

Abstract
Septins are GTP-binding cytoskeletal proteins that contribute to cell polarity, vesicle trafficking, cytokinesis and cell morphogenesis. Here we have characterised the six septins encoded by the genome of the model filamentous fungus Neurospora crassa. Analysis of septin null mutants demonstrated that septins limit the sites of emergence of germ tubes and are important for septation and conidiation in N. crassa. Septins constituted a range of different higher-order structures in N. crassa – rings, loops, fibres, bands, and caps – which can co-exist within the same cell. They showed different patterns of localisation at germ tube tips, with GFP-CDC-10 and CDC-11-GFP forming a subapical collar with lower signal intensity at the tip apex, CDC-3-GFP and CDC-12-GFP organized as a cap at the tip apex and GFP-ASP-1 forming an extended subapical collar. Purification of the septin complex and mass spectrometry of isolated proteins revealed that the septin complex consists predominantly of CDC-3, CDC-10, CDC-11 and CDC-12. Immunoprecipitation of the putative septin ASP-1 revealed that this protein interacts with the core septin complex.
Introduction
Septins are found in a range of eukaryotes from animals to yeasts but are absent from higher plants and certain protists [1], [2]. The septins were first discovered in the budding yeast Saccharomyces cerevisiae in a mutant screen by Hartwell and colleagues [3]. They are a family of GTP-binding proteins that interact with actin, microtubules and membranes and are involved in regulating cytoskeletal dynamics, secretion and membrane remodelling [4]. Septins form polarised, highly organised, oligomeric filaments which assemble into a variety of higher order (HO) structures [5]–[7]. The incorporation of a single alternative subunit, which in turn can be regulated by phosphorylation, allows the formation of different HO assemblies [8].
In vitro, septins purified from Drosophila, mammals, and budding yeast form short (∼32 nm long) filaments, which can assemble end to end into >1500 nm long paired filaments upon dialysis into physiological (75 mM) salt [9]. Purified mammalian septins self-assemble into rings 0.6 µM in diameter and in the presence of the adaptor protein anillin organises actin filaments into bundles while recruiting septin filaments to the bundles [10]. Ultrastructural analysis of yeast sphaeroplasts has demonstrated that septins are found as rings and as ordered gauzes at the cell cortex [11].
In the budding yeast, septins demarcate areas of the plasma membrane and scaffold proteins, which are crucial for the proper assembly of protein complexes such as the contractile actomyosin ring (CAR) and polarisome [12]–[15]. The bud ring acts as a diffusion barrier for polarisome components in the bud. During cytokinesis the septin ring scaffolds the CAR and related cytokinetic proteins and acts as a diffusion barrier to maintain diffusible exocyst and polarisome proteins at the cleavage site [12]–[14]. Septins have been shown to limit diffusion of plasma membrane protein-encoding mRNA (Ist2) thereby compartmentalising the plasma membrane [16]. Additionally, a septin-dependent diffusion barrier forms in the nuclear envelope and limits the movement of extant nuclear pores into the bud [17]. The barrier also contributes to the segregation of other ageing factors, such as carbonylated proteins and DNA circles, thereby ensuring the asymmetric inheritance of age during cell division [17].
Septins have been well studied in budding yeast and to a lesser extent in the fission yeast, Schizosaccharomyces pombe. Filamentous fungi, however, provide a range of cell types and morphologies and tractable experimental systems with which to decipher the roles of septins and elucidate novel functions [18]. Unlike budding yeast, various HO septin assemblies can coexist within hyphae of the filamentous fungus Ashbya gossypii and these structures utilise different signalling pathways for their assembly and maintenance [7]. For example deletion of Gin4 or Elm1 kinases completely abolished the formation of inter-region septin rings (IR rings - septin rings in the middle of a hyphal cell) but not tip-associated septin filaments or septin rings at branch points [7].
In the trimorphic pathogen C. albicans, cdc3 and cdc12 are essential whereas Δcdc10/Δcdc10 and Δcdc11/Δcdc11 null mutants were viable but displayed conditional defects in cytokinesis, cell wall deposition and bud morphology [19]. Candida albicans Δcdc10/Δcdc10 and Δcdc11/Δcdc11deletion mutants formed abnormal hyphae and the Δcdc11/Δcdc11 and Δcdc10/Δcdc10 null mutants were defective for invasive growth and virulence [19], [20]. Interestingly in C. albicans, septin localisation in hyphae is different from that observed during budding or pseudohyphal growth. Septins localise to a tight ring at the bud and pseudohyphal necks whereas in emerging germ tubes septins localise as a diffuse collar or cap at the site of outgrowth [19]. Additionally, a faint cap of septins is also present at the tips of C. albicans hyphae but not pseudohyphae [19]. Analysis of septin gene expression in C. albicans has demonstrated that none of the septins are hyphal specific, which suggests that the change in septin localisation is due to changes in septin regulation during morphogenetic transitions [19].
In the dimorphic plant pathogen Ustilago maydis none of the four septin genes are essential, however, all single septin deletion mutants show conditional lethality when grown at 34°C [21]. At restrictive temperatures septin null mutants were swollen and regularly lysed; both defects could be rescued by the addition of sorbitol [21]. Additionally, septin deletion mutants were hypersensitive to compounds known to affect cell wall integrity (caffeine, calcofluor white and chlorpromazine) suggesting that the primary defect in these mutants is in cell wall construction and possibly osmoregulation [21]. Septins in U. maydis were also shown to have roles in infection [21], [22] similar to septins in C. albicans and Cryptococcus neoformans [20], [23]. The septins formed three HO structures in U. maydis: bud neck collars, band-like structures at the growing hyphal tip and long septin fibres that partially colocalised with microtubules [21]. Only Sep4 (S. cerevisiae cdc10 ortholog) formed long septin fibres in U. maydis [21]. Cdc10 septin fibres have also been shown to occasionally colocalise with microtubules (MTs) in dikaryotic hyphae of C. neoformans [23]. Interestingly in the plant pathogen Magnaporthe oryzae, the location of the appressorium septum is determined by the site of septin ring assembly [24]. During appressorium formation the septin ring functions as both a scaffold for Tea-1-mediated F-actin assembly and as a diffusion barrier for the Rvs167 I-BAR protein, which is thought to induce membrane curvature at the tip of the emerging penetration peg [25].
The genome of filamentous fungus Aspergillus nidulans encodes a complement of five septins, four of which, aspA, aspB, aspC, aspD, are orthologous to the S. cerevisiae septins cdc11, cdc3, cdc12 and cdc10, respectively. The aspE gene is a member of group 5 septins and orthologs are restricted to other filamentous fungi. All five septins are expressed during vegetative growth, demonstrating a lack of sporulation-specific septins, with aspB (cdc3 ortholog) having the highest expression level [26]. Deletion of aspA, aspB, or aspC in A. nidulans leads to delayed septation, increased emergence of germ tubes and hyphal branches and decreased asexual sporulation [27], [28]. These findings suggest that one of the key function of septins in filamentous fungi is to prevent the inappropriate emergence of germ tubes and branches, which is in keeping with their known role as a diffusion barrier and scaffold in budding yeast; presumably cell polarity components are concentrated at a specific site by HO septin structures and are prevented from diffusing freely. Defects in conidiation and septation in septin null mutants demonstrate that septins play a minor role in cytokinesis. In A. nidulans and A. fumigatus, septins localise as rings in forming septa and emerging branches and germ tubes [27]–[29].
To further understand the role of septins in filamentous fungi we analysed the entire septin complement of the model filamentous fungus Neurospora crassa using deletion mutants, live-cell imaging and mass spectrometry of purified septin complexes. The genome of N. crassa encodes six putative septins [2]. Four ORFS (NCU8207, NCU3515, NCU2464 and NCU3795) are orthologs of the core S. cerevisiae septins, cdc3, cdc10, cdc11, and cdc12 and two hypothetical proteins (ORFs NCU1998 termed asp-1 and NCU6414 termed asp-2) contain canonical septin sequences but are only found in filamentous fungi (Pan et al., 2007).
Here we show that the N. crassa septins CDC-3, CDC-10, CDC-11, and CDC-12 participate in septation, cell polarity, and conidiation. Septins formed a range of HO assemblies: rings, loops, fibres, bands and caps. Intriguingly, septins displayed distinct patterns of localisation in germ tube tips suggesting they may perform different roles during tip growth. Mass spectrometry analysis of purified septin complexes revealed that the core complex consisted predominately of CDC-3, CDC-10, CDC-11, and CDC-12, with ASP-1 only detectable in immunoprecipitated complexes when ASP-1 was used as a bait. We were unable to detect a phenotype for the asp-2 null mutant, localise GFP-ASP-2 or find evidence of its expression during vegetative growth.
Results
Septins Limit the Emergence of Germ Tubes and are Involved in Conidiation
Bioinformatic and phylogenetic analysis of the N. crassa genome has revealed the presence of six putative septins [2]. Four ORFS (NCU8207, NCU3515, NCU2464 and NCU3795) are orthologs of the core S. cerevisiae septins, cdc3, cdc10, cdc11, and cdc12, respectively, so we adopted this nomenclature as recommended by Pan et al. (2007). Two hypothetical proteins were found to contain canonical septin sequences but are only found in filamentous fungi [2]. These two ORFs, NCU1998 and NCU6414, were named ASP-1 and ASP-2, respectively. To determine the role of septins in N. crassa we analysed strains in which individual septins were deleted or where a combination of core septins were deleted. Previous studies in A. nidulans have shown that septins delay and limit the emergence of germ tubes and are involved in septation [28]. To understand if this is a general function of septins in filamentous fungi we initially assessed the rates of germination and cell fusion, septation and the morphology of septin null mutants and compared these with the wild type (Figures 1 and 2). A slight but significant (p = <0.05) decrease in the rate of germination in Δcdc-10 and Δcdc-11 strains was observed, which suggests that in these strains germ tubes emerge later than in the wild-type. No significant differences in germination and fusion rates were observed for cdc-3, cdc-12, asp-1 and asp-2 deletion mutants (Figure 2A).
Figure 1. Septin deletion strains have altered morphologies.

Wild-type and single septin deletion strains were incubated in liquid VMM for up to 5 h then imaged by DIC microscopy to obtain representative images of germination and development. Septin deletion strains, with the exception of Δasp-1 and Δasp-2, fail to properly separate conidia and often appeared swollen and misshapen. Black arrows denote multiple germ tube emergence. White arrows show multiple CAT emergence. Scale bar, 5 µm.
Figure 2. Septin deletion strains display an increase in germ tube emergence and unseparated conidia and a reduction in septation compared to wild-type.

(A) The number of unseparated cells in was counted before incubation, the number of cells with more than two germ tubes was counted following 3 h of incubation in liquid VMM and the number of septate cells was determined with DIC microscopy after 5 h of incubation in liquid VMM (n = 300). There was a significant increase (p = <0.05) in the percentage of unseparated conidia and cells with multiple germ tubes compared to wild-type for all strains with the exception of Δcdc10, Δasp-1 and Δasp-2. Similarly, there was a significant decrease (p = <0.05) in the percentage of septate cells for all strains compared to the wild-type except for Δasp-1 and Δasp-2. (B) Strains were incubated in liquid VMM for 3 h then quantified for germination and cell fusion (n = 300). A black dot above bars indicate a significance difference (p = <0.05) compared to the wild-type control. The germination rates of Δcdc-10 and Δcdc-11 were slightly reduced compared to the wild-type.
Despite the relatively minor effects of the absence of individual septins on germination and fusion rates, the morphologies of the cdc-3, cdc-10, cdc-11, and cdc-12 deletion strains were markedly different from that of the wild-type. There were no obvious morphological defects for Δasp-1 and Δasp-2 strains (Figure 1). The major morphological defects displayed by strains lacking core septins included joined conidia, swelling, and multiple germ tube and CAT emergence (Figures 1 and 3). The presence of joined conidia was probably due to defects during conidiation and most likely stems from a failure of conidia to separate during budding due to the lack of a functional septin ring. We quantified the amount of conidia that were unseparated or exhibited more than one site of germ tube outgrowth and found a significant (p = <0.05) increase in Δcdc-3, Δcdc-10, Δcdc-11, and Δcdc-12 strains compared to wild-type (Figure 2). Strains lacking the core septins displayed a significant (p = <0.05) increase in the number of cells producing more than one germ tube and the proportion of unseparated cells than the wild-type, although the phenotype of the cdc-10 null mutant was not as severe. The Δasp-1 and Δasp-2 strains, however, were similar to wild-type.
Figure 3. CAT-mediated cell fusion is affected in septin null mutants.

Wild-type and single septin deletion strains were incubated in liquid VMM for up to 5 h then imaged by DIC microscopy to obtain representative images of cell fusion. In the wild-type each conidium most commonly formed a single CAT. In septin deletion strains, often several CATs are formed during cell fusion. Black arrows denote individual CATs prior to fusion. Scale bar, 5 µm.
We reasoned that an increase in sites of germ tube outgrowth might be accompanied by a reduction in radial extension due to hyperbranching so we measured the radial extension rates of the septin mutants and imaged branching in the mature colony (Table 1 and Figure 4). Indeed, strains lacking the core septins appeared to form more branches at the colony edge and showed a marked reduction in radial extension compared to the wild-type with Δcdc-3 colonies extending at a rate that was 25% slower than the wild-type. Again the Δcdc-10 strain showed a less severe phenotype, with an extension rate only 5% slower than wild type. The colony morphologies of Δcdc-3, Δcdc-11, and Δcdc-12 were slightly denser than the wild-type and showed areas of cell lysis whereas the Δcdc-10, Δasp-1, and Δasp-2 mutants appeared unaffected (Figure 5). Given the role for septins in septation and cytokinesis we assessed whether septation still occurred in the deletion mutants and if so whether there was any reduction in their formation. We found that septa still formed in the mutants and appeared identical to wild-type septa suggesting that septins are not required for septum formation per se; however, there was a significant reduction (p = <0.05) in the number of septate germ tubes formed after 5 h in the Δcdc-3, Δcdc-10, Δcdc-11 and Δcdc-12 mutants suggesting that septation is delayed. Research in U. maydis and C. albicans has shown that in the absence of septins the cell wall is abnormal. However, calcofluor white staining of N. crassa septin deletion mutants did not reveal any defects in cell wall formation at septa or the cell envelope (Figure S1).
Table 1. Radial extension of N. crassa septin mutants.
| Strain | Extension rate±S.d.(mm/hour)* |
| WT | 2.4±0.6 |
| Δcdc-3 | 1.8±0.4 |
| Δcdc-10 | 2.2±0.4 |
| Δcdc-11 | 1.7±0.6 |
| Δcdc-12 | 1.7±0.4 |
| Δasp-1 | 2.5±0.5 |
| Δasp-2 | 2.4±0.4 |
| Δcdc-10 Δcdc-11 | 1.6±0.3 |
| Δcdc-10 Δcdc-12 | 1.7±0.4 |
| Δcdc-11 Δcdc-12 | 1.7±0.4 |
| Δcdc-3 Δcdc-10 | 1.8±0.3 |
| Δcdc-3 Δcdc-11 | 1.6±0.4 |
| Δcdc-3 Δcdc-12 | 1.6±0.4 |
The radial extension of mycelia was assessed after 8 h growth on VMM plates at 35°C. Colony extension was measured along four randomly chosen radii on duplicate plates; n = 8 for each strain.
Figure 4. Branching is more prevalent at the colony edge in septin null mutants.

Wild-type and septin deletion strains were inoculated onto solid VMM, incubated for 18 d at 35°C then imaged using a stereomicroscope. Septin deletion strains, with the exception of Δasp1-1 and Δasp-2, appeared to form more branches at the colony edge.
Figure 5. Colony morphology is altered in septin deletion strains.

Wild-type and septin deletion strains were inoculated onto solid VMM, incubated for 3 d at 24°C then photographed. Septin deletion strains, with the exception of Δasp1-1 and Δasp-2, formed denser colonies, did not produce as many aerial hyphae as wild-type and occasionally regions of cell lysis were visible (black arrows).
To further dissect the functional roles of the N. crassa core septins we constructed double mutants lacking two core septins in different combinations. The resulting strains were all viable and exhibited similar germination and fusion rates to the wild-type (Figure 2A). Cell and colony morphologies of the double deletion strains were comparable to the single null mutants (Figures 5 and 6). However, the amount of unseparated cells and germ tubes was significantly (p = <0.05) increased and septation significantly reduced (p = <0.05) in the double mutants compared to single deletion strains suggesting that individual core septins may have non-overlapping roles (Figure 2B).
Figure 6. Septin double deletion strains have altered morphologies.

Wild-type and septin deletion strains were incubated in liquid VMM for up to 5 h then imaged by DIC microscopy to obtain representative images of germination and development. Septin double deletion strains were swollen and misshapen and failed to properly separate conidia. Black arrows denote multiple germ tube emergence. Scale bar, 5 µm.
To determine the effects of septin deletion on the actin cytoskeleton we generated a cdc-11 null mutant expressing GFP-Lifeact, which is a marker for F-actin [30], [31]. F-actin structures were unaffected in the absence of septin complexes although due to the polarity defects of the cdc-11 null mutant, F-actin accumulated at the growing germ tube tip and in the conidium body where new growth had presumably been initiated (Figure 7).
Figure 7. F-actin structures are unaffected in the absence of cdc-11 .

Lifeact-GFP-expressing wild-type and Δcdc-11 strains were incubated in liquid VMM for 3 h then imaged by widefield fluorescence microscopy. F-actin structures appeared as normal although due to the polarity defects of the cdc-11 null mutant, and actin accumulated at the tip and in the spore body. Scale bar, 5 µm.
Septins Form Rings, Fibres, Bar-like Structures and Caps
To visualise the septin cytoskeleton in N. crassa we made in-frame fusions of GFP to the 3′ ends of the open reading frames of CDC-3, CDC-10, CDC-11, CDC-12, ASP-1 and ASP-2 so that each resulting fusion protein was expressed from their endogenous promoter as the only copy of the gene. Fluorescent protein (FP)-expressing strains were imaged by wide-field fluorescence microscopy followed by iterative deconvolution. We were unable to visualise CDC-10, ASP-1 and ASP-2 by C-terminal integration of GFP or via overexpression of CDC-10-GFP, ASP-1-GFP and ASP-2-GFP from the ccg-1 promoter. Overexpression of cdc-10 and asp-1 tagged at their N-terminal ends with GFP was successful but we were still unable to visualise ASP-2 even when the expression of the ORF was driven by the strong ccg-1 promoter. Given that N-terminal overexpression of GFP-CDC-10 and GFP-ASP-1 was efficacious, we generated N-terminal GFP integration cassettes for these genes to ensure that expression level of the fusion construct was as close to wild-type levels as possible. Endogenous expression of GFP-CDC-10 gave an adequate signal-to-noise ratio so this strain was used for further experiments. However, the expression level of the GFP-ASP-1 fusion was too low to allow visualisation so we used the Pccg1-sgfp-asp-1 strain for imaging instead. To determine whether the GFP tag might interfere with septin function, we compared the phenotypes of the septin GFP fusion strains (cdc-3-gfp, gfp-cdc-10, cdc-11-gfp, cdc-12-gfp, and Pccg1-gfp-asp-1) with those of wild-type and septin-deleted strains. Germination, cell fusion, conidiation, and septation were all similar to wild-type in the septin-GFP strains, indicating that the GFP tags did not interfere with function (Figure S2).
All the septins localized at or close to the plasma membrane in germinating conidia at sites of symmetry breaking (Figure 8). Subsequently, at the tips of germ tubes we observed varied patterns of localisation for the different septins (Figure 8). CDC3-GFP and CDC12-GFP were present as a crescent at the growing germ tube tip whereas GFP-CDC10 and CDC11-GFP localised in the form of multiple bar-like structures in the tip region but the signal was noticeably weaker at the tip apex and more intense at the plasma membrane zone approximately 1 µM from the apex. GFP-ASP-1 localised to the plasma membrane but the fluorescent signal was evenly distributed forming a cap that extended ∼5 µM back from the tip. To our knowledge this is the first report of individual septins displaying varied localisation patterns in hyphal tips. Our findings suggest that given their variation in localisation patterns, the individual septins may play different roles in germ tube growth. The localisation pattern was broadly similar for each individual septin throughout the period of germ tube emergence and outgrowth.
Figure 8. Septins show different patterns of localisation at germ tube tips.

Septin-GFP strains were incubated in liquid VMM for 3 h then imaged with DIC and widefield fluorescence microscopy to obtain representative images of germination and development. At the hyphal tip septins localised as a cap (CDC-3-GFP and CDC-12-GFP), an extended cap (GFP-ASP-1) or as a bar-like structures (GFP-CDC-10 and CDC-11-GFP). Scale bar, 5 µm.
A near identical spatial organisation was observed for septins in CATs before and up to the point of making contact (Figure 9A and data not shown). After fusion, however, we observed that the characteristic bar-like structures for one septin, CDC-11, became concentrated around the fusion pore, indicating a possible role for septins in the terminal stages of cell fusion (Figure 9B). CDC-11-GFP became dispersed in the cytoplasm following fusion (data not shown).
Figure 9. CDC-11 localises to the tips of conidial anastomosis tubes (CATs) and becomes concentrated around the fusion pore.

Cells expressing CDC-11-GFP were incubated in liquid VMM for 3 h then imaged with DIC and widefield fluorescence microscopy. (A) CDC-11-GFP localised as bar-like structures within the tips of CATs before and after making contact with each other. (B) After cell fusion, CDC-11 became concentrated around the fusion pore. Scale bar, 5 µm.
All septins localised as rings at sites of septation suggesting that the heteromeric septin ring complex in N. crassa consists of five distinct septins (Figure 10A–E). Ring formation was preceded by an accumulation of septin fibres associated with the cell cortex which eventually concentrated as a ring (Figure 10F). We also observed two other HO septin assemblies in N. crassa: loops and fibres (Figures 10G–H).
Figure 10. Septins form diverse HO structures.

Cells expressing GFP-tagged septins were incubated in liquid VMM for 3 h then imaged with DIC and widefield fluorescence microscopy to obtain representative images of HO structures and septin ring formation. Septin rings formed by CDC-3-GFP (A), GFP-CDC-10 (B), CDC-11-GFP (C), CDC-12-GFP (D), and GFP-ASP-1(E). (F1, F2, F3) The CDC-11-GFP-labelled septin ring was formed by the accumulation of septin fibres. (G) Dense septin loops formed by CDC-11-GFP were associated with the cell cortex of an ungerminated conidium. (H, I) Septin fibres formed by GFP-CDC-10 (H) and CDC-11-GFP (I) were present in the cytoplasm. Scale bar, 5 µm.
Dense loops with a diameter of ∼1 uM were visualised with CDC-11-GFP but not with other GFP-tagged septins (Figure 10G). These structures were more commonly observed in ungerminated spores and did not appear to show an association with actively growing regions.
Fibres were approximately 0.2–0.5 µM thick and varied in length from 0.5–5 µM and were found throughout the cytoplasm with some being associated with the cell cortex. Septin fibres were observed with CDC-11-GFP or GFP-CDC-10 but not with other septins, and were more prevalent in ungerminated or slowly growing cells. The number of fibres increased drastically when GFP-CDC-10 was overexpressed by the ccg-1 promoter compared to endogenous levels (Figure 11). It has been suggested that fibres inhibit new growth [27]; however, we did not observe any inhibition of germination in strain Pccg-1-sgfp-cdc-10 compared to wild-type or sgfp-cdc-10 (Figure S2).
Figure 11. Septins fibres are more abundant when GFP-CDC-10 is overexpressed.

Conidia from two GFP-CDC-10 strains were harvested then imaged with fluorescence and DIC microscopy to obtain representative images of septin fibres. When GFP-CDC-10 is expressed from the Pccg-1 promoter instead of its native promoter then septin fibres are more abundant. The higher fluorescence background in sgfp-cdc-10 was due to the lower expression of the native cdc10 promoter compared with the ccg-1 promoter. Scale bar, 5 µm.
To determine if these fibres colocalise with MTs or F-actin, we attempted to generate strains co-expressing GFP-tagged septins and βtub-mCherry or lifeact-tagRFPT. However, the resulting strains exhibited a gross perturbation of cell morphology, inadequate co-expression, and mislocalisation of one or both polymers (data not shown). Similarly, we were unable to generate healthy strains expressing GFP-and RFP-tagged septins (data not shown).
To determine if HO septin structures form in the absence of an individual septin we analysed the ability of CDC-11-GFP and CDC-12-GFP to form rings in null mutants. In null mutants lacking cdc-3, cdc-11, and cdc-12, CDC-11-GFP and CDC-12-GFP were only localised to the cytoplasm and septin rings were completely absent (Figure 12). In Δcdc-10, Δasp-1 and Δasp-2 strains the localisation of CDC-11-GFP rings and tip-associated complexes were indistinguishable from wild-type (compare Figures 6 and 12). These findings suggest that CDC-3, CDC-11, and CDC-12 are essential for oligomerisation and HO structure assembly whereas CDC-10, ASP-1 and ASP-2 are not. Interestingly, although CDC-11-GFP and CDC-12-GFP display different patterns of localisation at germ tube tips, it appears that CDC-11 requires CDC-12 for proper tip distribution and conversely, CDC-12 requires CDC-11 for proper tip distribution (Figure 12).
Figure 12. Higher order septin structures fail to form in the absence of CDC-3, CDC-11, or CDC-12.

Septin deletion strains expressing CDC-11-GFP or CDC-12-GFP were incubated in liquid VMM for 5 h then imaged with DIC microscopy and widefield fluorescence to obtain representative images of septin ring formation in the absence of a core septin. Scale bar, 5 µm.
Purification and Analysis of Septin Complexes
We utilised tandem affinity purification (TAP) with magnetic beads to detect protein-protein interactions in septin complexes. Single native genomic loci were tagged by the introduction of an integration cassette containing the V5-HAT epitope and hygromycin selection marker. Confirmation of correct integration into the genome by homologous recombination was confirmed by genotyping PCR of transformants. Next, we examined expression of the different epitope-tagged septins. Western blot analysis of whole cell extracts with a monoclonal anti-V5 antibody demonstrated that all of the V5-HAT-tagged septins were expressed with the exception of V5-HAT-ASP-2 (Figure S3). Western analysis demonstrated the presence of two isoforms of CDC-3 and CDC-10 which could represent sumoylated or phosphorylated isoforms of these proteins as has been shown for S. cerevisiae Cdc3, Cdc11, and Shs1 [32]–[34].
Following confirmation of expression, we analysed the respective strains for phenotypic defects. In V5-HAT-expressing strains, germination, fusion, septation, and germ tube emergence was unchanged from wild-type (Figure S2), suggesting that the V5-HAT tag does not interfere with septin function. Immunoprecipitations (IPs) were carried out for the five V5-HAT-labelled septins CDC-3, CDC-10, CDC-11, CDC-12, and ASP-1. Following elution, the purified protein complexes were separated by SDS-PAGE and visualised by coomassie blue staining. Protein bands were excised then identified by tandem mass spectrometry by the SynthSys Centre at the University of Edinburgh. CDC-3, CDC-10, CDC-11, CDC-12 were detected in all five purifications. ASP-1 was only detected when V5-HAT-ASP-1 was used as the IP target. Figure 13 shows a SYPRO ruby-stained gel with protein profiles of the various IPs. No additional proteins were identified as specific components of septin complexes. These results demonstrate that the core septins form an oligomeric complex and suggests that ASP-1, although it probably does not constitute a core subunit of the septin polymer, interacts with it nonetheless.
Figure 13. Identification of constituents of septin complexes.
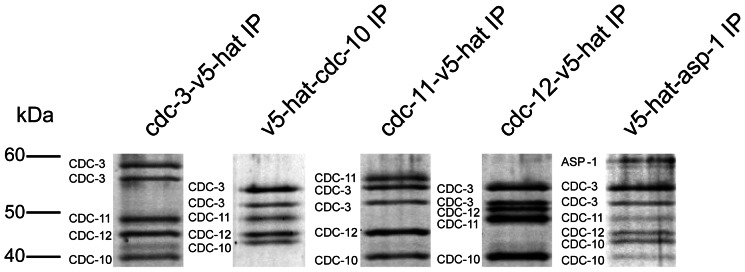
SYPRO Ruby-stained SDS PAGE gel of septin complexes. Protein were purified from septin-V5-HAT expressing strains and identified by mass spectrometry. The positions of the molecular mass markers (M, in kDa) are shown at the left of the panel.
Discussion
Septins have been well-studied in budding yeast and are known to have important roles in regulating cytokinesis, cell polarity, morphogenesis and cell wall synthesis [4]. The study of septins in filamentous fungi is still in its infancy but recent reports have revealed new roles for these cytoskeletal polymers. Of particular interest is the finding in filamentous fungi that septins form a variety of HO structures in a single cell and that these structures are regulated by different mechanisms and display different dynamics [7]. In addition, two septins specific to filamentous fungi have been identified in a recent phylogenetic study suggesting that septins may have evolved novel functions in these organisms [2]. To better define the roles of septins in filamentous fungi we studied the six septins present in the N. crassa genome using a combination of null mutants, microscopic analysis of GFP tagged alleles, and mass spectrometry of purified complexes.
We found that none of the N. crassa septins are essential for cell viability. Similarly, in other filamentous fungi such as A. nidulans, A. gossypii and C. albicans and the fission yeast S. pombe none of the septins studied were essential [7], [15], [19], [27], [28], [35]–[37]. This in the stark contrast to the situation in S. cerevisiae, where deletion of cdc3 and cdc12 was lethal, and cdc10 and cdc11 null mutants were viable only at low temperatures [3], [38]. Interestingly, the pathogenic basidomycetes C. neoformans and U. maydis which both grow by budding in their yeast phases, required core septins for growth at elevated temperatures. Cumulatively these findings highlight a seemingly important link between cell geometry and/or growth mode and the requirement of septins in a particular cell type or species. The phenotypes of N. crassa strains lacking the core septins (cdc-3, cdc-10, cdc-11, and cdc-12) were indistinguishable with all exhibiting reduced septation, increased germ tube emergence, defective separation of conidia, and swollen and misshapen germ tubes. Although the Δcdc-10 mutant showed the same phenotype as cdc-3, cdc-11, and cdc-12 deletion strains, quantification of multipolarisation, septation, and conidiation defects revealed these were not as greatly perturbed. The Δasp-1 and Δasp-2 null mutants did not show any detectable phenotype and behaved as wild-type. Calcofluor white staining of septin mutants showed that despite the misshapen and swollen phenotype of deletion strains, cell walls or septa did not appear to be thickened or abnormal compared to the wild-type suggesting that the phenotypes observed were not due to defective cell wall deposition. This contrasts with U. maydis where septin null mutants grown at a restrictive temperature showed a distinctly altered morphology associated with a thickening of the cell wall in the middle of the cell [21]. Similarly, in C. albicans, cell wall deposition is perturbed in the absence of cdc10 or cdc11 [19]. Taken together these results suggest that N. crassa, unlike U. maydis and C. albicans, does not share the strict requirement for septins in guiding the localisation of the cell wall synthesis machinery.
Given the similarity of the phenotypes for the cdc-3, cdc-10, cdc-11, and cdc-12 deletion mutants, it is likely that these core septins form a heteromeric complex whose function depends on the presence of these four septins. However, the less severe phenotype observed for the Δcdc-10 mutant suggests the role played by different septins in the assembly or function of a septin complex varies. Indeed, analysis of double mutants lacking two of the core septins in different combinations revealed an exacerbation of phenotypic defects compared to single deletion mutants, suggesting that some septin function is non-overlapping. It has been shown that in S. cerevisiae Cdc10 is phosphorylated by Cla4 and that Cdc10 is thought to link polymeric septin rods together [39], whereas Cdc3 is phosphorylated by the cyclin-dependent kinase Cdc28 and is thought to occupy a different part of the heteroctamer with different binding partners.
The distinct cell polarity defects in N. crassa core septins null mutants are reminiscent of the aberrant branching patterns observed in septin deletion strains of A. nidulans, and A. gossypii [28], [35]. We observed an increased number of germ tubes emerging from conidia and the formation of multiple CATs during cell fusion whereas typically only one CAT is formed per germling in the wild type [40]. These findings suggest that septins influence the maintenance and fidelity of the cell polarity machinery presumably by constraining and corralling this machinery to distinct locations on the plasma membrane, in keeping with the known function of septins functioning as a molecular boundary. The absence of a functional septin complex could feasibly alter the dynamics and/or localisation of important polarity components causing multiple germ tube and/or CAT emergence. Multiple bud emergence has been described in certain S. cerevisiae septin mutants [41] and has been ascribed to a failure to successfully corral the polarity machinery.
Although none of the core septins are essential in N. crassa, disruption of these genes singly or in combination produces pronounced morphological defects such as misshapen, wider germ tubes that are swollen at the tip. In C. albicans, deletion of either of the two non-essential core septins cdc10 and cdc11 produced hyphae that appeared as the wild-type but grew with a greater degree of curvature [19]. In N. crassa, the morphological defects do not seem to appear to stem from altered deposition of chitin. These morphological changes are therefore likely caused by defects in targeted secretion. Septins are known to interact with the exocyst and polarisome in S. cerevisiae [13], and, as such, it is possible that these components are mislocalised or improperly regulated in N. crassa in the absence of core septins.
Septation in N. crassa initially involves the assembly of a contractile actin ring (CAR) followed by septum formation and subsequent ingrowth, but does not result in complete cytokinesis and cell separation [30], [42], [43]. Neurospora crassa core septins are involved in but are not essential for septation – septation is delayed in their absence. The involvement of core septins in septation is unsurprising given their known roles in cytokinesis in other fungi [3], [15]. Saccharomyces cerevisiae has a strict requirement for core septins to form septa and complete cytokinesis, whereas in A. nidulans, A. gossypii, C. albicans and S. pombe core septin mutants, septa are still formed but in some instances they appear abnormal [19], [28], [35]–[37]. As with other phenotypic behaviour, N. crassa Δcdc-10 mutants did not display as severe a septation defect as Δcdc-3, Δcdc-11, and Δcdc-12 deletion strains again suggesting that it has a slightly different role to other core septins in septin complex formation.
The marked defect in cell separation during conidiation is in sharp contrast to the relatively mild defect in septation in N. crassa core septin deletion mutants. The majority of conidia in N. crassa are formed by macroconidiation which involves the formation of specialised aerial hyphae (conidiophores) and the subsequent formation of proconidial chains by repeated apical budding followed by sepation between proconidia and their separation into spores [44]. Neurospora crassa core septin deletion mutants produce chains of unseparated conidia which suggests that CAR contraction and cell wall/plasma membrane ingrowth fails to occur between proconidia. A role for septins in spore formation has been reported in A. nidulans and C. neoformans [23], [27], [28]. In these organisms the production of the specialised spore-forming structure, a conidiophore or basidium, respectively, is perturbed rather than the separation of spores. These differences are most likely due to differences in the sequence of events during macroconidium-formation in N. crassa compared to A. nidulans and C. neoformans. In the latter two organisms, septins are probably necessary for the initial cytokinetic events during the morphogenesis of the spore-forming structure and failure to undergo cytokinesis terminates the process. A fraction of cytokinetic events during spore-formation are successful hence the production of separated spores albeit at a reduced rate [23], [28].
We were able to label all of the N. crassa septins by integrating GFP at the endogenous locus of the respective genes, with the exception of ASP-2, which we were unable to visualise either by either integration of a knock-in cassette or overexpression. Due to the weak fluorescence of GFP-ASP-1, we used an overexpression fusion construct controlled by the ccg-1 promoter. In keeping with a previous proposition [45], septin localisation fell into three patterns: localisation to partition as septin rings, localisation to foci of new growth as caps and bar-like structures, and cytosolic localisation as loops and fibres. All septins localised as a ring and were all present at hyphal tips as has been shown for other fungal septins. However, at the tips of germ tubes and CATs we observed three specific patterns of septin localisation: a cap formed by CDC-3 and CDC-12, a band of bar-like structures formed by CDC-10 and CDC-11, and an extended cap formed by ASP-1. These structures/patterns marked the site of germ tube emergence prior to outgrowth and persisted during tip extension. We did not observe the formation of septin rings from these tip-associated complexes as has been found in A. gossypii. This is the first report demonstrating the co-existence of different patterns of septin localisation at a hyphal tip. It is possible that the different patterns observed represent different functional roles for septins in maintaining and/or regulating the tip growth apparatus in these hyphae. The presence of septins at the hyphal tip is consistent with their proposed roles as scaffold and boundaries at the plasma membrane. Interestingly, in C. albicans the exact pattern of septin localisation at hyphal tips is determined by the cell type; a diffuse band of Cdc10-GFP is found at the tips of germ tubes whereas in pseudohyphae the band is absent and instead Cdc10-GFP is organised into a cap. In this instance different patterns of septin organisation are due to changes in septin regulation during the elaboration of different hyphal types [19].
Another possible role for septins in N. crassa comes from their localisation during CAT-mediated cell fusion. During cell fusion, septin localisation at CAT tips is identical to that of germ tubes. Once contact between two CATs is made septins became enriched at the tip apex then gradually rearranged to form the perimeter of the nascent fusion pore, suggesting that they participate in the fusion process. A variety of other proteins are enriched in the tips of CATs growing chemotropically towards each other, remain in CAT tips following cell fusion, and are localized at the fusion pore [46]. These proteins include F-actin [30] which has been shown to interact with septins in mammalian cells (see discussion below).
In addition to rings, caps, and bar-like structures, we observed septins organised into loops and fibres. The dense septins loops, ∼1 µM in diameter, were in ungerminated conidia expressing CDC-11-GFP and a similar structure has been observed in A. nidulans, A. gossypii, A. fumigatus and in mammalian cells treated with anti-actin drugs [10]. In A. gossypii, an increase in the number of loops was associated with growth, arrest, heat shock, and treatment with anti-actin drugs led the authors to suggest that these structures are self-assembling reservoirs of septins that form above a certain concentration of free subunits [7]. In N. crassa, these loops are not observed in the presence of other HO septin structures or during growth and, as such, it is likely they serve a similar function.
Septin fibres were commonly observed in cells expressing CDC-11-GFP or GFP-CDC-10 but not other septin-GFP fusions. Septin fibres are present in a range of mammalian cells and have been recently found in A. nidulans, A. gossypii, A. fumigatus, C. neoformans, U. maydis, and chlamydiospores of C. albicans [7], [21], [23], [27], [29], [47], [48]. In mammalian, cells septin fibres colocalise with actin stress filaments and/or microtubules and in C. neoformans and U. maydis partially colocalise with microtubules, although colocalisation of fibres with actin cables was not carried out in these latter two organisms [21], [23]. Our attempts to generate strains co-expressing GFP-tagged septins and βtub-mCherry or Lifeact-tagRFPT were unsuccessful due to the resultant gross perturbation of cell morphology, inadequate co-expression, and mislocalisation.
The septin fibres observed in N. crassa appear very similar to those present in A. nidulans and A. fumigatus [27], [29]; they were 0.2–0.5 µM thick and 0.5–5 µM long, and are associated with the plasma membrane and found in the cytosol. When we overexpressed GFP-CDC-10 we saw a drastic increase in the number of fibres (Figure 11). The accumulation in septin fibres upon overexpression of GFP-CDC-10 suggests that these fibres could serve as storage reservoirs of free GFP-CDC-10 monomers, similar to the proposed role of the septin loops found in quiescent regions/cells [7]. Alternatively, based on the absence of septin fibres in budding yeast and the finding that yeast septin mutants do not produce extra growth foci, it has been proposed that septin fibres may have a role in suppressing new growth [27]. Indeed, the presence of fibres in non-growing regions and cells supports this idea and the notion that fibres are storage reservoirs [27]. However, as overexpression of GFP-CDC-10 in N. crassa does not supress germination or germ tube emergence (Figure S2), this implies that fibres are not a growth repressing structure.
To investigate the relationship of the various septins in the assembly of HO structures we analysed the ability of CDC-11-GFP or CDC-12-GFP to form rings and other structures in null mutants. Typically in S. cerevisiae, A. nidulans, and A. gossypii the absence of a single core septin abrogates the formation of HO structures [7], [28], [49], [50] whereas in U. maydis, C. albicans, and S. pombe these structures persist in septin null mutants [19], [21], [51]. We found that in N. crassa CDC-3, CDC-11, and CDC-12 are required for ring formation and proper localisation of septin complexes; in their absence the only fluorescence observed was cytoplasmic. Interestingly, although CDC-11-GFP and CDC-12-GFP display different patterns of organisation in the tip growth apparatus, it appears that CDC-11 requires CDC-12 for proper tip localisation and vice versa, CDC-12 requires CDC-11 for proper tip localisation. Conversely, deletion of cdc-10, asp-1, or asp-2 did not impede the formation and localisation of septin complexes. From these findings it can be expected that ASP-1 and possibly ASP-2 are “peripheral” septins that could modify or influence septin complexes but are not core constituents. It is possible that ASP-1 performs a similar regulatory role to Shs1 in S. cerevisiae, which binds to and interacts with the septin complex. It has been shown that Shs1 is phosphorylated [33] and that it plays a role in regulating the formation and stability of the septin ring complex [8]. Interestingly, A. nidulans mutants lacking AspE, the ASP-1 ortholog, showed a marked reduction of septin fibres while other structures are mostly unaffected, suggesting a specific role for this septin in fibre assembly [27]. The ability of cdc-10 null mutants to form HO structures is consistent with models of septin polymer formation in S. cerevisiae, where Cdc10 links septin oligomers together [38]. Similarly, in U. maydis the CDC-10 ortholog, Sep4, is not required to form HO assemblies. Septins have been shown to associate with actin in mammalian cells [10], [52]. To determine the effects of septin deletion on the actin cytoskeleton we generated a cdc-11 null mutant expressing GFP-lifeact, which is a marker for F-actin [30], [31]. F-actin structures were unaffected in the absence of septin complexes although the location of actin cable nucleation was dispersed in germ tube tips compared to the wild-type suggesting that septin complexes help to constrain regions of actin nucleation at the hyphal tip.
We were able to efficiently purify septin complexes from N. crassa cell extracts using five of the six septins as the target. The failure to detect expression of V5-HAT-tagged ASP-2 in western blots or in IP experiments, the lack of a phenotype for the asp-2 null mutant, the inability to express GFP fusions for ASP-2 by knock-in or by overexpression, and the failure to detect ASP-2 in subsequent IP of various septins, indicate that the asp-2 ORF is not expressed at detectable levels. Septin complexes were isolated in a convenient, rapid dynabead-based method with almost a complete absence of contaminants, confirming that the V5 epitope is highly efficacious for protein purification from N. crassa.
The composition of the purified N. crassa septin complex is similar to that obtained for S. cerevisiae and C. albicans; the septin oligomer is composed of CDC-3, CDC-10, CDC-11, and CDC-12 with ASP-1 present as an associating partner but is probably not an integral component of the septin complex.
Materials and Methods
Strains and Culture Conditions
Neurospora crassa strains used and generated in this study are listed in Table 2. Neurospora crassa strains generated during this study were derived from FGSC #2489 (WT 74-OR23-1 A), FGSC #6103 (his-3− A) FGSC #9717 (his-3− Δmus-51::bar+ A), FGSC #9718 (Δmus-51::bar+ a), and FGSC #9719 (Δmus-52::bar+ a) which were obtained from the Fungal Genetics Stock Center (FGSC; School of Biological Sciences, Kansas City, MO). Strains were maintained on Vogel’s minimal medium (VMM) plates with 2% sucrose and 1.5% agar and all manipulations were according to standard N. crassa techniques [53]. Hygromycin B (Calbiochem), nourseothricin (Werner BioAgents), and Ignite (phosphinothricin; Sigma-Aldrich) were used at concentrations of 200, 25, and 800 µg/ml, respectively.
Table 2. N. crassa strains used in this study.
| Strain | Genotype | Source |
| Wild-type | 74-OR23-1 A | FGSC #2489 |
| his-3− | his-3 A | FGSC #6103 |
| his-3−, Δmus-51 | his-3 Δmus-51::bar A | FGSC #9717 |
| Δmus-51 | Δmus-51::bar a | FGSC #9718 |
| Δmus-52 | Δmus-52::bar a | FGSC #9719 |
| lifeact-rfp | his-3+::Ptef-1-lifeact-tagrfpt Δmus-51::bar a | This study |
| Δcdc-3, Δmus-52 | Δcdc-3::hph Δmus-52::bar | This study |
| Δcdc-3 | Δcdc-3::hph | FGSC #11972 |
| Δcdc-3, Δcdc-10 | Δcdc-3::hph Δcdc-10::nat | This study |
| Δcdc-3, Δcdc-11 | Δcdc-3::hph Δcdc-11::nat | This study |
| Δcdc-3, Δcdc-12 | Δcdc-3::hph Δcdc-12::nat | This study |
| Δcdc-3, cdc-11-sgfp | Δcdc-3::hph cdc-11-sgfp::nat | This study |
| cdc-3-sgfp | cdc-3-sgfp::hph | This study |
| cdc-3-v5-hat | cdc-3-v5-hat::hph | This study |
| Δcdc-10, Δmus-51 | Δcdc-10::hph Δmus-51::bar | FGSC #11727 |
| Δcdc-10 | Δcdc-10::hph | This study |
| Δcdc-10, Δcdc-11 | Δcdc-10::hph Δcdc-11::nat | This study |
| Δcdc-10, Δcdc-12 | Δcdc-10::hph, Δcdc-12::nat | This study |
| Δcdc-10, cdc-11-sgfp | Δcdc-10::hph cdc-11-sgfp::nat | This study |
| sgfp-cdc-10 | sgfp-cdc-10::hph | This study |
| Pccg1-sgfp-cdc-10 | his-3+::Pccg-1-sgfp-cdc-10 | This study |
| sgfp-cdc-10, bml-mch | his-3+::Pccg-1-sgfp-cdc-10/Pccg-1-bml mch::nat | This study |
| v5-hat-cdc-10 | v5-hat-cdc-10::hph | This study |
| Δcdc-11, Δmus-51 | Δcdc-11::hph Δmus-51::bar | This study |
| Δcdc-11 | Δcdc-11::hph | FGSC #11971 |
| Δcdc-11, cdc-12-sgfp | Δcdc-11::hph cdc-12-sgfp::nat | This study |
| Δcdc-11, lifeact-sgfp | Δcdc-11::hph his-3+::Pccg-1-lifeact-sgfp | This study |
| cdc-11-sgfp | cdc-11-sgfp::hph | This study |
| cdc-11-v5-hat | cdc-11-v5-hat::hph | This study |
| Δcdc-12, Δmus-51 | Δcdc-12::hph Δmus-51::bar | This study |
| Δcdc-12 | Δcdc-12::hph | This study |
| Δcdc-12, cdc-11-sgfp | Δcdc-12::hph cdc-11-sgfp::nat | This study |
| cdc-12-sgfp | cdc-12-sgfp::hph | This study |
| cdc-12-sgfp, lifeact-rfp | cdc-12-sgfp::hph his-3+::Ptef-1-lifeact-tagrfpt | This study |
| cdc-12-v5-hat | cdc-12-v5-hat::hph | This study |
| Δasp-1, Δmus-51 | Δasp-1::hph Δmus-51::bar | This study |
| Δasp-1 | Δasp-1::hph | This study |
| Δasp-1, cdc-11-sgfp | Δasp-1::hph cdc-11-sgfp::nat | This study |
| Pccg1-sgfp-asp-1 | his-3+::Pccg-1-sgfp-asp-1 | This study |
| v5-hat-asp-1 | v5-hat-asp-1::hph | This study |
| Δasp-2, Δmus-51 | Δasp-2::hph Δmus-51::bar | This study |
| Δasp-2 | Δasp-2::hph | This study |
| Δasp-2, cdc-11-sgfp | Δasp-2::hph cdc-11-sgfp::nat | This study |
| v5-hat-asp-2 | v5-hat-asp-2::hph | This study |
Transformation and Transformant Selection
Electroporation of N. crassa was performed as described previously [54]. Transformants were selected either by growth on selection media containing either hygromycin or nourseothricin or by recovery of histidine prototrophy.
To create single and double gene deletion strains, knock-out cassettes were released from their respective plasmids by digestion with NotI and then transformed into strain FGSC #9718 (Δmus-51::bar). Genotyping analysis (PCR and Southern blotting) of deletion strains and others was carried out to confirm correct integration of constructs (data not shown). Selected transformants were back-crossed to the wild-type to restore the mus-51 or mus-52 gene and provide homokaryotic null mutants. One of the deletion strains obtained from the FGSC (FGSC #11727) was supplied as a heterokaryon and was back-crossed to the wild-type to acquire a homokaryotic deletion strain. Additionally, strain FGSC #11972 was crossed with FGSC #9719 strain to yield strain Δcdc-3, Δmus-52. FGSC strain #11971 was crossed to FGSC strain #9717 and to NCAB1721 to yield strains Δcdc-11, Δmus-51 and Δcdc-11, lifeact-sgfp, respectively.
To generate strains expressing epitope-tagged proteins from their native loci, plasmids containing knock-in cassettes were digested with NotI and transformed into strain FGSC #9718 (Δmus-51::bar) with the exception of cdc-3 integration cassettes which were digested with KpnI and transformed into strain FGSC #9719 (Δmus-52::bar). cdc-11-sgfp::nat and cdc-12-sgfp::sgfp knock-in cassettes were transformed into Δmus-51 or Δmus-52 septin null mutants to analyse their localisation in the absence of other septins.
C-terminal and N-terminal overexpression constructs for cdc-10, asp-1, and asp-2, were digested with NdeI (or SspI for pAB222 and pAB322) and targeted to the his-3 locus of strain FGSC #6103 (his-3−). To visualize septins simultaneously with microtubules, strain Pccg1-sgfp-cdc-10 was transformed with undigested pAB46BT to yield strain sgfp-cdc-10, bml-mch. An actin/septin co-labelled strain was generated by transforming FGSC #9717 with NdeI-digested pAB271 and then transforming the resulting lifeact-rfp strain with NotI-digested pAB427 (cdc-11-sgfp knock-in cassette) to yield strain cdc-11-sgfp, lifeact-rfp.
FP expression was examined in multiple transformants using a Nikon SMZ1500 stereomicroscope with a GFP (excitation 470/40 nm, 505 nm dichroic mirror, emission 530/40 nm) or RFP filter set (excitation 545/30 nm, 570 nm dichroic mirror, emission 620/60 nm). Intracellular expression patterns were analyzed in at least eight transformants per plasmid construct by widefield fluorescence microscopy. Transformants exhibiting FP expression were selected for subsequent live-cell imaging studies. Expression of V5-HAT-tagged proteins was determined by immunoblotting (see below).
Engineering of Deletion Cassettes and Epitope Tagging Cassettes
All primers and synthetic oligonucleotides used in this study are listed in Table S1. Plasmids constructed in this study are listed in Table 3. All PCRs were carried out using Phusion polymerase (Finnzymes), with the exception of genotyping PCRs which were carried out using Phire polymerase (Finnzymes). Cycling conditions for both polymerases were as follows: initial denaturation at 98°C for 45 s, followed by 30 cycles of 98°C for 10 s, annealing at 60–70°C (depending on tm of primer set) for 10 s, extension at 72°C for 20 s per kb of amplicon, and then a final extension at 72°C for 60 s. Wild-type genomic DNA was used as a template to amplify genomic sequences.
Table 3. Plasmids used in this study.
| Plasmid name | Vector backbone | Target/epitiope | Source |
| pAB415 | pRS426 | cdc-3-5f-hph-cdc-3-3f | This study |
| pAB425 | pRS426 | cdc-3-5f-sgfp-hph-cdc-3-3f | This study |
| pAB455 | pRS426 | cdc-3-5f-v5hat-hph-cdc-3-3f | This study |
| pAB416 | pRS426 | cdc-10-5f-hph-cdc-10-3f | This study |
| pAB616 | pRS426 | cdc-10-5f-nat-cdc-10-3f | This study |
| pAB426 | pRS426 | cdc-10-5f-sgfp-hph-cdc-10-3f | This study |
| pAB226 | pCCG::C-Gly::GFP | his-3-Pccg-1- cdc-10-sgfp | This study |
| pAB326 | pCCG::N-GFP | his-3-Pccg-1-sgfp-cdc-10 | This study |
| pAB526 | pRS426 | cdc-105f-hph-Pcdc-10-sgfp-cdc-10ff | This study |
| pAB556 | pRS426 | cdc-105f-hph-Pcdc-10-v5hat-cdc-10ff | This study |
| pAB417 | pRS426 | cdc-11-5f-hph-cdc-11-3f | This study |
| pAB617 | pRS426 | cdc-11-5f-nat-cdc-11-3f | This study |
| pAB427 | pRS426 | cdc-11-5f-sgfp-hph-cdc-11-3f | This study |
| pAB627 | pRS426 | cdc-11-5f-sgfp-nat-cdc-11-3f | This study |
| pAB647 | pRS426 | cdc-11-5f-mch-nat-cdc-11-3f | This study |
| pAB457 | pRS426 | cdc-11-5f-v5hat-hph-cdc-11-3f | This study |
| pAB418 | pRS426 | cdc-12-5f-hph-cdc-12-3f | This study |
| pAB618 | pRS426 | cdc-12-5f-nat-cdc-12-3f | This study |
| pAB428 | pRS426 | cdc-12-5f-sgfp-hph-cdc-12-3f | This study |
| pAB628 | pRS426 | cdc-12-5f-sgfp-nat-cdc-12-3f | This study |
| pAB458 | pRS426 | cdc-12-5f-v5hat-hph-cdc-12-3f | This study |
| pAB419 | pRS426 | asp-1-5f-hph-asp-1-3f | This study |
| pAB429 | pRS426 | asp-1-5f-sgfp-hph-asp-1-3f | This study |
| pAB229 | pCCG::C-Gly::GFP | his-3-Pccg-1-asp-1-sgfp | This study |
| pAB329 | pCCG::N-GFP | his-3-Pccg-1-sgfp-asp-1 | This study |
| pAB529 | pRS426 | asp-1-5f-hph-Pasp-1-sgfp-asp-1-3f | This study |
| pAB559 | pRS426 | asp-1-5f-hph-Pasp-1-v5hat-asp-1-3f | This study |
| pAB412 | pRS426 | asp-2-5f-hph-asp-2-3f | This study |
| pAB422 | pRS426 | asp-2-5f-sgfp-hph-asp-2-3f | This study |
| pAB222 | pCCG::C-Gly::GFP | his-3-Pccg-1- asp-2-sgfp | This study |
| pAB322 | pCCG::N-GFP | his-3-Pccg-1-sgfp-asp-2 | This study |
| pAB552 | pRS426 | asp-2-5f-hph-Pasp-2-v5hat-asp-2-3f | This study |
| pAB84BT | pRS426 | Pccg-1-bml-mch-nat | This study |
We used yeast recombinational cloning (YRC) to generate deletion cassettes (“knock-out”) and epitope-tagging cassettes (“knock-in”) [55], [56]. Three epitopes, GFP, mCherry and V5-HAT, were used to modify genes at their native loci. GFP and mCherry serve as markers for protein localisation whereas the V5-HAT epitope consists of two distinct epitopes (V5 is a viral epitope and HAT [histidine affinity tag] is a natural 19-aa poly histidine tag derived from chicken lactate dehydrogenase) and was utilised for tandem affinity purification of septin complexes. DNA fragments consisting of targeting flanks, a selection marker and, for knock-in constructs, an epitope were co-transformed into the yeast strain FY834 [57], along with the yeast shuttle vector pRS426 [58] digested with XhoI and EcoRI, for assembly in yeast via its endogenous recombination system [55], [56]. Yeast transformations were carried out with the lithium acetate/PEG method [59]. After 3 days of growth at 30°C, yeast colonies were harvested and pooled, then plasmid DNA recovered and resuspended in 20 µL of 10 mM Tris-Cl pH 8.0, as described previously [60]. Two microliters of the DNA suspension was electroporated into Escherichia coli DH5α. Following restriction digest screening and confirmation of the correct vectors by DNA sequencing, the cassette was cut out with the appropriate restriction enzymes and transformed into Δmus-51 or Δmus-52 N. crassa strains.
The final vectors consisted of targeting flanks specific for the gene of interest and functional elements (henceforth referred to as modules) such as dominant selection markers and epitopes (for integration cassettes). The procedure was simplified by generating standardised modules via PCR which contain defined regions of homology at the 5′ and 3′ end of the DNA fragment. This allows a variety of knock-out and knock-in cassettes to be created with the same targeting flanks by swapping in or out different modules (i.e. substituting the hph selection module for the nat selection module or substituting the GFP module for the V5-HAT module).
To generate targeting flanks, three ∼1 kb regions were amplified from the genes of interest (cdc-3, cdc-10, cdc-11, cdc-12, asp-1, and asp-2). Approximately ∼1 kb upstream of the gene of interest was amplified using primers P1 and P2 to yield flank A. Flank B consists of ∼1 kb of the region upstream of the gene’s stop codon and was amplified using primers P3 and P4. Flank C contains ∼1 kb of the UTR region downstream of the gene’s stop codon and was amplified using primers P5 and P6. Flanks A and C together with a marker module were used to create deletion cassettes whereas flanks B and C, in addition to a marker module and epitope module, were used to create C-terminal epitope tagging cassettes. For three genes (cdc-10, asp-1, and asp-2) C-terminal tagging by knock-in or overexpression was unsuccessful; we therefore created N-terminal knock-in cassettes. To generate N-terminal knock-in cassettes different targeting flanks were required. Flank D contains ∼1 kb of the region between base −2500 and −1500 upstream of the genes start codon and was amplified using primers N1 and N2. Approximately ∼1 kb directly upstream of the gene of interest was amplified using primers N3 and N4 to yield flank E. Flank F contains ∼1 kb of the region downstream of the gene’s start codon and was amplified using primers N5 and N6. N-terminal knock-in cassettes were constructed with these three flanks (D, E, and F), a marker module and an epitope module. All targeting flanks were amplified with a 30 bp extension which corresponds to a region of homology at the 5′- and 3′-end of the various modules or the linearised pRS426.
The two dominant selection markers used in YRC, the hph gene and nat gene, confer hygromycin resistance and nourseothricin resistance in N. crassa, respectively. The hph module was amplified from pGFP::hph::loxP [61] with the primers HPH YRC CC Fw and HPH YRC DD Rv. The nat module was amplified from pD-Nat [62] with the primers NAT YRC CC Fw and NAT YRC DD Rv. The GFP module for C-terminal tagging was amplified from pGFP::hph::loxP with the primers GFP YRC BB Fw and GFP YRC CC Rv. For N-terminal tagging the GFP module was amplified from pGFP::hph::loxP with the primers GFP YRC NT Fw and GFP YRC NT BB Rv. The mCherry module for C-terminal tagging was amplified from pRset-mCherry [63] with the primers MCH YRC BB Fw and MCH YRC CC Rv. The V5-HAT epitope was constructed de novo with N. crassa-codon optimized oligonucleotides V5HAT YRC A, V5HAT YRC B, and V5HAT YRC C which are partially overlapping and contain a 10× glycine linker at the 5′-end, the V5 and HAT sequences joined by a 5× glycine-alanine linker, and a region of defined homology at the 3′-end. After boiling for 5 min, the oligos were incubated at room temperature for 30 min to anneal them, then co-transformed into yeast along with cdc-3 targeting flanks B and C, the hph module and pRS426 linearised with XhoI and EcoRI. The resulting plasmid, pAB455, was checked by DNA sequencing and then used as a PCR template to generate the V5-HAT module for C-terminal tagging with primers V5HAT YRC BB Fw and V5HAT YRC CC Rv. For N-terminal tagging the V5-HAT module was amplified from pAB455 with primers V5HAT YRC NT Fw and V5HAT YRC NT BB Rv.
As the C-terminal GFP knock-in cassettes failed to provide any detectable signal for cdc-10, asp-1, and asp-2 (data not shown), we constructed C-terminal and N-terminal overexpression constructs using the In-Fusion® PCR cloning kit (Clontech). For C-terminal overexpression constructs, the cdc-10, asp-1, and asp-2 genes were amplified, without the stop codon, with the primers CDC10 IF Fw and CDC10 P4 Rv, ASP 1 IF Fw and ASP 1 P4, or ASP 2 IF Fw and ASP 2 P4, respectively. The PCR products were purified using a DNA clean and concentrator kit (Zymo Research) then integrated into XbaI- and PacI-digested pCCG::C-Gly::GFP [61] using the In-Fusion PCR cloning kit to give plasmids pAB226 (Pccg-1-cdc-10-sgfp), pAB229 (Pccg-1-asp-1-sgfp), and pAB222 (Pccg-1-asp-2-sgfp). Similarly, for N-terminal overexpression constructs, the cdc-10, asp-1, and asp-2 genes were amplified with the primers CDC10 IF NT Fw and CDC10 IF NT Rv, ASP 1 IF NT Fw and ASP 1 IF NT Rv, and ASP 2 IF NT Fw and ASP 2 IF NT Rv, respectively. The purified PCR products were then integrated into XbaI- and AscI-digested pCCG::N-GFP [61] using the In-Fusion PCR cloning kit to yield plasmids pAB326 (Pccg-1-sgfp-cdc-10), pAB329 (Pccg-1-sgfp-asp-1), and pAB322 (Pccg-1-sgfp-asp-2).
To visualise septins simultaneously with actin or microtubules we created a lifeact-tagRFP-T and a tubulin-mCherry fusion construct. The lifeact-tagRFP-T fusion was made from the plasmid pAB261 which contains Lifeact-GFP under the control of the tef-1 promoter [30]. The tagRFP-T ORF was amplified from pAL5 with the primers TAGRFPT IF Fw and TAGRFPT IF Rv. GFP was excised from pAB261 by digestion with PacI and EcoRI and the linearised plasmid was gel-extracted. The tagRFP-T PCR product was purified using a DNA clean and concentrator kit (Zymo Research) then integrated into PacI- and EcoRI-digested pAB261 with the In-Fusion® PCR cloning kit to yield plasmid pAB271 (Ptef-1-lifeact-tagrfpt). The tubulin-mCherry fusion was made with YRC. The 2647 bp Pccg-1-bml sequence was amplified from plasmid pMF309 [64] with the primers BTUB YRC Fw, which contains 30 bp of homology at the 3′-end of the linearised pRS426, and BTUB YRC Rv, which contains 30 bp of homology at the 5′-end of the mCherry module. The Pccg-1-bml sequence was co-transformed into yeast along with the mCherry module, the linearised pRS426 shuttle vector and a nat selection module modified to include a 30 bp region of homology to the 5′-end of the linearised pRS426. The resulting plasmid, pAB46BT, was isolated from yeast and screened as described above. DNA sequencing was carried out on all vectors to confirm in-frame cloning of the fusion constructs and correct amplification by PCR.
Protein Extraction, Affinity Purification and Immunoblotting
Antibody-conjugated magnetic beads were used for protein purification. To purify protein complexes we used anti-V5 antibody conjugated Dynabeads and charged His tag isolation (HTI) Dynabeads. We used two epitopes to label the N. crassa septin complexes; a V5 epitope, derived from the V5 simian virus and a poly-histidine sequence from chicken lactate dehydrogenase (HAT), which is known to be a superior alternative to the commonly used 6xHis tag [65]. The double epitope tag was created de novo using three overlapping synthetic oligonucleotides that were combined using YRC. We compared the efficacy of one-step and two-step affinity purification in immunoprecipitating CDC-12 and also differences in the eluted protein profiles using two buffers: 0.1 M glycine-HCl (pH 2.5) and hot (70°C) SDS sample loading buffer. The profiles of proteins isolated using the one-step purification with anti-V5 beads were identical to that obtained with the two-step method (Figure S4). In both cases negligible non-specific interactions were detected in untagged wild-type whole cell extract controls using anti-V5 beads, whereas HTI bead purification of whole cell extracts from wild-type and CDC-12-V5-HAT gave similar profiles. The HAT tag is functional, as demonstrated by the subsequent pulldown of the CDC-12-V5-HAT complex by anti-V5 beads. However, the large degree of non-specific contaminants bound to the HTI beads and the efficacy of the one-step anti-V5 bead IP, allowed us to exclude this step from the purification method.
To determine if the protein complex was completely eluted from the beads and to test for the elution of non-specific interactants from the beads, we monitored the protein profiles of immunoprecipitates eluted in 0.1 M glycine-HCl (pH 2.5) and hot (70°C) SDS sample loading buffer. Elution in glycine-HCl allows reuse of the beads but may leave proteins bound to the beads whereas elution in hot SDS sample loading buffer completely strips beads of protein but precludes reuse of the beads. A comparison of elution profiles is shown in Figure S4. The elution profiles for both buffers are essentially identical demonstrating that all bound proteins are eluted in both cases. However, an additional 25 kDa interactant is present in the SDS eluate in both the wild-type and cdc-12-v5-hat strains, which suggests that this is a non-specific interactant and binds to the bead surface rather than to the bead-bound complex. This contaminant was absent from the glycine-HCl eluate, and therefore all further elutions were carried out using 0.1 M glycine-HCl (pH 2.5).
All protein purification steps were performed at 4°C. Neurospora crassa strains expressing V5-HAT-tagged proteins were grown at 35°C for 2 days in 2 l flasks containing 500 ml of liquid VMM. The cultures were harvested by filtration through Mira-cloth (Calbiochem), washed twice with 250 ml ddH2O, and pressed between paper towels to remove excess water. The collected mycelium (∼10 g) was then frozen in liquid nitrogen and ground into a fine powder in a pre-cooled pestle and mortar. Ice-cold HG extraction buffer (50 mM HEPES [pH7.4], 137 mM NaCl, 10% glycerol freshly supplemented with a protease inhibitor cocktail [complete, EDTA-free tablet, Roche] and 1 mM PMSF) was added to the frozen grindate (5 ml of buffer/g of grindate) which was resuspended by vortexing for three 20 s pulses, inverted once every minute for 5 min and then centrifuged for 2370×g at 4°C for 10 min. The supernatant was carefully removed and used immediately for affinity purification. For affinity purification, two types of Dynabeads were used: His tag isolation (HTI) Dynabeads (Invitrogen) and anti-V5 conjugated Dynabeads (Invitrogen). Anti-V5 monoclonal antibody (Invitrogen) was coupled to M-270 epoxy Dynabeads (Invitrogen) at a concentration of 10 mg/ml according to the manufacturer’s instructions. Both types of Dynabeads were washed three times in HG extraction buffer before adding to supernatants. For tandem affinity purification 100 µl of HTI Dynabeads was added to 10 ml of supernatant and incubated on a roller at 4°C for 10 min. HTI Dynabeads were collected with a Dynal magnet (Invitrogen), washed three times in HTI wash buffer (50 mM HEPES [pH7.4], 300 mM NaCl, 0.01% Tween-20) and the complex eluted from the beads with two washes of 100 µl HTI elution buffer (50 mM HEPES [pH7.4], 150 mM NaCl, 300 mM imidazole, 0.01% Tween-20). The eluate was added to 100 µl anti-V5 Dynabeads and incubated on a roller at 4°C for 1 h. Anti-V5 Dynabeads Beads were collected with a Dynal magnet and washed twice in antibody Dynabead wash buffer (ADWB; 50 mM HEPES pH7.4, 150 mM NaCl, 0.02% Tween-20), once in ADWB minus Tween-20 and the proteins eluted twice in 20 µl 100 mM glycine-HCl (pH 2.5). For single step affinity purifications with anti-V5 Dynabeads, 250 µl anti-V5 Dynabeads were added to 4 ml of cell extract. The remaining purification steps were carried out exactly as described for the immunoprecipitation stage of the tandem purification. Purified protein samples were mixed with one-quarter volume of 4× sample buffer (0.2 M Tris-HCl, pH 6.8, 8% (w/v) SDS, 40% glycerol, 10% β-mercaptoethanol, 0.025% bromophenol blue) for SDS–PAGE. Samples were boiled for 5 min, and then separated by 10% SDS–PAGE. Gels were stained with either SYPRO Ruby (Invitrogen) or Safe blue (Invitrogen). For immunoblots, equal amounts of protein (100 µg) were separated by 10% SDS-PAGE, transferred to an Amersham Hybond nylon membrane in CAPS transfer buffer (10 mM CAPS [pH11], 10% methanol) for 80 min at 80 V and the membrane was then blocked in PBS, 2% skimmed milk and 0.2% Tween-20. V5-HAT-tagged proteins were probed using mouse monoclonal anti-V5 antibody (Invitrogen; diluted to 1∶10,000) and goat anti-mouse IgG- IRDye 800CW conjugate (LiCOR; diluted to 1∶20,000) in PBS, 2% skimmed milk, 0.02% Tween-20. Membranes were washed for 30 min in PBS then imaged using a LiCOR Odyssey scanner.
Mass Spectrometry
Destained SDS-PAGE gel bands were digested with sequencing grade modified porcine trypsin (Promega, ∼5–10 ng/µl) solutions in 50 mM ammonium bicarbonate, and evaporated to dryness by vacuum centrifugation. Micro-HPLC/MS/MS analyses were performed using an online system consisting of a micro-pump Agilent 1200 binary HPLC system (Agilent) coupled to a hybrid LTQ Orbitrap XL instrument (Thermo-Fisher). The LTQ was controlled through Xcalibur 2.0.7 and LTQ Orbitrap XL MS2.4SPI. Capillary Picotip columns (10 cm long, 360 mm outer diameter, 75 mm inner diameter) with a 15 mm tip opening and fitted with a borosilicate frit were obtained from New Objective (Presearch). Fused-silica tubing was from Composite Metal. Samples were analysed on a 2 h gradient for data dependent analysis. MS/MS data were searched using Mascot version 2.2 (Matrix Science Ltd.), against the Neurospora genome and annotations database (http://www.broad.mit.edu/annotation/fungi, accessed January 2012).
Live-cell Imaging
Conidia were collected from 3- to 5-day-old cultures and suspended in VMM. In all experiments, unless stated otherwise, cells were incubated in Lab-Tek 8-well chamber-slides (Nalge-Nunc International) for 3–5 h in 250 µl of VMM at a concentration of 106 cells/ml then imaged. Imaging was carried out at room temperature. Images were collected using a Nikon TE2000 microscope with: a Nikon100×/1.4 NA Plan-Apo objective; CoolLED PE-2 illuminator with LED excitation centred at 380 nm, 470 nm, and 550 nm; Chroma filter sets (for GFP: excitation 470/40 nm, 495 nm LP dichroic mirror, emission 525/50 nm; for RFP: excitation 535/50 nm, 565 nm LP dichroic mirror, emission 590 nm LP; for calcofluor white: excitation 355/50 nm, 400 nm LP dichroic mirror, emission 420 nm LP); Hammamatsu Orca-ER CCD camera; and MetaMorph software (Universal Imaging) for image acquisition. Exposure times ranged from 100–300 ms. To acquire 3D (x,y, and z) images, 10–15 widefield fluorescence images were captured at 0.4 µm steps using 2×2 camera binning. These images were processed through 10 iterative deconvolutions using AutoQuant image analysis software (Media Cybernetics). Projections and further processing steps were carried out with ImageJ software (rsbweb.nih.gov/ij/). To stain cell walls, cells were labelled for 30 min with 0.1 µg/ml calcofluor white (prepared from 1 mg/ml stock in ethanol; Sigma).
Quantification of Conidium Germination, Cell Fusion and Septation
Conidial germination was quantified as the percentage of conidia possessing one or more protrusions/hyphae whilst cell fusion was quantified as the percentage of conidia or conidial germlings involved in fusion. Cell fusion during colony initiation involves the fusion of specialized cell protrusions termed conidial anastomosis tubes (CATs) [40], [46]. The number of unseparated conidia was quantified as the percentage of cells attached to another cell at time 0 h. Septation was quantified as the percentage of cells with a septum and cells were scored as having multiple germ tubes if more than one germ tube had emerged from conidia. Statistical analysis (t-test) was performed using ‘R’ (http://www.r-project.org/).
Supporting Information
Cell wall deposition is unaffected in septin deletion strains. Wild-type and septin deletion strains were incubated in liquid VMM for up to 5 h, stained with calcofluor white then imaged with fluorescence microscopy. Scale bar, 5 µm.
(PDF)
Germination, cell fusion, septation, germ tube emergence and conidia formation in the septin-GFP or septin-V5-HAT strains are unaffected compared to wild-type. (A) Strains were incubated in liquid VMM for 3 h then counted for germination and cell fusion (n = 300). (B) The number of unseparated cells in various strains was counted before incubation, the number of cells with more than two germ tubes was counted following 3 h of incubation in liquid and the number of septate cells was determined with DIC microscopy after 5 h of incubation (n = 300). Differences in the assessed parameters between transformant strains and wild-type were statistically not significant (all p>0.1).
(PDF)
Western blot analysis of septin-V5-HAT-expressing strains. Whole-cell extracts from various strains (in italics) were separated by SDS-PAGE then probed with αV5. The positions of the molecular mass markers (M, in kDa) are shown at the left of the panel.
(PDF)
Comparison of one- and two-step purification and elution buffers. (A) Comparison of one- and two-step purification of whole-cell extracts from wild-type and cdc-12-v5-hat strains with elution in 0.1 M glycine-HCl (pH 2.5). Lanes 1 and 5, whole cell extract. Lanes 2 and 6, first step of two-step purification using HTI beads. Lanes 3 and 7, second step of two-step purification using αV5-beads. Lanes 4 and 8, one-step purification using αV5-beads. (B) Comparison of one- and two-step purification of whole-cell extracts from wild-type and cdc-12-v5-hat strains with elution in hot SDS buffer. Lanes 1 and 5, whole cell extract. Lanes 2 and 6, first step of two-step purification using HTI beads. Lanes 3 and 7, second step of two-step purification using αV5-beads. Lanes 4 and 8, one-step purification using αV5-beads. The positions of the molecular mass markers (in kDa) are shown at the left of the panel.
(PDF)
Oligonucleotide primers used in this study.
(PDF)
Acknowledgments
The authors would like to thank Martin Barrios-Llerena and Thierry Le Bihan from the SynthSys Centre at the University of Edinburgh for mass spectrometry analysis.
Funding Statement
Funding for this project was provided by a Biotechnology and Biological Sciences Research Council grant (# BB/E010741/1) to NDR and a Biotechnology and Biological Sciences Research Council Research Studentship to AB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nishihama R, Onishi M, Pringle JR (2011) New insights into the phylogenetic distribution and evolutionary origins of the septins. Biol Chem 392: 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan F, Malmberg RL, Momany M (2007) Analysis of septins across kingdoms reveals orthology and new motifs. BMC Evol Biol 7: 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hartwell LH (1971) Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res 69: 265–276. [DOI] [PubMed] [Google Scholar]
- 4. Spiliotis ET, Gladfelter AS (2012) Spatial guidance of cell asymmetry: septin GTPases show the way. Traffic 13: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sirajuddin M, Farkasovsky M, Hauer F, Kuhlmann D, Macara IG, et al. (2007) Structural insight into filament formation by mammalian septins. Nature 449: 311–315. [DOI] [PubMed] [Google Scholar]
- 6. Vrabioiu AM, Mitchison TJ (2006) Structural insights into yeast septin organization from polarized fluorescence microscopy. Nature 443: 466–469. [DOI] [PubMed] [Google Scholar]
- 7. DeMay BS, Meseroll RA, Occhipinti P, Gladfelter AS (2009) Regulation of distinct septin rings in a single cell by Elm1p and Gin4p kinases. Mol Biol Cell 20: 2311–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia G 3rd, Bertin A, Li Z, Song Y, McMurray MA, et al (2011) Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol 195: 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frazier JA, Wong ML, Longtine MS, Pringle JR, Mann M, et al. (1998) Polymerization of purified yeast septins: evidence that organized filament arrays may not be required for septin function. J Cell Biol 143: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kinoshita M, Field CM, Coughlin ML, Straight AF, Mitchison TJ (2002) Self- and actin-templated assembly of mammalian septins. Dev Cell 3: 791–802. [DOI] [PubMed] [Google Scholar]
- 11. Rodal AA, Kozubowski L, Goode BL, Drubin DG, Hartwig JH (2005) Actin and septin ultrastructures at the budding yeast cell cortex. Mol Biol Cell 16: 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barral Y, Mermall V, Mooseker MS, Snyder M (2000) Compartmentalization of the cell cortex by septins is required for maintenance of cell polarity in yeast. Mol Cell Biol 5: 841–851. [DOI] [PubMed] [Google Scholar]
- 13. Orlando K, Sun X, Zhang J, Lu T, Yokomizo L, et al. (2011) Exo-endocytic trafficking and the septin-based diffusion barrier are required for the maintenance of Cdc42p polarization during budding yeast asymmetric growth. Mol Biol Cell 22: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dobbelaere J, Barral Y (2004) Spatial coordination of cytokinetic events by compartmentalization of the cell cortex. Science 305: 393–396. [DOI] [PubMed] [Google Scholar]
- 15. Wu JQ, Ye Y, Wang N, Pollard TD, Pringle JR (2010) Cooperation between the septins and the actomyosin ring and role of a cell-integrity pathway during cell division in fission yeast. Genetics 186: 897–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takizawa PA, DeRisi JL, Wilhelm JE, Vale RD (2000) Plasma membrane compartmentalization in yeast by messenger RNA transport and a septin diffusion barrier. Science 290: 341–344. [DOI] [PubMed] [Google Scholar]
- 17. Shcheprova Z, Baldi S, Frei SB, Gonnet G, Barral Y (2008) A mechanism for asymmetric segregation of age during yeast budding. Nature 454: 728–734. [DOI] [PubMed] [Google Scholar]
- 18. Gladfelter AS (2010) Guides to the final frontier of the cytoskeleton: septins in filamentous fungi. Curr Opin Microbiol 13: 720–726. [DOI] [PubMed] [Google Scholar]
- 19. Warenda AJ, Konopka JB (2002) Septin function in Candida albicans morphogenesis. Mol Biol Cell 13: 2732–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Warenda AJ, Kauffman S, Sherrill TP, Becker JA, Konopka JB (2003) Candida albicans septin mutants are defective for invasive growth and virulence. Infect Immun 71: 4045–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alvarez-Tabares I, Perez-Martin J (2010) Septins from the phytopathogenic fungus Ustilago maydis are required for proper morphogenesis but dispensable for virulence. PLoS One 5: e12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyce KJ, Chang H, D'Souza CA, Kronstad JW (2005) An Ustilago maydis septin is required for filamentous growth in culture and for full symptom development on maize. Eukaryot Cell 4: 2044–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozubowski L, Heitman J (2010) Septins enforce morphogenetic events during sexual reproduction and contribute to virulence of Cryptococcus neoformans . Mol Microbiol 75: 658–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saunders DG, Dagdas YF, Talbot NJ (2010) Spatial uncoupling of mitosis and cytokinesis during appressorium-mediated plant infection by the rice blast fungus Magnaporthe oryzae . Plant Cell 22: 2417–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dagdas YF, Yoshino K, Dagdas G, Ryder LS, Bielska E, et al. (2012) Septin-mediated plant cell invasion by the rice blast fungus, Magnaporthe oryzae . Science 336: 1590–1595. [DOI] [PubMed] [Google Scholar]
- 26. Westfall PJ, Momany M (2002) Aspergillus nidulans septin AspB plays pre- and postmitotic roles in septum, branch, and conidiophore development. Mol Biol Cell 13: 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hernandez-Rodriguez Y, Hastings S, Momany M (2012) The septin AspB in Aspergillus nidulans forms bars and filaments and plays roles in growth emergence and conidiation. Eukaryot Cell 11: 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindsey R, Cowden S, Hernandez-Rodriguez Y, Momany M (2010) Septins AspA and AspC are important for normal development and limit the emergence of new growth foci in the multicellular fungus Aspergillus nidulans . Eukaryot Cell 9: 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Juvvadi PR, Fortwendel JR, Rogg LE, Steinbach WJ (2011) Differential localization patterns of septins during growth of the human fungal pathogen Aspergillus fumigatus reveal novel functions. Biochem Biophys Res Comm 405: 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berepiki A, Lichius A, Shoji JY, Tilsner J, Read ND (2010) F-actin dynamics in Neurospora crassa . Eukaryot Cell 9: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, et al. (2008) Lifeact: a versatile marker to visualize F-actin. Nat Methods 5: 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson ES, Blobel G (1999) Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. J Cell Biol 147: 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mortensen EM, McDonald H, Yates J 3rd, Kellogg DR (2002) Cell cycle-dependent assembly of a Gin4-septin complex. Mol Biol Cell 13: 2091–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takahashi Y, Iwase M, Konishi M, Tanaka M, Toh-e A, et al. (1999) Smt3, a SUMO-1 homolog, is conjugated to Cdc3, a component of septin rings at the mother-bud neck in budding yeast. Biochem Biophys Res Comm 259: 582–587. [DOI] [PubMed] [Google Scholar]
- 35. Helfer H, Gladfelter AS (2006) AgSwe1p regulates mitosis in response to morphogenesis and nutrients in multinucleated Ashbya gossypii cells. Mol Biol Cell 17: 4494–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berlin A, Paoletti A, Chang F (2003) Mid2p stabilizes septin rings during cytokinesis in fission yeast. J Cell Biol 160: 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tasto JJ, Morrell JL, Gould KL (2003) An anillin homologue, Mid2p, acts during fission yeast cytokinesis to organize the septin ring and promote cell separation. J Cell Biol 160: 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McMurray MA, Bertin A, Garcia G 3rd, Lam L, Nogales E, et al (2011) Septin filament formation is essential in budding yeast. Dev Cell 20: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weirich CS, Erzberger JP, Barral Y (2008) The septin family of GTPases: architecture and dynamics. Nat Rev Mol Cell Biol 9: 478–489. [DOI] [PubMed] [Google Scholar]
- 40. Roca MG, Arlt J, Jeffree CE, Read ND (2005) Cell biology of conidial anastomosis tubes in Neurospora crassa . Eukaryot Cell 4: 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, et al. (2000) Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae . Mol Cell Biol 20: 4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berepiki A, Lichius A, Read ND (2011) Actin organization and dynamics in filamentous fungi. Nat Rev Microbiol 9: 876–887. [DOI] [PubMed] [Google Scholar]
- 43. Calvert ME, Wright GD, Leong FY, Chiam KH, Chen Y, et al. (2011) Myosin concentration underlies cell size-dependent scalability of actomyosin ring constriction. J Cell Biol 195: 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Springer ML, Yanofsky C (1989) A morphological and genetic analysis of conidiophore development in Neurospora crassa . Genes Dev 3: 559–571. [DOI] [PubMed] [Google Scholar]
- 45. Lindsey R, Momany M (2006) Septin localization across kingdoms: three themes with variations. Curr Opin Microbiol 9: 559–565. [DOI] [PubMed] [Google Scholar]
- 46. Read ND, Lichius A, Shoji JY, Goryachev AB (2009) Self-signalling and self-fusion in filamentous fungi. Curr Opin Microbiol 12: 608–615. [DOI] [PubMed] [Google Scholar]
- 47. Kaufmann A, Philippsen P (2009) Of bars and rings: Hof1-dependent cytokinesis in multiseptated hyphae of Ashbya gossypii . Mol Cell Biol 29: 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin SW, Douglas LM, Konopka JB (2005) Cell cycle dynamics and quorum sensing in Candida albicans chlamydospores are distinct from budding and hyphal growth. Eukaryot Cell 4: 1191–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Byers B, Goetsch L (1976) A highly ordered ring of membrane-associated filaments in budding yeast. J Cell Biol 69: 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim HB, Haarer BK, Pringle JR (1991) Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC3 gene product and the timing of events at the budding site. J Cell Biol 112: 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. An H, Morrell JL, Jennings JL, Link AJ, Gould KL (2004) Requirements of fission yeast septins for complex formation, localization, and function. Mol Biol Cell 15: 5551–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kinoshita M, Kumar S, Mizoguchi A, Ide C, Kinoshita A, et al. (1997) Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes Dev 11: 1535–1547. [DOI] [PubMed] [Google Scholar]
- 53.Davis RH (2000) Neurospora : contributions of a model organism. New York: Oxford University Press. xii, 333 p. p.
- 54. Margolin BS, Freitag M, Selker EU (1997) Improved plasmids for gene targeting at the his-3 locus of Neurospora crassa by electroporation. Fungal Genet Newsl 44: 112. [Google Scholar]
- 55. Collopy PD, Colot HV, Park G, Ringelberg C, Crew CM, et al. (2010) High-throughput construction of gene deletion cassettes for generation of Neurospora crassa knockout strains. Methods Mol Biol 638: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oldenburg KR, Vo KT, Michaelis S, Paddon C (1997) Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res 25: 451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Winston F, Dollard C, Ricupero-Hovasse SL (1995) Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11: 53–55. [DOI] [PubMed] [Google Scholar]
- 58. Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gietz RD, Woods RA (2001) Genetic transformation of yeast. BioTechniques 30: 816–820, 822–816, 828 passim. [DOI] [PubMed]
- 60. Robzyk K, Kassir Y (1992) A simple and highly efficient procedure for rescuing autonomous plasmids from yeast. Nucleic Acids Res 20: 3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Honda S, Selker EU (2009) Tools for fungal proteomics: multifunctional neurospora vectors for gene replacement, protein expression and protein purification. Genetics 182: 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kück U, Hoff B (2010) Application of the nourseothricin acetyltransferase (nat1) as dominant marker for the transformation of filamentous fungi. Fungal Genet Newsl 53: 9–11. [Google Scholar]
- 63. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, et al. (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572. [DOI] [PubMed] [Google Scholar]
- 64. Freitag M, Hickey PC, Raju NB, Selker EU, Read ND (2004) GFP as a tool to analyze the organization, dynamics and function of nuclei and microtubules in Neurospora crassa . Fungal Genet Biol 41: 897–910. [DOI] [PubMed] [Google Scholar]
- 65. Chaga G, Bochkariov DE, Jokhadze GG, Hopp J, Nelson P (1999) Natural poly-histidine affinity tag for purification of recombinant proteins on cobalt(II)-carboxymethylaspartate crosslinked agarose. J Chromatogr A 864: 247–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell wall deposition is unaffected in septin deletion strains. Wild-type and septin deletion strains were incubated in liquid VMM for up to 5 h, stained with calcofluor white then imaged with fluorescence microscopy. Scale bar, 5 µm.
(PDF)
Germination, cell fusion, septation, germ tube emergence and conidia formation in the septin-GFP or septin-V5-HAT strains are unaffected compared to wild-type. (A) Strains were incubated in liquid VMM for 3 h then counted for germination and cell fusion (n = 300). (B) The number of unseparated cells in various strains was counted before incubation, the number of cells with more than two germ tubes was counted following 3 h of incubation in liquid and the number of septate cells was determined with DIC microscopy after 5 h of incubation (n = 300). Differences in the assessed parameters between transformant strains and wild-type were statistically not significant (all p>0.1).
(PDF)
Western blot analysis of septin-V5-HAT-expressing strains. Whole-cell extracts from various strains (in italics) were separated by SDS-PAGE then probed with αV5. The positions of the molecular mass markers (M, in kDa) are shown at the left of the panel.
(PDF)
Comparison of one- and two-step purification and elution buffers. (A) Comparison of one- and two-step purification of whole-cell extracts from wild-type and cdc-12-v5-hat strains with elution in 0.1 M glycine-HCl (pH 2.5). Lanes 1 and 5, whole cell extract. Lanes 2 and 6, first step of two-step purification using HTI beads. Lanes 3 and 7, second step of two-step purification using αV5-beads. Lanes 4 and 8, one-step purification using αV5-beads. (B) Comparison of one- and two-step purification of whole-cell extracts from wild-type and cdc-12-v5-hat strains with elution in hot SDS buffer. Lanes 1 and 5, whole cell extract. Lanes 2 and 6, first step of two-step purification using HTI beads. Lanes 3 and 7, second step of two-step purification using αV5-beads. Lanes 4 and 8, one-step purification using αV5-beads. The positions of the molecular mass markers (in kDa) are shown at the left of the panel.
(PDF)
Oligonucleotide primers used in this study.
(PDF)