

Abstract
The nematode worm C. elegans is an ideal model organism for relatively simple, low cost neuronal imaging in vivo. Its small transparent body and simple, well-characterized nervous system allows identification and fluorescence imaging of any neuron within the intact animal. Simple immobilization techniques with minimal impact on the animal's physiology allow extended time-lapse imaging. The development of genetically-encoded calcium sensitive fluorophores such as cameleon 1 and GCaMP 2 allow in vivo imaging of neuronal calcium relating both cell physiology and neuronal activity. Numerous transgenic strains expressing these fluorophores in specific neurons are readily available or can be constructed using well-established techniques. Here, we describe detailed procedures for measuring calcium dynamics within a single neuron in vivo using both GCaMP and cameleon. We discuss advantages and disadvantages of both as well as various methods of sample preparation (animal immobilization) and image analysis. Finally, we present results from two experiments: 1) Using GCaMP to measure the sensory response of a specific neuron to an external electrical field and 2) Using cameleon to measure the physiological calcium response of a neuron to traumatic laser damage. Calcium imaging techniques such as these are used extensively in C. elegans and have been extended to measurements in freely moving animals, multiple neurons simultaneously and comparison across genetic backgrounds. C. elegans presents a robust and flexible system for in vivo neuronal imaging with advantages over other model systems in technical simplicity and cost.
Keywords: Neuroscience, Issue 74, Physiology, Biophysics, Neurobiology, Cellular Biology, Molecular Biology, Anatomy, Developmental Biology, Biomedical Engineering, Medicine, Caenorhabditis elegans, C. elegans, Microscopy, Fluorescence, Neurosciences, calcium imaging, genetically encoded calcium indicators, cameleon, GCaMP, neuronal activity, time-lapse imaging, laser ablation, optical neurophysiology, neurophysiology, neurons, animal model
Introduction
Here we present practical methods for in vivo calcium imaging in C. elegans neurons. The development of genetically encoded calcium-sensitive fluorophores with high signal-to-noise ratio makes C. elegans a comparatively straightforward and cost effective system for measurement of neurophysiology and activity. Our imaging is done with a standard compound microscope using wide-field fluorescence imaging of commonly available fluorophores. We present several techniques employing various fluorophores and different sample preparations, discussing the strengths and weakness of each. Data is then presented from two example experiments. An excellent additional resource on the techniques described here can be found in WormBook, "Imaging the activity of neurons and muscles" by R. Kerr, (http://www.wormbook.org) 3.
Two major classes of genetically encoded fluorescent calcium reporters are commonly used in C. elegans: single channel GCaMP and FRET-based cameleon. We will describe methods and show examples for data generated by each.
GCaMP is based on a modified Green Fluorescent Protein (GFP) that is sensitive to the surrounding calcium concentration. This is accomplished by fusion of GFP and the high calcium affinity protein calmodulin, such that the binding of calcium by calmodulin brings the GFP molecule into an efficient fluorescent confirmation 2. The recent advancements in these fluorophores generate exceptional signal size with up to 500% increase in fluorescence intensity over a physiological range of calcium levels and reasonably fast kinetics of ~95 msec rise time and ~650 msec decay time 4. Over relatively short time periods (mins), these large signals can allow for lower resolution imaging (lower magnification) and, given a well-behaved initial baseline measurement, negate the necessity for continuous baseline or comparative measurements.
Cameleon has the advantage of being a FRET-based fluorophore that generates a ratiometric measurement comparing two independent channels or wavelengths 1. It consists of two separate fluorophores (cyan- and yellow-emitting fluorescent proteins, CFP and YFP) linked by a calmodulin protein. The complex is illuminated with blue light (440 nm) that excites the CFP. Binding of calcium brings the fluorophores closer together, increasing fluorescence resonance energy transfer (FRET) from the CFP (donor) to the YFP (acceptor) and causing the CFP emission (480 nm) to decrease and the YFP emission (535 nm) to increase. Relative calcium levels are measured as the ratio of the YFP/CFP intensity. Cameleon kinetics are slower than that of GCaMP, measured in vivo to have a rise time of ~1 sec and a decay time of ~3 sec 5. However, the ratio of oppositely moving signals increases the signal size and compensates for a number of possible artifacts due to changes in fluorophore concentration, motion or focus drift and bleaching.
Genetically encoded fluorescent reporters negate much of the sample preparation required with exogenously administered probes and C. elegans small transparent body allows imaging within the intact animal using simple wide field fluorescence. The main technical challenge in sample preparation is therefore to safely immobilize the animals. There are a number of different commonly used techniques each with advantages and disadvantages. Using a pharmacological agent to paralyze the animals is easy to implement and allows the mounting of multiple animals on one preparation (Levamisole, a cholinergic agonist that causes muscle tissue to seize is typically used 6). C. elegans can also be physically immobilized by mounting them on stiff 10% agarose 7, 8. This minimizes impact on animal physiology, allows long-term imaging (hours) and recovery of multiple animals but is more technically difficult. Both of these techniques restrict physical access to the animals (which are under a cover slip) and can therefore only be used with certain experimental stimuli (such as light, temperature, electric field or laser damage). For stimuli where physical access is required, such as touch or administration of chemicals, many studies have successfully glued C. elegans in place (using veterinary grade glue) 9. This is technically more challenging, is a single animal preparation and does not allow animal recovery. Finally, numerous microfluidic devices have been employed that physically restrain C. elegans, preserving animal physiology, allowing exposure to most types of stimuli (depending on the device design) and can enable rapid exchange and recovery of the animals 10, 11. However microfluidics require additional technical skills and capabilities in design, fabrication and implementation. In immobilized animals activity and stimulus response can generally be measured in sensory and interneurons. Activity of motor neurons requires more sophisticated techniques for imaging in moving animals. Here we will present detailed methods employing the two most straightforward techniques of pharmacological paralyzation and immobilization with stiff agarose.
The methods presented here can be used to measure neuronal activity and cell physiology in C. elegans. We give an example of each: using GCaMP to measure the sensory response of the ASJ neuron to an external electric field, and using cameleon to measure the physiological calcium response to laser damage of a neuron. These examples show the benefits and drawbacks of the two types of fluorophores and illustrate what is possible with the system.
Protocol
1. Optical Setup
Use a standard compound microscope with epifluorescence imaging capabilities. We use a Nikon Eclipse Ti-U inverted microscope with an Intensilight HG Illuminator.
For best image and signal quality use a high magnification, high numerical aperture objective. We typically use a Nikon X60 1.4 N.A. oil immersion objective. In some cases it is possible to use lower magnification (X40, X20) depending on expression level of the fluorophore and signal strength.
Use a high sensitivity cooled CCD camera, such as an Andor Clara Camera to maximize image sensitivity and minimize background noise.
For single color fluorophores use the standard microscope fluorescence filter set designed for the particular wavelength of the fluorophore. For GCaMP use a standard GFP filter set optimized for excitation at 488 nm and emission at 525 nm. No further optical modifications are required.
- For ratiometric fluorophores, such as cameleon, use a dual imaging system such that identical images can be simultaneously captured at two separate wavelengths. This is accomplished by projecting the dual images side-by-side onto the camera. A simple setup to accomplish this, using basic optical components on an optical grade breadboard, is illustrated in Figure 1. It consists of:
- A microscope filter set with 440 ±20 nm excitation filter and 455 nm long pass dichroic mirror.
- An image mask placed at the initial image plane immediately outside the microscope imaging port (where the camera CCD sensor would usually sit). This restricts the field of view such that it will fill only half of the camera sensor. Placing this mask at the first image plane results in sharp image borders and no overlap when the two images are projected side-by-side.
- The first relay lens set one focal length away from the initial image plane such that it collimates the light.
- A dichroic mirror that splits the light into the two separate imaging wavelengths or channels reflecting one color (>515 nm, yellow) while transmitting the other (<515 nm, cyan).
- Emission filters (band-pass filters) further restricting the transmitted light to the specific imaging wavelengths (cyan at 485 ± 40 nm and yellow at 535 ± 30 nm),
- The second relay lenses (one for each channel) placed in the beam paths such that it refocuses the light to a second image plane at the CCD camera.
- A pair of mirrors and a second dichroic mirror directs the beam paths such that the two images recombined and are projected side-by-side onto the camera CCD sensor. Figure 4A shows an example of the resulting image.
Initial Optical Alignment: Initiate setup and alignment of the optics with a high contrast image using a low magnification lens and transmitted wide-field light illumination (a black sharpie drawn on a microscope slide works well). Bring the image to focus through the microscope oculars and then switched to the microscope port to be used. With high light illumination view the image on an index card at the initial focal plane (a few cm outside the microscope port). Place the first relay lens (C) in the beam path such that it is one focal distance from the initial focal plane and collimates the light while not deflecting the beam path (i.e. is directly centered in the beam path). Place the mask at the first focal plane, adjusted to restrict half the field of view and produce a sharp boarder in the image. Position the mirrors and filters (D, E, G) such that the beam path remains parallel to the table surface and runs in two parallel paths side-by-side, as shown in Figure 1B. Center the second relay lenses (F) in their respective beam paths and move them back and forth along the paths to bring the images to a focus on an index card placed were the camera CCD sensor will be. If done correctly the two images should be parfocal with the microscope oculars and other camera ports. Finally place the camera in position and fine-tune the alignment.
Fine Tuning Optical Alignment: Use the dual image viewed by the CCD camera to fine tune the optical alignment. Use a high contrast image and start with lower magnification, switching to higher magnification for final alignment. Position the transmitted image (cyan channel in Figure 1B) to cover one half of the camera's field of view by moving the camera in X and Y. Position the reflected image (yellow channel in Figure 1B) to cover the other half of the field of view by adjusting the last two mirrors (G) in its beam path. Optimize focus by moving the second relay lens for each channel back and forth along the beam path. If left unaltered the optical setup should be stable and only require rare fine tune adjustment.
2. Sample Preparation and Data Acquisition.
Acquire animals expressing a calcium sensitive fluorophore in the neuron of interest from the Caenorhabditis Genetics Center (C.G.C.) or other sources. Alternatively transgenic animals can be constructed using standard C. elegans techniques. Single cell expression is not necessary (and sometimes not desirable if multiple neurons are to be investigated, see 12) as long as images and measurements from multiple cells can be readily separated. If the fluorophore transgene is not integrated into the genome, significant variation in expression can occur from animal to animal. In this case, preselecting animals with higher expression levels using a fluorescent dissecting microscope increases the signal-to-noise ratio and improves the measurements. Fluorophore expression levels need to be sufficient to be readily imaged with the camera under the acquisition parameters required by the experiment.
Make the agarose pads required for the desired immobilization technique (see 2.3 below) by pressing a small drop of molten agarose between two glass slides spaced by two pieces of laboratory tape. If necessary, agarose pads can be transferred to a glass cover slip before mounting the animals. See Figure 2.
- Immobilize C. elegans for imaging using one of the following techniques:
- Pharmacological Paralyzation: Add 0.05% Levamisole to 2% agarose pads (mixing it into the molten agarose before making the pads in 2.2). Pick 5-10 animals onto the pad and cover with a cover slip. Apply small amounts of hot wax mixture (50% paraffin wax and 50% petroleum jelly) to the corners of the cover slip to hold it in position. Allow 5-10 min for the Levamisole to take effect and the animals to become completely still.
- Stiff Agarose Immobilization: C. elegans can be physically immobilized using stiff 10% agarose pads and 0.05 μm diameter polystyrene nanoparticles. Make 10% agarose pads (10% agarose in NGM buffer by weight) as in 2.2. Add ~3 μl of bead solution (1:10 polystyrene microspheres into NGM by volume) to the top of the pad. Quickly, pick 5-10 C. elegans into the pool of bead solution on the pad and gently cover with a cover slip. Use molten wax applied to the corners of the cover slip to hold it in position. (For additional details on this technique see 7 as well as a previous JoVE article 8)
- Gluing: C. elegans can be glued in place using small amounts of veterinary grade adhesive applied to one side of the animal.
- Microfluidic Devices: Numerous microfluidic devices have been designed to physically hold C. elegans in place for imaging.
Place the prepared slide on the microscope and focus on the desired neuron. It is often easiest to find the C. elegans using bright field illumination and low magnification. Then switching to high magnification and fluorescence imaging to find and focus on the specific neuron.
Stimulus: If a neuronal response to a specific stimulus is desired (light, electric field, gustatory and olfactory cues, temperature etc.), the stimulus apparatus must be designed into the microscope setup, such that it may be administered to the animals in the sample preparation while imaging.
Acquire images using the microscope CCD camera. Exposure times and excitation light level will depend on the baseline signal and expression level of the fluorophore, with weaker signals requiring longer exposure times with concordantly less time resolution. We typically use ~300 msec exposure times.
Acquire images either individually or as a time-lapse movie. Because C. elegans neurons generally display slow activation dynamics (i.e. they lack sodium based action potentials), relatively slow frame rates of ~1 sec are often sufficient to measure neuron dynamics. Acquisition rates up to ~90 frames per second have been used although these may be limited by the integration time necessary for weak fluorescence. Individual movies of a single neuron can easily extend for several minutes (~5-10 min) or longer if the immobilization preparation is stable and the frame rate relatively slow.
3. Data Analysis
There are a number of different fluorescent imaging software packages available with dynamic and ratiometric analysis capabilities including ImageJ and NIS-Elements. We use a simple custom MATLAB program that measures the average fluorescent signal over a selected region of interest.
For individual images (or time-lapse movies that remain perfectly still) select a region of interest (ROI) within the imaged neuron and a separate nearby region for the background measurement. For ratiometric images select similar ROI and background regions for each of the dual images.
Slight movement of the imaged neuron within a time-lapse movie requires additional analysis to automatically select the ROI in each frame, which can be accomplished using numerous object tracking schemes. Our program displays a compressed image of all images in the movie from which a rough boundary (encompassing the object of interests in all frames) is manually selected and an independent background region selected. For the first frame, an ROI is manually selected from within the boundary region. For each subsequent frame, the program selects the brightest pixels from within the boundary region to generate an ROI of the correct size (i.e. the same number of pixels as the manual first frame ROI). For images where the object of interests is sufficiently brighter then the surrounding background within the boundary region this protocol selects a reasonable ROI for each frame.
The fluorescent signal, F, for each frame is calculated as the average pixel intensity in the ROI minus the average intensity in the background region. Ratiometric values are calculated as the ratio of the two signals (YFP-YFPbackground)/(CFP-CFPbackground)-0.65. The factor of 0.65 compensates for bleed through of the CFP channel into the YFP channel, which will be dependant on the fluorophores as well as the particular filter set used (this value is typically ~0.6 for cameleon setups).
The relative calcium level for each frame is calculated as the percent change in intensity normalized to an initial baseline value measured from the first frame (or series of frames): ΔF/F=(F(t)-Fo)/ Fo, where F(t) is the fluorescence at time t and Fo is the initial baseline value.
A ROI selecting the cell body generates the strongest most robust signals but it is also possible to select and measure signals from a ROI along the nerve processes.
4. Problem Solving
Bleaching: Exposure to excitation light can cause bleaching of the fluorophore and is marked by a steady decrease in signal that is dependent on exposure levels. Ratiometric measurements help to compensate for this. However selective bleaching of one wavelength can still occur (typically CFP bleaches quicker then YFP). If bleaching occurs reduce the light exposure level by reducing exposure times and/or excitation intensity (with neutral density filters in the illumination pathway). If necessary, one can compensate for bleaching by running mock trials (with the same excitation exposure but no neuronal stimulus) and quantifying the time constant of signal decay.
Immobilization: For the simple image analysis described here it is critical the animal remains relatively still throughout the recording period. C. elegans respond to light exposure and experimental stimuli, making them most likely to move at the time of recording. Slight movement can introduce significant noise into the measurement especially when measuring from axon processes. Ratiometric analysis helps to mitigate these effects to some extent and measurements from the cell body are more robust. Careful execution and refinement of the immobilization procedures will result in completely still animals.
Background Selection: Careful selection of a background region is necessary for successful image analysis. Background regions should be reasonably uniform and darker then the imaged neuron. It must be sufficiently removed to avoid scattered light (and thus pickup a signal) from the imaged neuron. We find a uniform region 20-50 μm from the ROI works well in most cases.
Ratiometric Optics: Correct alignment of the imaging optics is critical for generating a clear dual image. Commercial microscope attachments for ratiometric imaging are available. An alternative technique is to employ rapid sequential imaging using a fast filter wheel to rapidly switch between emission wavelengths while recording separate images for each.
Ratiometric Analysis: The analysis described here is a relatively simple technique that generates robust signals by virtue of averaging over a ROI. It requires a recognizably bright object to select the ROI. More sophisticated ratiometric analysis is possible by precisely aligning and mapping the dual images such that the fluorescent ratio can be calculated pixel by pixel.
Representative Results
Here we present results from two separate experiments. The first employs GCaMP to measure the response of a specific sensory neuron to a defined external stimulus, giving a good example of how fluorescent calcium reporters can be used to optically monitor neuronal activity in intact C. elegans. The second employs cameleon to measure the intracellular calcium transient triggered within a neuron in response to specific laser damage, thus illustrating how calcium physiology can be measured within a single cell in vivo. To focus on the technical aspects of each measurement individual trial results are presented and discussed in detail. Often the average response over time (calculated as the average response at each time point with respect to the stimulus) or a specific metric (such as the average amplitude of the response) are calculated across numerous trials. Typically 10-20 trials are necessary to generate an acceptable average measurement but this number will depend in the inherent variability of the response. Such data analysis for the experiments discussed here can be found in12 and 13.
GCaMP measurement of sensory response: When subjected to a strong external electric field (≥3 V/cm),C. elegans actively crawl toward the negative pole of the field with precise directed movement. We previously showed that this electrotactic behavior is primarily mediated by the left and right ASJ neurons, amphid sensory neurons located in the animal's head12. To visualize this response we used a transgenic strain expressing GCaMP3 specifically in the left and right ASJ neurons under the gpa-9 promoter. We immobilized animals for imaging using agarose pads containing 0.05% Levamisole sandwiched between two cover slips (as described in the procedures). The electrical stimulus was administered using a custom built imaging chamber that fits on the stage of an inverted Nikon Ti microscope (see12). In brief, the cover slip preparation is placed (with the worm side down) over a hole in a small plastic chamber allowing access for the objectives from below and standard imaging through the cover slip. The chamber was filled with 0.25 mM NaCl and 50 mM glycerol buffer and the electric field stimulus applied using two platinum electrodes lining the ends of the chamber. As described above, a single ASJ neuron expressing CGaMP was imaged using a X100 1.4 N.A. oil immersion objective and standard GFP filter set. A time-lapse movie was acquired for 80 sec (1 frame/sec, 300 msec exposure time), while subjecting the animal to an external 3 V/cm electric field for three separate trials each lasting 10 sec (Figure 3). We measured fluorescence intensity at the cell body using the image analysis described above. The video displays slight faults of both movement and focus drift over the course of the experiment. The strength of the GCaMP signal remains robust however showing large a ~250% increase in fluorescence in response to both the 1st and 3rd stimuli. The result from the second stimuli is substantially reduced demonstrating variability in the neuronal response. Bleaching is minimal as evident by the return to a consistent baseline level.
Cameleon measurement of cellular calcium physiology: Traumatic cellular injury triggers a large calcium transient within a neuron that plays an essential role in the physiological response dictating cellular fate (i.e. cell death vs. initiation of repair processes). We can measure this damage-induced response in vivo using a femtosecond laser to sever individual C. elegans neurons14 while simultaneously measuring cellular calcium signals using cameleon YC3.60 13, 15. Animals expressing cameleon YC3.60 in the six mechanosensory neurons (under the mec-4 promoter), were immobilized using 10% agarose and polystyrene nanoparticles as described in the procedures. We employed dual imaging optics to record signals from both the CFP and YFP fluorescence channels as described in the procedures. We imaged a single ALM neuron and laser targeted the axon ~20 μm from the cell body (Figure 4A). The axon was severed by a brief (<1 sec) exposure to light from a femtosecond pulsed infrared laser focused at the target point along the axon by the imaging objective (see13 for details on laser surgery). Time-lapse images at a frame every 3 sec, with 400 msec exposure times were recorded for 320 sec while performing laser surgery and calcium levels calculated using ratiometric analysis.
Signals were measured independently at the cell body (Figure 4B) and for the axon segment within 5 μm of the cut point (Figure 4C). The figures show both the CFP and YFP intensities as well as the resulting FRET ratio. The measurement at the cell body is well behaved with the CFP rapidly decreasing and the YFP increasing in response to laser damage at t = 0 sec (red arrow). This results in an immediate ~200% increase in the ratiometric signal (ΔF/F) which is sustained for ~90 sec before falling back to near baseline levels. Bleaching is minimal as evident from the consistent baseline.
In the axon segment close to the cut point, the local amount of fluorophore varies over the course of the experiment, complicating the signal. Laser surgery severs the axon and briefly ruptures the membrane, allowing fluorophore to escape and temporarily reducing its local cytoplasmic concentration. This is evident from an initial decrease in the YFP trace in Figure 4C and a comparison of the axon segment near the damage point in the YFP images at times t = 0 sec and t = 6 sec, Figure 4A. At later time points, the severed end swells as part of its continued recovery, resulting in more localized fluorophore and an increase in intensity. This is most evident in the slowly increasing CFP trace in Figure 4C and a comparison of the axon segment in CFP images at times t = 0 sec and t = 270 sec, Figure 4A. However these variations affect both channels equally and the FRET ratio effectively compensates. The resulting measurement shows a response similar to the cell body with an immediate ~150% increase in signal (ΔF/F), a dramatic recovery to near baseline at ~90 sec and then an additional smaller secondary response at ~150 sec. The ratiometric analysis is critical to this measurement as it would be extremely difficult to separate the calcium signal from the other effects, which can vary widely from neuron to neuron and surgery to surgery. The axon signal has more noise compared to the cell body signal primarily due to dimmer fluorescence and the smaller ROI in the narrower axon.
 Figure 1. Basic setup and ratiometric optics. A) The photograph shows the experimental setup consisting of an inverted compound microscope and ratiometric imaging optics. B) A schematic diagram illustrating the imaging optics needed for the ratiometric FRET-based measurements. The image is split into the two wavelengths or channels, which are projected side-by-side on the CCD array.
Figure 1. Basic setup and ratiometric optics. A) The photograph shows the experimental setup consisting of an inverted compound microscope and ratiometric imaging optics. B) A schematic diagram illustrating the imaging optics needed for the ratiometric FRET-based measurements. The image is split into the two wavelengths or channels, which are projected side-by-side on the CCD array.
 Figure 2. Sample preparation. C. elegans are mounted on thin agarose pads for imaging. A) Agarose pads are made by sandwiching a small drop of molten agarose between two microscope slides spaced by two pieces of laboratory tape. B) Animals are transferred onto the agarose pad and covered with a coverslip, held in place by a small amount of wax at each of the four corners.
Figure 2. Sample preparation. C. elegans are mounted on thin agarose pads for imaging. A) Agarose pads are made by sandwiching a small drop of molten agarose between two microscope slides spaced by two pieces of laboratory tape. B) Animals are transferred onto the agarose pad and covered with a coverslip, held in place by a small amount of wax at each of the four corners.
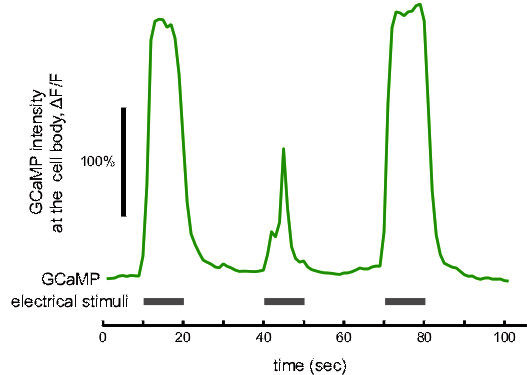 Figure 3. GCaMP example data. The GCaMP signal, ΔF/F, recorded in vivo from the cell body of the ASJ neuron responding to an alternating on/off electric field (green trace). Thick grey lines indicate periods when the external electrical field (3 V/cm) was applied to the animal. Scale bar represents 100% increase in fluorescence intensity.
Figure 3. GCaMP example data. The GCaMP signal, ΔF/F, recorded in vivo from the cell body of the ASJ neuron responding to an alternating on/off electric field (green trace). Thick grey lines indicate periods when the external electrical field (3 V/cm) was applied to the animal. Scale bar represents 100% increase in fluorescence intensity.
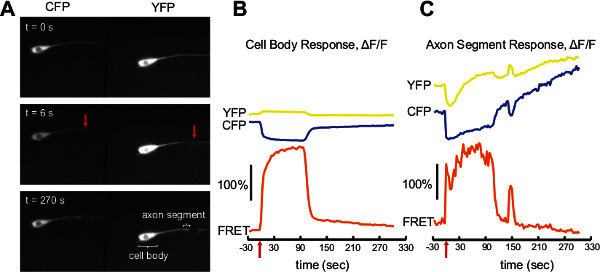 Figure 4. Cameleon example data. A) Three separate frames showing the dual image view of an ALM neuron before and after laser surgery of the axon 20 μm from the cell body. The red arrow in the middle panel indicates the cut point. The bottom panel shows the cell body and axon segment producing the signals in B and C. B) The cameleon YC3.60 signal measured at the cell body of the ALM neuron before and after laser surgery (red arrow). C) The cameleon YC3.60 signal measured at the axon segment within 5 μm of the cut point before and after laser surgery. Yellow traces indicate YFP signals, blue traces indicate CFP signals and orange traces are the resulting FRET ratio. All times are relative to time of laser surgery. Scale bars represent a 100% increase in fluorescence intensity. Click here to view larger figure.
Figure 4. Cameleon example data. A) Three separate frames showing the dual image view of an ALM neuron before and after laser surgery of the axon 20 μm from the cell body. The red arrow in the middle panel indicates the cut point. The bottom panel shows the cell body and axon segment producing the signals in B and C. B) The cameleon YC3.60 signal measured at the cell body of the ALM neuron before and after laser surgery (red arrow). C) The cameleon YC3.60 signal measured at the axon segment within 5 μm of the cut point before and after laser surgery. Yellow traces indicate YFP signals, blue traces indicate CFP signals and orange traces are the resulting FRET ratio. All times are relative to time of laser surgery. Scale bars represent a 100% increase in fluorescence intensity. Click here to view larger figure.
Discussion
Genetically encoded calcium indicators have been widely utilized in C. elegans neurobiology. Numerous groups have employed these techniques to study response of primary sensory neurons to external stimuli as demonstrated here with the ASJ response to an electrical field. Prominent examples include sensation of mechanical touch, specific chemicals, temperature and an electric field 12, 16-19. Activity of interneuron and muscle cells have also been monitored both in response to stimuli and in control of animal behavior. Many of these studies have pushed these techniques a step further using image recognition and tracking techniques to enable recording from neurons in partially restrained or even freely moving animals as well as recording from multiple neurons simultaneously. Additional studies have investigated other aspects of calcium physiology such as the response to neuronal damage 13, 20 as presented here as well as neuronal degeneration on longer time scales21.
In designing a particular experiment, each of the alternative techniques regarding commonly used calcium reporters and animal immobilization should be carefully considered. Immobilization techniques will be dictated by technical aspects such as the need for physical access to the animal, the ability to recover animals following imaging or the need for high throughput. Different fluorescent calcium reporters have specific advantages and disadvantages as well. GCaMP employs straightforward single channel image analysis and the large signal-to-noise ratio makes it applicable to many situations. Cameleon on the other hand requires more complex imaging and analysis but the resulting ratiometric measurement can be critical in situations with potentially large artifacts. For example, fluctuations in the amount of fluorophore (as in the laser damage example) can also occur over long time periods (hrs) due to changes in expression levels. Additional dual imaging strategies are also possible such as GCaMP co-expressed with (or physically linked to) a RFP fluorophore. While not FRET-based this would take advantage of the large dynamic range of modern GCaMP variants while using the red channel as a real-time baseline measurement.
It is important to note that numerous two-channel imaging microscope systems and analysis software are commercially available which, while often more expensive, may be attractive to researchers reluctant to construct the home built system described here. Commercial dual imaging systems (DualView2-DV2, Photometrics) typically use optics similar to our home built system but are contained within a single microscope attachment with standardized alignment procedures. Alternatively, fast filter exchange systems (Lambda DG-4/DG-5 Plus, Sutter Instruments) allow rapid real-time sequential imaging of two color channels without additional imaging optics but are also expensive and require precise synchronization between the filter exchange and camera acquisition.
In summary, C. elegans presents a number of advantages as an in vivo imaging system. The genetically encoded fluorophores are often cell specific and can eliminate the need for injecting or administration of exogenous fluorophores. C. elegans optical accessibility allows imaging within intact animals that are easy and cost effective to maintain, do not require complicated dissection or neuronal culturing and preserve cell physiology. In addition, C. elegans has great capabilities for genetic analysis with a large number of available genetic reagents and well-established techniques. There are of course disadvantages to the system, the primary concern perhaps being that it is an invertebrate. In addition, fluorophores report relative changes in calcium concentration rather than an absolute value and the time resolution of measurements can be restricted by both the dynamics of the fluorophore but also the necessary integration times of weak fluorescence signals. Finally, while calcium is an integral part of neuronal activity and signaling the fluorophores do not directly report the membrane voltage potential as measured with electrophysiological techniques. Nonetheless illustrated by the procedures presented here, C. elegans is an attractive, relatively simple, cost-effective system for a wide range of neuronal imaging studies.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
Several people contributed to the work described in this paper. CVG built the experimental setup, and LS, SHC, and CVG performed the experiments. CVG and SHC wrote the manuscript. All authors subsequently took part in the revision process and approved the final copy of the manuscript. We thank Paul Sternberg for the GCaMP strain. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the NIH National Center for Research Resources (NCRR). The MATLAB image analysis program was adapted from that used in 18. The authors were supported by Boston University and The Massachusetts Life Sciences Center.
References
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Kerr R. WormBook. The C. elegans Reseach Community; 2006. Imaging the activity of neurons and muscles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods. 2009;6:875. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Ihring A, Guerrero G, Isacoff EY, Joesch M, Nakai J, Borst A. In vivo performance of genetically encoded indicators of neural activity in flies. J. Neurosci. 2005;25:4766–4778. doi: 10.1523/JNEUROSCI.4900-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JB. WormBook. The C. elegans Reseach Community; 2005. Acetylcholine. [Google Scholar]
- Kim E, Sun L, Gabel CV, Fang-Yen C. Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization. PLoS ONE. 2013;8(1):e53419. doi: 10.1371/journal.pone.0053419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne AB, Edwards TJ, Hammarlund M. In vivo Laser Axotomy in C. elegans. J. Vis. Exp. 2011. p. e2707. [DOI] [PMC free article] [PubMed]
- Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR. Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans. Neuron. 2000;26:583–594. doi: 10.1016/s0896-6273(00)81196-4. [DOI] [PubMed] [Google Scholar]
- Chronis N, Zimmer M, Bargmann CI. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat. Methods. 2007;4:727–731. doi: 10.1038/nmeth1075. [DOI] [PubMed] [Google Scholar]
- Hulme SE, Shevkoplyas SS, Apfeld J, Fontana W, Whitesides GM. A microfabricated array of clamps for immobilizing and imaging C. elegans. Lab. Chip. 2007;7:1515–1523. doi: 10.1039/b707861g. [DOI] [PubMed] [Google Scholar]
- Gabel CV, Gabel H, Pavlichin D, Kao A, Clark DA, Samuel AD. Neural circuits mediate electrosensory behavior in Caenorhabditis elegans. J. Neurosci. 2007;27:7586–7596. doi: 10.1523/JNEUROSCI.0775-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinan-Lucarre B, Gabel CV, Reina CP, Hulme SE, Shevkoplyas SS, Slone RD, Xue J, Qiao Y, Weisberg S, Roodhouse K, et al. The Core Apoptotic Executioner Proteins CED-3 and CED-4 Promote Initiation of Neuronal Regeneration in Caenorhabditis elegans. PLoS Biol. 2012;10:e1001331. doi: 10.1371/journal.pbio.1001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SH, Clark DA, Gabel CV, Mazur E, Samuel AD. The role of the AFD neuron in C. elegans thermotaxis analyzed using femtosecond laser ablation. BMC Neurosci. 2006;7:30. doi: 10.1186/1471-2202-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt KS, Quast KB, Giles AC, De S, Hendrey D, Nicastro I, Rankin CH, Schafer WR. Dopamine mediates context-dependent modulation of sensory plasticity in C. elegans. Neuron. 2007;55:662–676. doi: 10.1016/j.neuron.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, Goodman MB, Bargmann CI. Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature. 2007;450:63–70. doi: 10.1038/nature06292. [DOI] [PubMed] [Google Scholar]
- Clark DA, Gabel CV, Gabel H, Samuel AD. Temporal activity patterns in thermosensory neurons of freely moving Caenorhabditis elegans encode spatial thermal gradients. J. Neurosci. 2007;27:6083–6090. doi: 10.1523/JNEUROSCI.1032-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron D, Shibuya M, Gabel C, Wasserman SM, Clark DA, Brown A, Sengupta P, Samuel AD. A diacylglycerol kinase modulates long-term thermotactic behavioral plasticity in C. elegans. Nat. Neurosci. 2006;9:1499–1505. doi: 10.1038/nn1796. [DOI] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu ZL, Goncharov A, Jin YS, Chisholm AD. Calcium and Cyclic AMP Promote Axonal Regeneration in Caenorhabditis elegans and Require DLK-1 Kinase. Journal of Neuroscience. 2010;30:3175–3183. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi L, Gerstbrein B, Frokjaer-Jensen C, Royal DC, Mukherjee G, Royal MA, Xue J, Schafer WR, Driscoll M. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat. Neurosci. 2004;7:1337–1344. doi: 10.1038/nn1347. [DOI] [PubMed] [Google Scholar]