

Abstract
Hyperbranched polyesters (HPE) have a high efficiency to encapsulate bioactive agents, including drugs, genes and proteins, due to their globe-like nanostructure. However, the use of these highly branched polymeric systems for tissue engineering applications has not been broadly investigated. Here, we report synthesis and characterization of photocrosslinkable HPE hydrogels with sustained drug release characteristics for cellular therapies. These HPE can encapsulate hydrophobic drug molecules within the HPE cavities, due to the presence of hydrophobic inner structure that is otherwise difficult to achieve in conventional hydrogels. The functionalization of HPE with photocrosslinkable acrylate moieties renders the formation of hydrogels with highly porous interconnected structure, and mechanically tough network. The compressive modulus of HPE hydrogels was tunable by changing the crosslinking density. The feasibility of using these HPE networks for cellular therapies was investigated by evaluating cell adhesion, spreading and proliferation on hydrogel surface. Highly crosslinked and mechanically stiff HPE hydrogels have higher cell adhesion, spreading, proliferation compared to soft and complaint HPE hydrogels. Overall, we showed that hydrogels made from HPE could be used for biomedical applications that require control cell adhesion and control release of hydrophobic clues.
Keywords: Hyperbranched Polyester, Hydrogels, Photocrosslinking, Drug Release, Tissue Engineering
1. Introduction
Engineering tissues holds great promise for treating organ failures associated with disease, injury, and degeneration.1–4 However, numerous challenges remain, such as the inability to mimic the complex cell-microenvironmental interactions to regulate the formation of a functional tissue.5–7 Designing advanced biomaterials with controlled release properties and cell adhesion characteristics can be beneficial to precisely control cellular behavior and facilitate the formation of specific functional tissues.8–12 Synthetic dendritic macromolecules have received a great deal of attention in drug delivery applications due to their unique nanosized molecular structure and physicochemical properties.13–15 Dendritic macromolecules (such as dendrimers and hyperbranched polymers (HPE)) have a highly branched, globe-like molecular shape with a multitude of functional groups at their periphery.15–18 The globular shaped macromolecules have no chain entanglement, so it is easy for the outer functional groups to interact with other chemical moieties, resulting in a higher reactivity and loading efficiency over their conventional linear polymer counterparts.14, 17, 19 Additionally, due to their unique nanostructure, dendritic polymers can encapsulate drug molecules into their interior cavities or form polymer-drug complexes/conjugates through a host-guest chemistry.20, 21 Although dendritic polymeric macromolecules have been proposed as a carrier for various bioactive agents, including drugs17, genes22 and proteins23, relatively few studies have investigated these highly branched polymeric macromolecules for tissue engineering applications.24, 25
Recent studies have proposed using these dendritic macromolecules by combining them with linear polymers or protein hybrids for various biomedical applications.24–29 For example, Zhong et al. had incorporated polyamidoamine (PAMAM) dendritic macromolecules within collagen scaffolds to improve mechanical properties, bio-stability, and the structural integrity of the crosslinked network.30 They observed that the addition of PAMAM dendritic macromolecules to collagen networks resulted in a significantly higher proliferation of human conjunctival fibroblasts compared to the collagen scaffold. In another approach, Desai et al. developed PEGylated PAMAM dendritic macromolecules (G3) with different lengths of PEG chains.24 The free ends of the PEG chains were further modified with acrylate groups to obtain photocrosslinked hydrogels for tissue engineering and drug delivery applications. They reported that long chain PEG is required to obtain crosslinked hydrogel network, whereas shorter PEG chains did not results in hydrogel formation.24 They attributed this shortcoming to the formation of cluster/aggregation of PEG and dendrimers that spatially restricts acrylate groups from photoinitiator molecules.24
In a similar study, Carnahan et al. proposed photopolymerized hybrid dendritic-linear polyester-ethers hydrogels as sealants for corneal repair.19, 27 They observed that the hybrid linear-dendritic copolymers were able to withstand high pressure and stress and thus, were able to seal the wound better compared to the conventional sutures.19, 27 In another study, Söntjens et al. proposed to use photocrosslinkable derivatives of the poly(glycerol-succinic acid)-polyethylene glycol dendritic–linear copolymers for cartilage tissue engineering.25 The mechanical properties, degradation characteristics, and hydration degree were tuned by changing the dendrimer concentration. They were able to encapsulate chondrocytes within the hydrogel network and showed that the dendrimers enhanced the production of extracellular matrix (ECM) containing collagen (type II) and proteoglycans.25 In a different approach, Wang et al. proposed to use a dendritic macromolecule (aliphatic ester dendrimers with PEG as a spacer) with inorganic nanoplatelets to obtain hydrogels non-covalently.31 This study resulted in unique self-healing properties and mechanical stability of the hybrid hydrogels.
Although hydrogels based on dendritic macromolecules have been reported recently, most of them are hybrid copolymers with linear polymers as crosslinking agents. Moreover, most of these hybrid structures are synthesized from low generation (G1 and G2) dendrimers/hyperbranched polymeric macromolecules. The drawbacks of these types of hybrid structures include low mechanical strength, poor drug loading efficiency, and poor physiological stability. More recently, Zhang et al. proposed a novel hydrogel system composed of hyperbranched poly(amine-ester) macromolecules for the delivery of anticancer drugs.28, 29 They showed that hydrogels made from high generation dendritic macromolecules have a prolonged and sustained drug release ability due to the formation of dense crosslinked networks. Being different from conventional hydrogels made from hydrophilic linear polymer, dendrimer hydrogels consist of dendritic and globular macromolecules, which have hydrophobic inner structure and hydrophilic exterior groups. The structure of dendrimers enables the entrapment of hydrophobic therapeutic agents and their subsequent controlled release, which solves some of the existing problems of conventional hydrogels.
Here, we propose photocrosslinkable and degradable hyperbranched polyester (HPE) hydrogels made by crosslinking hyperbranched macromers with sustained drug release ability for engineering cell-matrix interactions. We synthesized photocrosslinked generation 4 (G4) HPE macromolecules by functionalizing them with acrylate groups. The presence of ester groups in the backbone can be used to tune the biological properties and degradability of the scaffolds. Additionally, the hydroxyl groups on periphery can be used to further functionalize or immobilize macromolecules. The chemical and physical properties of HPE hydrogels obtained by crosslinking HPE macromers were evaluated by investigating swelling kinetics, in vitro degradation, drug release kinetics, mechanical properties, and inner microstructure conformation. The biological properties of these hydrogels were evaluated by investigating cell adhesion, spreading and proliferation on the hydrogel surface. The feasibility of using HPE hydrogels for tissue engineering applications was also investigated.
2.0 Experimental Section
Materials
1,1,1-trimethylolpropane (TMP), 2,2-bis(hydroxymethyl)propionic acid (bis-MPA), acryloyl chloride, p-toluene sulphonic acid (p-TSA), tetrahydrofuran (THF) and sodium dodecyl sulfate (SDS) were purchased from Sigma-Aldrich (Wisconsin, USA). All reagents and solvents were used without further purification.
Synthesis of hydroxyl terminated HPE (Generation 4)
HPE (Generation 4) was synthesized according to the pseudo-one-step procedure.16 Briefly, 1.34 g TMP (0.01 mol, central core), 12.07 g bis-MPA (0.09 mol, repeating unit) (in stoichiometric correspondence to the second generation) and 60.4 mg p-TSA (catalyzer) were mixed in a 250 mL reactor flask. The mixture was allowed to react for 2 hour at 140 °C. The water produced in the reaction was removed under reduced pressure for 1 hour. Then 16.10 g bis-MPA (0.12 mol) (correspondence to the third generation) and 80.5 mg p-TSA were added into the flask and the water was removed under reduced pressure for 1 hour. The same procedure was repeated to obtain fourth generation HPE.
Synthesis of acrylated HPE (HPE-A)
Hydroxyl terminated HPE (4.288 g, theoretically [OH]=38.4 mmol) was fully dissolved in 50 mL THF, and then 1.63 g anhydrous Na2CO3 (1.54 mmol) was added. The flask was cooled in an ice/water bath. Thereafter, acryloyl chloride (30%, 40% or 50% molar ratio to the hydroxyl groups) was added drop wise. The acrylation reaction was carried out at 37 °C for 5 hour under argon blanket. The THF was then evaporated using a rotavapor and the prepared acrylated HPE (HPE-A) was stored at 4 °C until further use.
Characterization of HPE and acrylated HPE
HPE and HPE-A were dissolved in DMSO-D6 and nuclear magnetic resonance (1H NMR) spectra were recorded on a DMX-400 spectrometer (Bruker, Rheinstten, Germany) operating at 400 MHz [1H NMR (DMSO-D6): δ (ppm) 6.36, 6.21 and 6.10 (−CH=CH2), 1.15 (−CH3)]. Fourier transform infrared spectrometer (Alpha Bruker) was used to record the evidence of characteristic functional groups in HPE and acrylated HPE [FTIR wavenumbers (cm−1): 3400 (−OH), 1724 (>C=O), 1635 (−C=C) and 1040 (−C−O−)]. The hydrodynamic size, zeta potential and electrophoretic mobility of HPE and HPE-A were recorded using Zetasizer Zen3600 (Malvern, UK). The molecular weight of HPE and HPE-A was determined by using gel permeation chromatography (GPC, Waters, Milford, MA). The samples were dissolved in THF (0.5% w/v) and injected at the flow rate of 1 ml/min.
Preparation of HPE hydrogel
The prepolymer solution was prepared by dissolving HPE-A macromer into Dulbecco's phosphate buffered saline (DPBS; GIBCO) containing 0.25% (w/v) 2-hydroxy-1-(4-(hydroxyethoxy)phenyl)-2-methyl-1-propanone (Irgacure 2959, CIBA Chemicals) as a photoinitiator. To fabricate the hydrogel, 60 μL of prepolymer solution was pipetted between a polytetrafluoroethene sheet and a 3-(trimethoxysilyl)propyl methacrylate (TMSPMA) coated glass slide separated by a 1 mm spacer and exposed to UV light (360–480 nm, 7.0 mW/cm2, Omnicure S2000, Canada) for 60 sec. The samples with three different HPE-A concentrations namely 40% (w/v) labeled as HPE40, 50% (w/v) labeled as HPE50 and 60% (w/v) labeled as HPE60, were prepared using medium acrylated HPE and followed for further analysis.
Swelling behavior of HPE hydrogels in DI and DPBS
The swelling behavior of the HPE hydrogels was measured over two days at 37 °C. HPE hydrogel samples were weighed (W0) immediately following hydrogel formation and immersed in 2 mL of either DPBS or deionized water. At predetermined time intervals, the swollen hydrogels were taken out from the immersion medium and weighed (Wt) after wiping excess water carefully. The swelling ratio was calculated by Wt/W0. All experiments were performed in triplicates.
Unconfined compression testing
The samples for mechanical testing were prepared as mentioned previously and allowed to equilibrate in DPBS for 24 hour before testing. After excess water was blotted out, the samples were tested at the rate of 1 mm/min on an Instron 5542 mechanical tester fitted with 50N load cell. The compressive modulus was determined using initial 5–15% linear region of related stress strain curve.
In vitro degradation of HPE hydrogels
Degradation of HPE hydrogels was investigated in (a) DPBS and (b) 0.01M NaOH solution at 37 °C. The samples were freeze dried to get the initial dry weight. At regular intervals, the samples were removed from degradation buffer, washed in deionized water, freeze dried and weighed to determine the mass loss. All experiments were performed in triplicate.
Morphology of HPE hydrogels
Scanning electron microscopy (SEM) was employed to analyze the interior morphology of HPE hydrogels. Fully swollen hydrogel samples were quickly frozen in liquid nitrogen and lyophilized. The freeze dried hydrogel samples were fractured to expose the cross-sections. The fractured samples were mounted on the aluminum stubs and sputtered with gold at 15 mA for 2 min using Hummer 6.2 sputtering system. The morphology of pores was captured using a scanning electron microscope (JSM 5600LV, JEOL USA Inc., Peabody, MA) at an accelerated voltage within 5–10 mm working distance.
Drug release analysis of PEG and HPE hydrogels
Osteogenic inducer dexamethasone acetate (DA) was selected as a model agent for drug release study. Drug loaded PEG hydrogels were prepared from PEGDA1000 (Mw≈1000) solution with physically dispersed DA. For drug loaded HPE hydrogels, DA and HPE were first dissolved in methanol and stirred for 24 h to insure encapsulation of DA into HPE macromers. The DA encapsulated HPE macromers were then obtained after evaporating the methanol. The content of DA used in this study was 14 mg/g of HPE-A. Hydrogels (PEG hydrogels and HPE hydrogels) with the same concentration of macromers contained the same amount of DA. Drug release from both hydrogels was carried out in 2 mL PBS solution at 37 °C. At different time intervals, 1 mL of medium was pipetted out for analysis and 1 mL of fresh PBS was replenished. The concentrations of released drugs were quantified using a UV-visible spectrophotometer (V-570, JASCO, Japan) and the absorption wavelength of DA analysis was 286 nm.
Cells adhesion and proliferation
To evaluate whether the HPE hydrogels allow for cell adhesion, NIH-3T3 cells were seeded on HPE hydrogels. Briefly, the cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco, USA), supplemented with 10% of heat-inactivated fetal bovine serum (HiFBS, Gibco-USA) and 1% antibiotic (penicillin/streptomycin, Gibco, USA), at 37 °C, in a humidified atmosphere with 5% of CO2. When reached 80% confluency, cells were trypsinized (0.05% Trypsin/EDTA, Gibco, USA), resuspended in DMEM and seeded on HPE hydrogels at a density of 2.5×105cells/10μL/ sample. Cells were allowed to adhere for 1 hour and then media was replenished. The viability of the cells at day 4 was evaluated using a Live/Dead assay kit (Invitrogen, USA). Samples with the cells were incubated for 20 min with calcein AM/ethidium homodimer and washed three times with PBS. All samples were examined under an inverted fluorescence microscope (Nikon, Eclipse TE 2000U, Japan). The proliferation rate of NIH-3T3 cells seeded on HPE hydrogels was assessed using an Alamar Blue assay (Invitrogen, USA) after 1, 4 and 10 days of culture. Briefly, media was replaced with 10% (v/v) of Alamar Blue reagent and incubated at 37 °C, 5% CO2 humidified atmosphere for 4 hours. At the end of the incubation period the supernatant of the cultures was aliquoted into 96-well plate in triplicates and a colorimetric reading was performed using a microplate reader (Epoch, Biotek, USA) at 570 nm and 600 nm. Culture medium with 10% (v/v) Alamar Blue was used as negative control, whereas cells that were cultured on tissue culture plastic surface (TCPS) were used as positive control. After supernatant removal, cells were washed in PBS and fresh medium was added.
Statistical evaluation
Experimental data were presented as mean ± standard deviation (n=3). Statistical differences between the groups were analyzed using one-way ANOVA using post-hoc. P-value less than 0.05 was considered statistically significant.
3. Results and discussion
3.1 Synthesis and Characterization of Hyperbranched Polyester (HPE) and Acrylated HPE
The synthesis and molecular structures of HPE and acrylated HPE (HPE-A) macromolecules are schematically presented in Figure 1. HPE macromolecules were synthesized according to conventional acid-catalyzed esterification reactions which was reported before16, and HPE-A was prepared by the acrylation of HPE using acryloyl chloride. To ensure the water solubility and crosslinkability of HPE macromers, only 30% (low), 40% (medium), and 50% (high) of theoretical hydroxyl groups were targeted for acrylation. According to the 1H-NMR results (Figure 2a), the acrylation degrees of HPE obtained were 23.8% (low), 31.7% (medium), and 36.3% (high) respectively. A relatively low experimental acrylation degree of HPE compared to theoretical acrylation might be attributed to the inefficiency of acrylation as a result of steric hindrance.
Figure 1. Synthesis of HPE and HPE-A macromolecules.

HPE (Generation 4) was synthesized y pseudo-one-step procedure using 1,1,1-trimethylolpropane (TMP) as a core and 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) as a repeating unit.
Figure 2. Chemical characterization of HPE and HPE-A macromolecules.

(a) 1H-NMR spectra of HPE and HPE-A with different degrees of acrylation are shown. Strong peaks related to acrylate groups at 6.36, 6.21 and 6.10 ppm were observed in HPE-A. (b) Strong peaks at 3400 cm−1, 1730 cm−1 and 1050 cm−1 are related to OH, C=O and C-O groups respectively; whereas a weak peak of methylene group is observed at 1635 cm−1 after acrylation of HPE macromolecules. (c) Size exclusion spectra of HPE and HPE-A dissolved in THF were obtained using GPC. The GPC curve demonstrated an increase in the hydrodynamic volume of HPE in THF after acylation.
A prepolymer solution containing acrylated HPE was subjected to UV light to obtained covalently crosslinked hydrogel network. At a low acrylation degree of HPE, a weak and unstable hydrogel network was obtained after subjecting it to light. HPE-A macromers with a medium acrylation degree (31.7%) can be well dispersed in deionized water and photocrosslinked to form stable hydrogels. At a high acrylation degree of HPE, insoluble aggregates were formed in the precursor solution. The formation of aggregates is mainly attributed to the strong hydrophobic nature from the presence of a large number of acrylate groups on the surface of HPE-A macromers. Based on NMR results, we selected HPE macromers with a medium acrylation degree for synthesis and characterization of HPE hydrogels.
The stability of HPE macromolecules in physiological conditions before and after acrylation was investigated by monitoring zeta potential and hydrodynamic diameter. The zeta potential and mobility of HPE macromolecules were −26.1 ± 0.56 mV and −2.05 ± 0.04 μmcm/Vs respectively. After acrylation (HPE-A), zeta potential increased to −9.8 ± 0.28 mV, while the mobility increased to −0.77 ± 0.02 μmcm/Vs. The increase in zeta potential and mobility after acrylation was due to a decrease in the surface hydroxyl groups. The mean hydrodynamic diameter of HPE in physiological was 141 nm (PDI ~ 0.4), while after acrylation, it was reduced to 104 nm (PDI ~ 0.21) (Figure S1). The decrease in hydrodynamic diameter can be attributed to the decrease in surface hydrophilicity of HPE macromolecules after the partial acrylation of hydroxyl groups. The hydrodynamic diameter of HPE as determined by DLS was larger than that of a single molecule, which suggested aggregation of HPE molecule due to hydrogen bonding. The aggregation was weakened after acrylation of HPE as the number of −OH groups was reduced in HPE-A. To confirm the presence of aggregates, we dissolved HPE in dimethylformamide (DMF) and determined hydrodynamic diameter. (Figure S1c) The results indicated bimodal distribution of HPE in DMF with peak at 13.3 nm and 178 nm. This indicates that individual HPE molecules were ~10–15 times smaller compared to the aggregates. The size of individual HPE molecules and HPE aggregates were in correlation with the previously published literatures.32, 33
To further verify the acrylation of HPE macromolecules, chemical structures of HPE and HPE-A were investigated using Fourier Transform Infra-Red (FTIR) spectroscopy. As shown in Figure 2b, strong peaks at 1730 cm−1 and 1050 cm−1 were attributed to C=O and C-O groups of ester linkage, respectively. The non-modified HPE macromolecules did not exhibit any peaks between 1500–1700 cm−1, whereas a weak peak was observed at 1634 cm−1 (C=C) for HPE-A macromolecules, corresponding to the acrylates groups (C=C). Both 1H-NMR and FTIR spectra further suggests that the HPE-A macromolecules contains crosslinkable C=C bonds along with hydrophilic hydroxyl groups, that makes it suitable for in situ hydrogel formation.
The molecular weight distribution and elution time of HPE before and after the acrylation process was monitored by gel permeation chromatography (GPC) using tetrahydrofuran (THF) as a solvent. In GPC, smaller analytes spend more time in the porous column and thus have higher retention times, whereas larger analytes spend little time in the column and elutes quickly. The results indicated that the retention time of HPE-A was decreased when compared to the HPE, suggesting an increase in molecular weight after acrylation (Figure 2c). This was attributed to the presence of acrylate groups on HPE-A that readily swells in THF. The weight average molecular weight (Mw) and the number average molecular weight (Mn) of HPE and HPE-A were determined by GPC, calibrated with linear polystyrene standards. Due to the different structures of dendritic polymers and linear ones, hydrodynamic radius of HPE or HPE-A is smaller than that of its linear counterpart of the same molar mass. Thus, the molar masses of HPE and HPE-A determined by GPC are lower compared to the theoretical value. Mw and Mn of HPE were 4231 Da and 2938 Da respectively, while for HPE-A were 5345 Da and 3404 Da respectively. The polydispersity index (PDI) of HPE and HPE-A calculated was 1.44 and 1.57, respectively. The results reported here are comparable to the previously published literature.16 By comparing the Mw of HPE (4231 Da) and HPE-A (5345 Da), the number of acrylate groups were calculated to be ~15.5 per HPE molecules.
To determine the extend of final conversion of acrylate groups upon UV exposure, sol content of crosslinked hydrogels was investigated. The as prepared hydrogels were freeze dried to determined the dry weight. The freeze dried HPE samples were allowed to hydrate in deionized water. In fully hydrated condition, unreacted HPE was allowed to leach out for 24 hours. After 24 hours, the fully swollen hydrogel network was freeze dried to obtain the gel content. The results indicated that almost 20–30% of HPE (compared to initial mass) was not crosslinked within the network. This might be attributed due to: (a) aggregation of HPE macromers in the solution due to hydrogen bonding that limits diffusion of photoinitiator within HPE aggregates, (b) due to hydrophobic nature of acrylate groups that tend to fold back to hydrophobic inner and might not be available for crosslinking, or (c) intra crosslinking of acrylate group present on same HPE macromer. As expected, the degree of conversion of acrylate group upon UV exposure was independent of initial concentration of HPE and no significant difference between HPE40, HPE50 and HPE60 was observed in terms of total sol content.
3.2 Microstructural evaluation of HPE Hydrogels
In tissue engineering, an ideal hydrogel matrix should have a porous structure and controlled physical and chemical properties. An evaluation of the scaffold microstructure is essential in designing mechanically robust hydrogels with tunable physicochemical properties. For example, pore size directly influences the mechanical performance of the hydrogel network and the interconnectivity of those pores determines the physicochemical properties of the network.34–36 We synthesized hydrogels containing 40, 50, and 60 % of HPE-A macromers with a medium degree of acrylation and hereafter referred to HPE40, HPE50 and HPE60 respectively. The fully swollen hydrogels were frozen in liquid nitrogen and lyophilized. The freeze dried hydrogels were used to determine the effect of crosslinking density on the microstructure. The microstructure analysis shows that all the freeze dried HPE networks have highly porous and interconnected structures (Figure 3). We calculated the pore size of the freeze-dried networks by determining the major axis of individual pores using ImageJ. An increase in HPE concentration resulted in a decrease in the pore size. The average pore diameter of freeze dried HPE40 networks was determined to be 50±16 μm. With an increase in HPE concentrations, pore size decreased to 17±5 μm and 16±6 μm for HPE50 and HPE60, respectively. The decrease in the pore size can be attributed to the increase in crosslinking density due to an increase in HPE concentration and as a consequence, an increase in the covalent bonds that are formed.
Figure 3. Structural and physical characterization of HPE hydrogels made by crosslinking HPE macromers.

Effect of HPE macromolecules concentration on microstructure of freeze dried hydrogel network was evaluated using scanning electron microscopy. At low HPE concentration (40% HPE), highly porous and interconnected structure was observed. With an increase in HPE concentration, pore size decreased due to increase in crosslinked density. The bars represent mean ± standard deviation (n=3), (*p < 0.05).
3.3 Mechanical properties of HPE hydrogels
A hydrogel scaffold for tissue engineering applications should be able to structurally support tissue in-growth while withstanding in vivo mechanical stress.37 Mechanical properties of HPE hydrogels were determined by unconfined compression tests. Figure 4a demonstrates stress-strain curves of hydrogels with different HPE concentrations. All the hydrogels showed biphasic behavior with a toe and a linear region. The stiffness of the hydrogel networks was determined by evaluating compressive modulus from the toe region of the stress-strain curve (5–15% strain). The increase in HPE macromer concentration results in an increase in the stiffness of the hydrogel network. The compressive modulus increased from 3.4±1.8 kPa to 30.4±7 kPa when HPE concentration increased from 40% to 60% (Figure 4b).
Figure 4. Mechanical properties of HPE hydrogels was evaluated using unconfined compression test.

(a) Representative stress strain curves for different concentration of HPE hydrogels are shown. (b) Increase in HPE concentration resulted in an increase in compressive modulus indicating formation of highly crosslinked network. The error bars represent mean ± standard deviation (n=3), (*p < 0.05).
The increase in the mechanical properties can be attributed to the formation of dense network that can sustain high mechanical deformation. The microscopic images indicate a decrease in pore size from HPE40 to HPE50 (Figure 3). Whereas, no significant difference in pore size was observed between HPE50 and HPE60, however mechanical properties were different. The increase in the mechanical strength of HPE60 compared to HPE50 might be attributed to the thicker pore wall and heterogeneous pore size distribution. Thus, the low crosslinking density and high swelling ratio of HPE40 hydrogels resulted in significant deformation at low compression stress, while at high HPE concentration (HPE60), relatively strong and compact networks were formed with the ability to withstand relatively high compressive loads.
3.4 Swelling characteristics of HPE hydrogels
Mechanical properties and surface characteristic of hydrogels are strongly influenced by water content and the degree of crosslinking.36, 38 Moreover, swelling characteristics of polymer networks are critical in tissue engineering applications, as it correlates with the diffusivity of oxygen, drugs, nutrients and other water soluble metabolites. The swelling behavior of the HPE hydrogels made by crosslinking HPE macromers was investigated over a period of 48 hours in DPBS and deionized water at 37 °C (Figure 5a and S1). The dynamic swelling profiles of HPE hydrogels exhibited a fast swelling behavior in the first hour and achieved the equilibrium state within 22 hours in both deionized water and DPBS. The initial faster swelling of HPE hydrogels was due to the osmotic pressure difference. We also observed a slight de-swelling of hydrogel networks in deionized water after the initial swelling (Figure S2). This may be attributed to the Donnan effect that was balanced by the diffusion of unreacted HPE macromers.39 This effect was more predominant in HPE40 hydrogels. On the other hand, the Donnan effect was suppressed in DPBS, due to the presence of free ions that counter-balanced the osmotic pressure.
Figure 5. Stability of HPE hydrogels in physiological conditions.

(a) Stability of HPE hydrogels was investigated by evaluating swelling behavior and in vitro degradation of 40%, 50% and 60% HPE hydrogels in physiological environment. All the hydrogels reached equilibrium hydration degree within 24 hours. A significant decrease in the swelling ratio of HPE hydrogels due to increase in HPE concentration was observed. (b) In degradation study, an initial mass loss (Phase I) observed was due to leaching out of sol content. A slow and linear degradation after initial mass loss was observed (Phase II). The bars represent mean ± standard deviation (n=3), (*p < 0.05).
The effect of concentration was also observed on the equilibrium swelling ratios of HPE hydrogels. For example, the swelling ratio of HPE hydrogels was decreased with an increase in monomer concentration in both deionized water and DPBS (Figure 5a and S1). This is mainly attributed to the increase in crosslinking density of hydrogel networks as demonstrated by SEM images. Additionally, a significantly lower equilibrium swelling ratio of HPE hydrogels was observed in DPBS compared to deionized water (Figure 5a and S1). This is because of the “salting-out” effect, which is a characteristic of the aqueous solutions of many polymers.40 The addition of salts in polymer aqueous solutions results in a partial dehydration of polymer chains and decreases the hydrophilicity of the polymer chains.41 Thus, the presence of salt reduces the hydrophilicity and equilibrium swelling ratio of HPE hydrogels.
3.5 Degradation characteristic of HPE hydrogels
For most tissue engineering applications, biomaterials with a tunable degradation behavior are desirable. The in vitro degradation profiles of HPE hydrogels in DPBS and a sodium hydroxide (NaOH, 0.01M) solution at 37 °C were investigated (Figure 5b and S2). A biphasic degradation profiles was observed for the HPE hydrogels. The hydrogels showed considerable mass loss within the first 24 hours (Phase I) followed by a relatively slow degradation rate (Phase II). The initial mass loss (Phase I) was due to the leaching out of low molecular weight sol contents (unreacted and intramolecular crosslinked HPE precursors), while the subsequent controlled degradation (Phase II) mainly resulted from the hydrolysis of esters backbone. The degradation due to hydrolysis of HPE hydrogels made by crosslinking HPE macromers followed a linear profile. This might be attributed to the unique globular molecular structure and hydrophobic interior of HPE macromolecules that resulted in a gradual degradation of the crosslinked network.
The HPE hydrogels had a similar mass loss profile in both DPBS and 0.01M NaOH solutions. The hydrogels with low HPE concentration had a faster degradation rate compared to that of high HPE concentration. For example, remaining mass for HPE40 was 48.3±4.7 %, whereas for HPE60 remaining mass was 60.7±1.9 % after 14 days in DPBS (Figure 5b). The significant difference between HPE40 and HPE60 can be attributed to the difference in crosslinking density.
An accelerated degradation of HPE hydrogels was observed in 0.01M NaOH solutions (Figure S3), due to the enhanced hydrolysis of ester groups in the alkaline medium. HPE40 hydrogels fully degraded within seven days. On day 7, the remaining mass ratio was 3.4% in 0.01M NaOH solution, while in DPBS it was of 52.8%. The degradation of HPE hydrogels, especially the complete degradation of HPE40 hydrogel in 0.01M NaOH solution, revealed that HPE hydrogels are biodegradable. Based on the degradation studies in PBS and 0.01M NaOH solution, we believe that HPE hydrogels can be completely degraded under physiological condition (PBS, 37 °C).The exact degradation mechanisms of HPE hydrogel structure are not known. However, it might be possible that the degradation of HPE hydrogels may be due to the hydrolysis of ester groups that are present in the HPE macromer (intra-molecular) and between HPE macromolecules (inter-molecular).
When compared to the bulk degradation behavior exhibited by most of the polymers, the controlled degradation of HPE hydrogels can be beneficial in tissue engineering.42 The degradation of hydrogel network will enable tissue in-growth and the formation of ECM that are of utmost importance in functional tissue regeneration. Moreover, by entrapping biochemical cues within hydrogel network to achieve a controlled release profile will facilitate the regulation of cell behaviors like migration, proliferation, and/or controlled differentiation, accompanied by ECM biosynthesis.
3.6 Sustained Release of Dexamethasone from HPE Hydrogels
The stable and sustained release of therapeutic agents within scaffolds is an important factor for engineering next generation tissue scaffolds. This is especially preferable for tissue regeneration and stem cell differentiation that requires the use of growth factors and other bioactive cues to direct cell-matrix interactions. However, many of the useful growth factors and bioactive agents are hydrophobic, which limits the practical application of hydrogels as a delivery vehicle. As a conventional hydrogel consists of crosslinked hydrophilic linear polymers, it is difficult to incorporate hydrophobic agents within such a hydrophilic platform and to obtain sustained release of the entrapped agents. To overcome these limitations, hydrogels with complex structures were designed, including hydrogel/micelle composites,43 hydrogel/microparticle composites,44, 45 and alginate bead-embedded silk fibroin scaffolds46. However, most of these composite materials involve complicated synthesis and processing steps that result into the low loading of therapeutic agents within a composite structure.
Here, we investigated drug release behaviors from the hydrogels with building blocks of hyperbranched macromolecules expecting to achieve a sustained release of a hydrophobic agent. For this purpose, the hydrophobic osteogenic inducer dexamethasone acetate (DA) was uniformly distributed and encapsulated into PEG hydrogels and HPE hydrogels to compare the sustained release over the course of eight days (Figure 6a). The hydrophilic nature of linear PEG macromers limits the uniform dispersion of DA within the prepolymer solution and results in phase separation (Figure 6b). Whereas, DA can be easily entrapped within HPE-A macromer due to the presence of hydrophobic cavities. The release of DA from PEG hydrogels and HPE hydrogels showed very different release profiles (Figure 6b). For PEG hydrogels, the limited release of DA was observed after a relatively fast release in the first two days. The cumulative drug releases from PEG40, PEG50, and PEG60 hydrogels were 31.9 %, 23.7 %, and 18.5 % respectively. The slow and inefficient drug release from PEG hydrogels can be attributed to the low solubility of DA in PEG solutions that resulted in the formation of microscale crystals and phase separation. Whereas, HPE hydrogels showed a sustained release of entrapped DA over a period of eight days (Figure 6c). The cumulative release ratios of DA from HPE40, HPE50 and HPE60 on the day 8 were 76.7 %, 53.4 %, and 44.6 %, respectively. Therefore, HPE hydrogel systems hold the potential of being a platform in tissue engineering for sustained release of hydrophobic drugs.
Figure 6. Sustain release of hydrophobic drug from HPE hydrogels made by crosslinking HPE macromers.

(a) Schematic showing encapsulation of dexamethasone acetate (DA) within hydrophobic cavities of HPE-A macromolecules. After photocrosslinking, DA loaded HPE-A macromers forms covalently crosslinked network. (b) Image showing PEG-DA and HPE-DA solution. Due to hydrophobic nature of DA, HPE macromolecule is able to uniformly entrap DA and result in a stable solution. Whereas, PEG-DA solution shows phase separation due to precipitation of DA aggregates. The release profile of DA was investigated from PEG and HPE hydrogels. PEG hydrogels reached a plateau phase after initial release of entrapped DA. (c) Whereas, HPE hydrogels made by crosslinking HPE macromers showed sustain release of entrapped DA. The sustain release of DA can also be attributed to linear degradation profile of HPE hydrogels.
3.7 Microfabricated HPE Hydrogels
Compared to linear polymers, hyperbranched polymeric macromolecules show higher surface functionality, reactivity, and encapsulation efficiency. Due to their structural organization at the molecular level, the viscosity of the precursor solution containing hyperbranched polymeric macromolecules is always lower than that of linear polymers having similar molecular weight. We observed that even at a high concentration of acrylated HPE (HPE-A) macromolecules, the prepolymer solution could be easily injected using a 22-gauge surgical needle (Figure 7a). The injection of photocrosslinkable materials has been proposed for minimally invasive therapies as structurally stable hydrogel networks can be formed at the site of injection after light exposure.47–49
Figure 7. Microfabricated photocrosslinkable HPE hydrogels made by crosslinking HPE macromers.

(a) Precursor solution containing 50% HPE macromers showed low viscosity and can be injected using 22-gauge needle. When subjected to UV radiation the prepolymer solution quickly formed covalently crosslinked network. The optical images representing the flexibility of 150 μm thin methylene blue dyed HPE hydrogel sheet. The HPE hydrogel sheet can be easily handled without breaking. After folding, the hydrogel sheet easily unfolds and regains its original shape in the DPBS bath. (b) Schematic presentation for developing patterned HPE hydrogel structures using photolithography. Fluorescent images of different micropatterned structures obtained using photolithography (scale bar = 500 μm).
Microfabrication techniques have been extensively used to pattern cell-laden hydrogels for studying fundamental cell biology.4, 9 Several techniques have been developed for microfabrication including photolithography, microcontact printing, microfluidics, micropatterning and microassembly. The recent development in photocrosslinked polymers have provided the impetus to engineer micropatterned hydrogels for various biomedical applications.4, 9, 50 We have utilized photolithography to fabricate microgels by crosslinking HPE macromers (Figure 7b). In this process, precursor solution consisting of photocrosslinkable polymer along with initiator is exposed to ultraviolet radiation through a mask. The selective exposure of the precursor solution to UV results in crosslinked polymer patterns. Compared to linear polymers, HPE have highly branched star-like structure and exhibit sustained release profile. By utilizing HPE as nanobuilding blocks for microfabrication structure, it is possible to tune and regulate spatial distribution of cells within a 3D network, as well as guiding and controlling cellular behavior.
3.8 In vitro Cell Adhesion and Proliferation
Along with tunable physical and chemical properties, cell adhesion, spreading, and proliferation are equally important properties while designing a new material for biomedical applications.51 Earlier studies have indicated that cell adhesion can be tuned by incorporating biological molecule52, 53, or nanoparticles54, 55. The initial cells adhesion and spreading play important role in cellular functions and tissue regeneration.2, 51, 56 We investigated initial cell adhesion and spreading HPE hydrogels using NIH-3T3 fibroblasts-like cells. The cells were seeded on the HPE hydrogels, and after day 4, their morphology as well as viability, were evaluated. At low HPE concentrations (HPE40), cells remained round-shaped and failed to adhere to the surface. As the HPE concentration was increased, enhanced cell adhesion and spreading was observed (Figure 8a). NIH ImageJ software was used to quantify the cell adhesion and spreading rates (Figure 8b). The increase in cell adhesion may be attributed to the increase in the stiffness of hydrogels or protein adsorption on hydrogel surface.
Figure 8. Control cells adhesion and proliferation on HPE hydrogels.
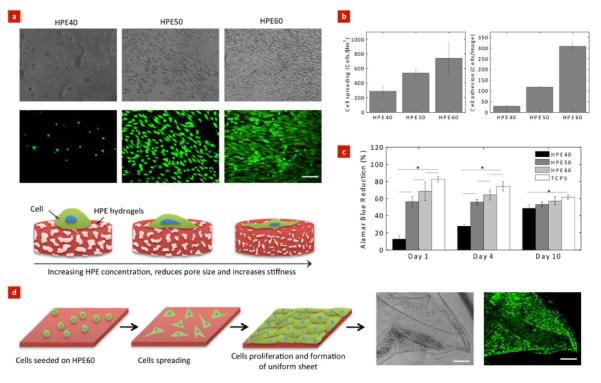
(a) Representative phase contrast and fluorescent images of NIH-3T3 fibroblasts on the surface of the HPE hydrogels. Cells readily attached and spread on hydrogels with higher HPE concentration. (Scale bar = 100 μm). Schematic showing the increase in cells adhesion and spreading can be attributed to higher stiffness of hydrogels resulting from an increase in HPE concentration. (b) Cells adhesion and spreading on the hydrogels surface was quantified using ImageJ. Increase in HPE concentration results in an increase in cells adhesion and spreading. (c) The metabolic activity of cells strongly depends on the HPE concentration. Tissue culture polystyrene was used as a positive control. (d) Controlled cell adhesion properties of HPE60 can be utilized to fabricate uniform cell layer. We observed after 4 day, cells uniformly covered the surface of hydrogels and the cell layer can be easily handled using forceps (Scale bar = 500 μm). The bars represent mean ± standard deviation (n=3), (*p < 0.05).
The effect of change in cell adhesion on their metabolic activity was investigated using Alamar Blue assay. Alamar Blue is an indicator dye, which incorporates an oxidation-reduction reaction within proliferative cells. An active ingredient of Alamar Blue is Resazurin, which is a non-toxic, cell-permeable compound and is virtually non-fluorescent. Upon entering cells, viable cells reduce resazurin to resorufin, which produces a very bright red fluorescence. The results indicated that the metabolic activity of cells seeded on hydrogel surfaces directly depend on the HPE concentration (Figure 8c). The metabolic activity of the cells increases with an increase in the HPE concentration. On days 1 and 4, HPE40 showed lower cell proliferation compared to HPE50, HPE60 and TCPS. This might be attributed to weak cell-matrix interactions. However on day 10, the proliferation rate of cells seeded on HPE40 was enhanced. These results show a direct correlation between HPE concentrations and cell adhesion and spreading, indicating that different cellular responses can be achieved by varying the HPE hydrogel concentrations.
Materials with controlled cellular adhesion can be used in a range of biomedical and biotechnological applications.10, 57, 58 Here, we showed that by controlling HPE hydrogel formulation, it is possible to tailor cell adhesion properties. Earlier studies have showed that by controlling cell adhesion and proliferation on biomaterials surface, it can be used to design scaffolds for a range of tissue engineering applications and cell therapy.38, 56 To showcase the feasibility of using HPE hydrogels for cellular therapies, we prepared HPE hydrogel sheets with a 150 μm thickness (Figure 8d). The thin sheets of HPE hydrogels made by crosslinking HPE macromers show high flexibility and can be easily retrieved and handled. We further demonstrated that cells readily adhere and proliferate on the hydrogel sheet to form uniform cell layer covering the entire hydrogel surfaces. We were able to successfully transfer the cell-laden hydrogels without breaking them. This is the first report that shows that HPE hydrogels made by crosslinking HPE macromers support controlled cell adhesion and can potentially be used for cellular therapies.
4.0 Conclusion
Hyperbranched polyester hydrogels were made by crosslinking hyperbranched polyester macromers with sustained drug release characteristic. Formation of covalently crosslinked hyperbranched polymeric network results in mechanically tough hydrogels with controlled cell adhesion properties. By utilizing microfabrication approaches, spatially controlled geometries and patterns can be obtained that render the utility of these hybrid hydrogels for microscale tissue engineering approaches. We successfully encapsulated a hydrophobic drug (dexamethasone) within the hydrophobic cavity of HPE and demonstrated the sustained release profile of the entrapped drug. Controlled cell adhesion, spreading, and proliferation was also obtained by tuning HPE formulation thus enabling the development of cell-material platforms that can be applied and tailored to specific functionalities in tissue engineering applications.
Supplementary Material
ACKNOWLEDGMENTS
HZ and HY would like to acknowledge National Natural Science Fund for Distinguished Young Scholar (Grant No. 51025313). AP would like to acknowledge postdoctoral fellowship awarded by Natural Science and Engineering Research Council, Canada. AKG acknowledges financial support through the MIT-Portugal Program (MPP-09Call-Langer-47). SMM thanks the Portuguese Foundation for Science and Technology (FCT) for the personal grant SFRH/BD/42968/2008 (MIT-Portugal Program) and the financial support of MIT/ECE/0047/2009 project. This research was funded by the US Army Engineer Research and Development Center, the Institute for Soldier Nanotechnology, the NIH (EB009196; DE019024; EB007249; HL099073; AR057837), and the National Science Foundation CAREER award (AK).
Footnotes
Supporting Information
Included in the Supporting Information are hydration and degradation studies of HPE hydrogels. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
HZ, AP and AKG contributed equally. HZ, AP, AKG, HY and AK conceived the idea and designed the experiments. HZ and AP synthesized hyperbranched polymer and performed hydration kinetics, degradation study and microfabricated hydrogel structures. AKG performed FTIR, mechanical testing, zeta potential and hydrodynamic size, scanning electron microscopy. SMM performed cells adhesion, spreading and proliferation studies. SM and GI helped in image analysis and data quantification. HZ, AP, AKG, HY and AK analyzed experimental data and wrote the paper. All authors discussed the results and commented on the manuscript.
References
- 1.Khademhosseini A, Langer R. Biomaterials. 2007;28(34):5087–5092. doi: 10.1016/j.biomaterials.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 2.Khademhosseini A, Vacanti JP, Langer R. Sci. Am. 2009;300(5):64–71. doi: 10.1038/scientificamerican0509-64. [DOI] [PubMed] [Google Scholar]
- 3.Langer R, Vacanti JP. Science. 1993;260(5110):920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 4.Zorlutuna P, Annabi N, Camci-Unal G, Nikkhah M, Cha JM, Nichol JW, Manbachi A, Bae H, Chen S, Khademhosseini A. Adv. Mater. 2012;24(14):1782–804. doi: 10.1002/adma.201104631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutolf MP. Nat. Mater. 2009;8(6):451–453. doi: 10.1038/nmat2458. [DOI] [PubMed] [Google Scholar]
- 6.Oliveira JM, Sousa RA, Kotobuki N, Tadokoro M, Hirose M, Mano JF, Reis RL, Ohgushi H. Biomaterials. 2009;30(5):804–813. doi: 10.1016/j.biomaterials.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 7.Schuldiner M, Yanuka O, Itskovitz-Eldor J, Melton DA, Benvenisty N. Proc. Natl. Acad. Sci. U S A. 2000;97(21):11307–11312. doi: 10.1073/pnas.97.21.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher OZ, Khademhosseini A, Langer R, Peppas NA. Acc. Chem. Res. 2010;43(3):419–28. doi: 10.1021/ar900226q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khademhosseini A, Langer R, Borenstein J, Vacanti JP. Proc. Natl. Acad. Sci. U S A. 2006;103(8):2480–7. doi: 10.1073/pnas.0507681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS. Adv. Mater. 2010;22(31):3484–3494. doi: 10.1002/adma.200904179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Adv. Mater. 2009;21(32–33):3307–29. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu CJ, Gaharwar AK, Schexnailder PJ, Schmidt G. Materials. 2010;3(5):2986–3005. doi: 10.1021/am100609t. [DOI] [PubMed] [Google Scholar]
- 13.Hirao A, Yoo H-S. Polym. J. 2011;43(1):2–17. [Google Scholar]
- 14.Joshi N, Grinstaff M. Curr. Top. Med. Chem. 2008;8(14):1225–1236. doi: 10.2174/156802608785849067. [DOI] [PubMed] [Google Scholar]
- 15.Gao C, Yan D. Prog. Polym. Sci. 2004;29(3):183–275. [Google Scholar]
- 16.Malmström E, Johansson M, Hult A. Macromolecules. 1995;28(5):1698–1703. [Google Scholar]
- 17.Gillies E, Frechet J. Drug Discov. Today. 2005;10(1):35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- 18.Huang F, Scherman OA. Chem. Soc. Rev. 2012 [Google Scholar]
- 19.Mintzer MA, Grinstaff MW. Chem. Soc. Rev. 2011;40(1):173. doi: 10.1039/b901839p. [DOI] [PubMed] [Google Scholar]
- 20.Cheng Y, Xu Z, Ma M, Xu T. J. Pharm. Sci. 2008;97(1):123–143. doi: 10.1002/jps.21079. [DOI] [PubMed] [Google Scholar]
- 21.Irfan M, Seiler M. Ind. Eng. Chem. Res. 2010;49(3):1169–1196. [Google Scholar]
- 22.Dufes C, Uchegbu I, Schatzlein A. Adv. Drug Deliver. Rev. 2005;57(15):2177–2202. doi: 10.1016/j.addr.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 23.Dong ZQ, Hamid KA, Gao Y, Lin YL, Katsumi H, Sakane T, Yamamoto A. J. Pharm. Sci. 2011;100(5):1866–1878. doi: 10.1002/jps.22428. [DOI] [PubMed] [Google Scholar]
- 24.Desai PN, Yuan Q, Yang H. Biomacromolecules. 2010;11(3):666–673. doi: 10.1021/bm901240g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sontjens SHM, Nettles DL, Carnahan MA, Setton LA, Grinstaff MW. Biomacromolecules. 2006;7(1):310–316. doi: 10.1021/bm050663e. [DOI] [PubMed] [Google Scholar]
- 26.Wang DK, Hill DJT, Rasoul FA, Whittaker AK. Journal of Polymer Science Part A: Polymer Chemistry. 2012;50(6):1143–1157. [Google Scholar]
- 27.Carnahan MA, Middleton C, Kim J, Kim T, Grinstaff MW. J Am. Chem. Soc. 2002;124(19):5291–5293. doi: 10.1021/ja025576y. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Dong Y, Wang L, Wang G, Wu J, Zheng Y, Yang H, Zhu S. J. Mater. Chem. 2011;21(35):13530. [Google Scholar]
- 29.Zhang H, Zhao C, Cao H, Wang G, Song L, Niu G, Yang H, Ma J, Zhu S. Biomaterials. 2010;31(20):5445–5454. doi: 10.1016/j.biomaterials.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 30.Zhong SP, Yung LYL. J. Biomed. Mater. Res. Part A. 2009;91A(1):114–122. doi: 10.1002/jbm.a.32188. [DOI] [PubMed] [Google Scholar]
- 31.Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T. Nature. 2010;463(7279):339–343. doi: 10.1038/nature08693. [DOI] [PubMed] [Google Scholar]
- 32.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Macromol. Biosci. 2008;9(5):515–524. doi: 10.1002/mabi.200800269. [DOI] [PubMed] [Google Scholar]
- 33.Santra S, Kaittanis C, Perez JM. Langmuir. 2009;26(8):5364–5373. doi: 10.1021/la9037843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaharwar AK, Dammu SA, Canter JM, Wu C-J, Schmidt G. Biomacromolecules. 2011;12(5):1641–1650. doi: 10.1021/bm200027z. [DOI] [PubMed] [Google Scholar]
- 35.Anseth KS, Bowman CN, Brannon-Peppas L. Biomaterials. 1996;17(17):1647–1657. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- 36.Gaharwar AK, Kishore V, Rivera C, Bullock W, Wu C-J, Akkus O, Schmidt G. Macromol. Biosci. 2012;12(6):779–793. doi: 10.1002/mabi.201100508. [DOI] [PubMed] [Google Scholar]
- 37.Ma PX. Mater. Today. 2004;7(5):30–40. [Google Scholar]
- 38.Hoffman AS. Adv. Drug Deliver. Rev. 2002;54(1):3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 39.Muscatello MM, Stunja LE, Asher SA. Anal. Chem. 2009;81(12):4978–86. doi: 10.1021/ac900006x. [DOI] [PubMed] [Google Scholar]
- 40.Wang DK, Hill DJT, Rasoul FA, Whittaker AK. J. Polym. Sci. A1. 2012;50(6):1143–1157. [Google Scholar]
- 41.Wang J, Satoh M. Polymer. 2009;50(15):3680–3685. [Google Scholar]
- 42.Hutson CB, Nichol JW, Aubin H, Bae H, Yamanlar S, Al-Haque S, Koshy ST, Khademhosseini A. Tissue Eng. Part A. 2011;17(13–14):1713–23. doi: 10.1089/ten.tea.2010.0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim B-S, Smith R. e. C., Poon Z, Hammond PT. Langmuir. 2009;25(24):14086–14092. doi: 10.1021/la9017618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baumann MD, Kang CE, Stanwick JC, Wang Y, Kim H, Lapitsky Y, Shoichet MS. J. Controlled Release. 2009;138(3):205–213. doi: 10.1016/j.jconrel.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Singh A, Suri S, Roy K. Biomaterials. 2009;30(28):5187–5200. doi: 10.1016/j.biomaterials.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mandal BB, Kundu SC. Biomaterials. 2009;30(28):5170–5177. doi: 10.1016/j.biomaterials.2009.05.072. [DOI] [PubMed] [Google Scholar]
- 47.Elisseeff J, Anseth K, Sims D, McIntosh W, Randolph M, Langer R. Proc. Natl. Acad. Sci. U S A. 1999;96(6):3104–3107. doi: 10.1073/pnas.96.6.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kretlow JD, Klouda L, Mikos AG. Adv. Drug Deliver. Rev. 2007;59(4–5):263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 49.Yu L, Ding J. Chem. Soc. Rev. 2008;37(8):1473–1481. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 50.Mihaila SM, Gaharwar AK, Reis RL, Marques AP, Gomes ME, Khademhosseini A. Adv. Healthcare Mater. 2013 doi: 10.1002/adhm.201200317. DOI: 10.1002/adhm.201200317. [DOI] [PubMed] [Google Scholar]
- 51.Lutolf M, Hubbell J. Nat. Biotechnol. 2005;23(1):47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 52.Hersel U, Dahmen C, Kessler H. Biomaterials. 2003;24(24):4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 53.Benoit DSW, Anseth KS. Acta Biomater. 2005;1(4):461–470. doi: 10.1016/j.actbio.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Gaharwar AK, Rivera CP, Wu CJ, Schmidt G. Acta Biomater. 2011;7(12):4139–4148. doi: 10.1016/j.actbio.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 55.Schiraldi C, D'Agostino A, Oliva A, Flamma F, De Rosa A, Apicella A, Aversa R, De Rosa M. Biomaterials. 2004;25(17):3645–3653. doi: 10.1016/j.biomaterials.2003.10.059. [DOI] [PubMed] [Google Scholar]
- 56.Liu WF, Chen CS. Mater. Today. 2005;8(12):28–35. [Google Scholar]
- 57.Gaharwar AK, Schexnailder PJ, Kline BP, Schmidt G. Acta Biomater. 2011;7(2):568–577. doi: 10.1016/j.actbio.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 58.Wischerhoff E, Uhlig K, Lankenau A, Börner HG, Laschewsky A, Duschl C, Lutz J-F. Angew. Chem. Int. Edit. 2008;47(30):5666–5668. doi: 10.1002/anie.200801202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.