modulating cellular homeostasis. Prolonged stimulation leads to cell death, probably through necrosis (17, 18). To prevent epithelial cell death after noxious stimulation, it is important to reverse the dying process while it is still reversible. However, the precise cellular response to hyperoxia remains unknown. Autophagy, at a basal level, maintains cellular homeostasis (19–21). In previous studies, we showed that the autophagy marker protein LC3b directly interacts with the apoptotic signaling molecule Fas after oxidative stress (22, 23). LC3b, along with other autophagic proteins, maintains cellular homeostasis in the presence of noxious stimulations such as hyperoxia (22, 23). Therefore, we identified this event as cytoprotective. However, the overexpression or deletion of LC3b only slightly modulates cell survival (16), indicating other critical elements in this process. This prompted us to investigate the role of p62/SQSTM1 further in cellular homeostasis and cellular responses to oxidative stress.
Recent studies suggest that p62 acts as a signaling adaptor during autophagy, apoptosis, and necrosis (24). p62 carries the ability to recruit and oligomerize signaling molecules to control the fate of cells (25, 26). This function is performed through controlling the autophagy–lysosome pathway. The autophagy–lysosome pathway is a highly conserved protein degradative system in eukaryotic cells (27). p62 was found to be a receptor for ubiquitinated proteins, to deliver them selectively into the autophagosome, and subsequently degrade them into lysosomes (27). This process requires the autophagic marker protein LC3b. LC3b interacts with p62 via the p62 LC3–interacting region (the LIR domain) (13–15). Thus, p62 may constitute an important element in the selective degradation of harmful cellular components upon noxious stimulation (27, 28). We therefore, hypothesized that p62, along with LC3b, constantly eliminates proapoptotic protein tBID, and subsequently prevents apoptosis.
In this study, we observed increased p62 expression, along with autophagy marker protein LC3b, in lung alveolar epithelial cells that were subjected to experimental hyperoxia. This finding implies an inducible adaptive effect in response to oxidative stress in these cells. Hyperoxia has been found to activate Fas-mediated apoptosis in lung epithelial cells (6–9) and to induce BID truncation (intrinsic apoptotic pathway) (6–9). BID is localized in the cytosolic fraction of cells as an inactive precursor (29–31). After proteolytic cleavage by caspase-8 in response to hyperoxia, a truncated COOH-terminal cleavage fragment is produced, referred to as tBID (29–31). tBID translocates to the mitochondria and induces cytochrome c release (29–31). BID is a long-lived proapoptotic protein. However, it is not active under basal conditions. Its active form, tBID, was found to be a ubiquitinated protein (31). tBID was previously believed to be degraded solely by the proteasome-mediated pathway (32). The degradation of tBID is well-accepted as limiting apoptosis in living cells (29–31). Apparently, the hyperoxia-induced formation of tBID leads to apoptosis, if not salvage. Our data in this study indicate that p62 modulates tBID concentrations in alveolar cells. The p62/LC3b complex probably functions as a carrier leading tBID to lysosomal degradation. A devastating effect on lung epithelial cells induced by hyperoxia involves the disruption by hyperoxia of this transport line. p62 directly interacted with BID, and this interaction was eliminated by hyperoxia after only a short period of exposure (4 h). This result is not surprising, given that p62 carries a ubiquitinated protein–binding domain (UBA domain) on its C-terminal. Further investigation showed that PB1 domain mutants caused similar effects on cell death and tBID formation, implying that the PB1 domain is also crucial. Our results are concordant with previous studies reporting that autophagic degradation requires the PB1 domain–mediated oligomerization of p62 (33). We believe that the oligomerized p62/BID/LC3b complex may function as a transport system to degrade ubiquitinated tBID and protect epithelial cells from apoptosis, in the absence or presence of hyperoxia. After hyperoxia, the integrity of the p62/BID/LC3b complex is disrupted, resulting in a significantly less efficient degradation of tBID. Previous work has repeatedly demonstrated that hyperoxia induces BID truncation/tBID formation. However, the mechanisms behind this induction were not entirely understood. In this study, we provide novel insights into hyperoxia-induced tBID accumulation. Hyperoxia augments tBID in two regards: (1) less degradation via disrupting the p62/LC3b/BID complex, as shown in this work; and (2) increased production by hyperoxia-induced caspase-8 activation, as shown in previous work (7–9).
Consistent with our previous report (9, 16), Cav-1 is required to facilitate the interactions among p62, BID, and LC3b. We have shown that hyperoxia caused the dissociation of the Cav-1/LC3b complex in a time-dependent manner (16). However, hyperoxia-induced p62/Cav-1 dissociation occurs much sooner (4 h versus 24 h), compared with LC3b/Cav-1 dissociation. As soon as after 4 hours of hyperoxia, p62 may have lost its protective function. In this report, we further demonstrated that p62 and LC3b trafficked in an opposite direction from the lipid raft portion to the nonraft portion after hyperoxia.
We conclude that hyperoxia-induced p62 confers cytoprotection against cell death, probably during the initial phase after hyperoxia. As proposed in Figure 7, this protective effect is achieved by directly interacting with LC3b/tBID, leading to the degradation of tBID via the LC3b-mediated lysosomal pathway. Prolonged hyperoxia not only dissociates the p62/LC3b/BID complex, but also suppresses p62 protein concentrations (Figure 1; late time points, hyperoxia for 48–72 h). Thus, after prolonged hyperoxia, epithelial cells will move toward unsalvageable cell death. The detailed mechanisms by which prolonged hyperoxia suppresses p62 protein require further investigation. In addition, our previous study showed mild but not robust protection after the manipulation of LC3b concentrations in epithelial cells (16). The present work provides an explanation, namely, that the deletion of p62 and LC3b together led to much more significant increases in caspase-3 activities, cleaved PARP, and tBID (Figures 3C and 3D). LC3b may only function as an assistant to p62 in this process. LC3b along with p62, but not by itself alone, maintains cellular homeostasis after hyperoxia.
Figure 7.

Schemata of proposed mechanisms. p62 confers cytoprotection after hyperoxia in lung epithelial cells. In the absence of stress (hyperoxia; left), p62 interacts with tBID via its UBA domains. LC3b also interacts with p62. p62 forms a polymer via its PB1 domains. This polymerized p62/LC3b/tBID complex is assembled using Cav-1–related lipid raft as a platform. Cellular homeostasis requires the constant formation of the p62/LC3b/tBID complex, and this complex is degraded via lysosome-mediated pathways. After hyperoxic stress stimulation (right), the interactions between p62 and Cav-1, and between p62 and LC3b, p62, and tBID, are disrupted. Furthermore, hyperoxia induces p62 to migrate in the opposite direction compared with LC3b. The hyperoxia-induced trafficking of p62 and LC3b leads to dissociation of the p62/LC3b/tBID complex. The dissociation of the p62/LC3b/tBID complex stops translocating tBID into lysosome for degradation. Thus, increased tBID translocates to the mitochondria and leads to cytochrome c release and subsequent caspase-dependent cell death.
In conclusion, p62 SQSTM1 plays critical roles in maintaining cellular homeostasis and cytoprotection after oxidative stress. The manipulation of p62 may provide additional therapeutic targets in the management of epithelial cell death during acute lung injury.
Acknowledgments
The authors thank Dr. Maria Diaz-Meco from Sanford-Burnham Medical Research Institute for providing the p62 wild-type and mutant clones, Dr. Chang Hyeok An for his help with the isolation of Type II cells, and Emeka Ifedigbo for the coordination of animal work.
Footnotes
This work was supported by National Institutes of Health grants K08 HL085601 (Y.J.) and R01 HL102076 (Y.J.).
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2012-0017OC on January 18, 2012
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 1967;2:319–3234143721 [Google Scholar]
- 2.Bernard GR. Acute respiratory distress syndrome: a historical perspective. Am J Respir Crit Care Med 2005;172:798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingbar DH. Mechanisms of repair and remodeling following acute lung injury. Clin Chest Med 2000;21:589–616 [DOI] [PubMed] [Google Scholar]
- 4.Pagano A, Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Ann N Y Acad Sci 2003;1010:405–416 [DOI] [PubMed] [Google Scholar]
- 5.Meyrick B. Pathology of the adult respiratory distress syndrome. Crit Care Clin 1986;2:405–428 [PubMed] [Google Scholar]
- 6.Lee PJ, Alam J, Sylvester SL, Inamdar N, Otterbein L, Choi AM. Regulation of heme oxygenase–1 expression in vivo and in vitro in hyperoxic lung injury. Am J Respir Cell Mol Biol 1996;14:556–568 [DOI] [PubMed] [Google Scholar]
- 7.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998;94:491–501 [DOI] [PubMed] [Google Scholar]
- 8.Kim TH, Zhao Y, Barber MJ, Kuharsky DK, Yin XM. BID-induced cytochrome c release is mediated by a pathway independent of mitochondrial permeability transition pore and Bax. J Biol Chem 2000;275:39474–39481 [DOI] [PubMed] [Google Scholar]
- 9.Zhang M, Lee SJ, An C, Xu JF, Joshi B, Nabi IR, Choi AM, Jin Y. Caveolin-1 mediates Fas–BID signaling in hyperoxia-induced apoptosis. Free Radic Biol Med 2011;50:1252–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azakir BA, Desrochers G, Angers A. The ubiquitin ligase Itch mediates the antiapoptotic activity of epidermal growth factor by promoting the ubiquitylation and degradation of the truncated C-terminal portion of BID. FEBS J 2010;277:1319–1330 [DOI] [PubMed] [Google Scholar]
- 11.Tait SW, de Vries E, Maas C, Keller AM, D’Santos CS, Borst J. Apoptosis induction by BID requires unconventional ubiquitination and degradation of its N-terminal fragment. J Cell Biol 2007;179:1453–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tung YT, Hsu WM, Lee H, Huang WP, Liao YF. The evolutionarily conserved interaction between LC3 and p62 selectively mediates autophagy-dependent degradation of mutant huntingtin. Cell Mol Neurobiol 2010;30:795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirano Y, Yoshinaga S, Ogura K, Yokochi M, Noda Y, Sumimoto H, Inagaki F. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J Biol Chem 2004;279:31883–31890 [DOI] [PubMed] [Google Scholar]
- 14.Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor–induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem 2005;280:35625–35629 [DOI] [PubMed] [Google Scholar]
- 15.Ito T, Matsui Y, Ago T, Ota K, Sumimoto H. Novel modular domain PB1 recognizes PC motif to mediate functional protein–protein interactions. EMBO J 2001;20:3938–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol 2011;46:507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verhagen AM, Coulson EJ, Vaux DL. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol 2001;2:reviews3009.1–reviews3009.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mantell LL, Kazzaz JA, Xu J, Palaia TA, Piedboeuf B, Hall S, Rhodes GC, Niu G, Fein AF, Horowitz S. Unscheduled apoptosis during acute inflammatory lung injury. Cell Death Differ 1997;4:600–607 [DOI] [PubMed] [Google Scholar]
- 19.Ryter SW, Chen ZH, Kim HP, Choi AM. Autophagy in chronic obstructive pulmonary disease: homeostatic or pathogenic mechanism? Autophagy 2009;5:235–237 [DOI] [PubMed] [Google Scholar]
- 20.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005;115:2679–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004;6:463–477 [DOI] [PubMed] [Google Scholar]
- 22.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule–associated protein 1 light chain–3B (LC3B) activates extrinsic apoptosis during cigarette smoke–induced emphysema. Proc Natl Acad Sci USA 2010;107:18880–18885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 2010;183:649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci 2007;32:95–100 [DOI] [PubMed] [Google Scholar]
- 25.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1–TRAF6 pathway. EMBO J 2000;19:1576–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009;137:721–735 [DOI] [PubMed] [Google Scholar]
- 27.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 2006;103:4952–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000;290:1717–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 1999;96:615–624 [DOI] [PubMed] [Google Scholar]
- 30.McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Cowburn D. Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell 1999;96:625–634 [DOI] [PubMed] [Google Scholar]
- 31.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. BID, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998;94:481–490 [DOI] [PubMed] [Google Scholar]
- 32.Breitschopf K, Zeiher AM, Dimmeler S. Ubiquitin-mediated degradation of the proapoptotic active form of BID: a functional consequence on apoptosis induction. J Biol Chem 2000;275:21648–21652 [DOI] [PubMed] [Google Scholar]
- 33.Leitner D, Wahl M, Labudde D, Krause G, Diehl A, Schmieder P, Pires JR, Fossi M, Wiedemann U, Leidert M, et al. The solution structure of an N-terminally truncated version of the yeast CDC24p PB1 domain shows a different beta-sheet topology. FEBS Lett 2005;579:3534–3538 [DOI] [PubMed] [Google Scholar]
- 34.Bals R, Beisswenger C, Blouquit S, Chinet T. Isolation and air–liquid interface culture of human large airway and bronchiolar epithelial cells. J Cyst Fibros 2004;2:49–51 [DOI] [PubMed] [Google Scholar]