

Abstract
The PGBD3 piggyBac transposon inserted into CSB intron 5 early in the primate lineage. As a result of alternative splicing, the human CSB gene now encodes three proteins: CSB, a CSB-PGBD3 fusion protein that joins the N-terminal CSB domain to the C-terminal PGBD3 transposase domain, and PGBD3 transposase. The fusion protein is as highly conserved as CSB, suggesting that it is advantageous in health; however, expression of the fusion protein in CSB-null cells induces a constitutive interferon (IFN) response. The fusion protein binds in vivo to PGBD3-related MER85 elements, but is also tethered to c-Jun, TEAD1, and CTCF motifs by interactions with the cognate transcription factors. The fusion protein regulates nearby genes from the c-Jun (and to a lesser extent TEAD1 and CTCF) motifs, but not from MER85 elements. We speculate that the fusion protein interferes with CSB-dependent chromatin remodeling, generating double-stranded RNA (dsRNA) that induces an IFN response through endosomal TLR or cytoplasmic RIG-I and/or MDA5 RNA sensors. We suggest that the fusion protein was fixed in primates because an elevated IFN response may help to fight viral infection. We also speculate that an inappropriate IFN response may contribute to the clinical presentation of CS.
Keywords: interferon (IFN), piggyBac-derived element 3 (PGBD3), fusion protein, AP-1 family transcription factor (c-Jun), CCCTC-binding factor (CTCF)
1. Hiding in plain sight: the (re)discovery of the CSB-PGBD3 fusion protein
In late spring of 2005, while checking the NCBI Entrez Gene portal for sequence updates, John Newman noticed an annotated PGBD3 piggyBac transposon embedded in intron 5 of the human CSB gene (also known as ERCC6). According to the spliced EST and mRNA tracks on the UCSC browser, this PGBD3 element appeared to be functioning as an alternative 3′ exon that could be spliced either to CSB exons 1–5 or to a fragment of CSB exon 5 transcribed from a cryptic promoter within that exon (Fig. 1A). It did not take long to discover that the PGBD3 element contained a continuous ORF encoding the PGBD3 transposase flanked upstream by a 3′ splice site and downstream by a polyadenylation site; and most strikingly, the transposase ORF was in frame with CSB exon 5 suggesting, as we soon confirmed, that the human CSB locus generated not one but three proteins: full length CSB by default splicing of CSB exons 1–22, a CSB-PGBD3 fusion protein by alternative splicing of CSB exons 1–5 to the 3′ terminal PGBD3 exon, and solitary PGBD3 transposase by splicing of the exon 5 fragment to the 3′ terminal PGBD3 exon (Fig. 1B). Perhaps most surprisingly, the CSB-PGBD3 fusion protein was not a curiosity of the human genome, but had first arisen in the common ancestor of humans and marmoset about 43 million years ago (Mya) and had been highly conserved ever since — strongly implying that the fusion protein conferred a selective advantage on our primate ancestors and continues to do so today. Finally, as might be expected from such extraordinary conservation, the two new proteins generated by alternative splicing and polyadenylation were abundant: In a survey of 7 cell lines, the CSB-PGBD3 fusion protein was typically 4-fold more abundant than full length CSB protein, and the solitary PGBD3 transposase 4-fold more abundant than the fusion protein (Newman et al., 2008).
Fig. 1. The PGBD3 transposon functions as an alternative 3′ terminal exon, enabling the human CSB gene to generate three proteins instead of one.

Splicing of CSB exons 1–22 generates functional CSB (1493 residues); alternative splicing of CSB exons 1–5 (residues 1–465) to the piggyBac PGBD3 transposase ORF generates the CSB-PGBD3 fusion protein (1061 residues); and transcription driven by a cryptic promoter in exon 5 generates solitary PGBD3 transposase (593 residues). We assume translation of solitary PGBD3 transposase begins at the first methionine in PGBD3 because initiation at the last methionine in CSB exon 5 would noticeably increase the predicted gel mobility from 68 kDa to 80 kDa (Newman et al., 2008). An appropriate balance between normal splicing of the host gene and alternative splicing to the PGBD3 3′ terminal exon is probably maintained in different host gene contexts by synergy between the 3′ splice site and polyadenylation signal (Niwa et al., 1990) that flank the PGBD3 ORF. Note also that all CSB mutations downstream of exon 5, and most compound heterozygous mutations seen in CS, allow synthesis of the CSB-PGBD3 fusion protein in the absence of functional CSB. The 889 MER85s are 140 bp mobile elements that were derived from PGBD3 by internal deletion of the transposase ORF, and subsequently mobilized by a PGBD3 transposase in trans (Gray et al., 2012; Lander et al., 2001). Key: (A) blue lines, boxes, and arrows, CSB DNA; black and gray arrows, CSB and cryptic transcription start sites, respectively; red lines, mRNA splicing; orange arrow, PGBD3 piggyBac element; pA, poly(A) sites; 5′ ss and 3′ ss, donor and acceptor splice sites. (B) heavy blue lines and boxes, CSB protein sequences and motifs; heavy orange line, PGBD3 transposase; light blue and orange lines, 5′ untranslated region. The C-terminal ubiquitin binding domain (UBD) may be part of a quality control mechanism that prevents incision at the site of DNA damage until the TCR-NER complex is correctly assembled (Anindya et al., 2010; Gray and Weiner, 2010). The CSB-PGBD3 fusion protein is also known as CPFP for CSB-piggyBac transposable element derived 3 fusion protein (Horibata et al., 2011). Adapted from Fig. 1 of Gray et al. (2012).
We were convinced from the beginning that the CSB-PGBD3 fusion protein was likely to play a role in CS because it was difficult to believe that an abundant protein which shares a 465 residue N-terminal domain with CSB and has been conserved for 43 My would not have some effect on CSB functions — whether as a modulator of CSB activity in normal individuals, a modulator of disease in CS individuals, or in some other capacity we could not yet imagine. We also wanted ensure that the inconvenient truth (3 proteins from 1 gene) would not remain hidden in plain sight from the CS community on the UCSC browser. Still, we wondered about our debt to the unnamed bioinformaticist (or algorithm) who discovered and annotated the presumptive CSB-PGBD3 fusion protein. Was this unsung curator fully aware of the provocative implications of the genomic and transcriptional anatomy, or was it all in a day’s work and quickly forgotten? And how could we publish our (re)discovery of the fusion protein without including the invisible benefactor as coauthor? When all was said and done, it took more work at the bench than we anticipated to prove the browser right, and the experimental value-added satisfied the demands of conscience and reviewers.
2. MER85s, a large family of internally deleted PGBD3 elements, complicate the CSB-PGBD3 story
PGBD3, like many other inverted terminal repeat transposons, has given rise to a family of Miniature Inverted Terminal Repeat Transposable Elements or MITEs (Feschotte and Pritham, 2007; Feschotte, 2008). These 140 bp MER85s are nonautonomous, internally-deleted PGBD3 elements that have lost the transposase ORF, but retain all terminal sequences required for mobilization in trans by the PGBD3 transposase (Fig. 1A); the essential sequences include not only the perfect 13 bp inverted repeats at the ends of the element, but internal sequences that are distinct in structure and function, and confer polarity on MER85s as is the case for most other MITEs (Feschotte and Pritham, 2007; Feschotte, 2008). MER85s arose in the common ancestor of Old World and New World Monkeys about 40–45 Mya and multiplied to over 889 dispersed copies (Gray et al., 2012) before mobility declined about 35 Mya (Lander et al., 2001) -— perhaps because the transposase activity was lost to mutation or selected against to prevent runaway multiplication. Since the PGBD3 transposase ORF is highly conserved from marmoset to human, we initially speculated that the CSB-PGBD3 fusion protein might regulate genes by binding to nearby MER85s through the C-terminal PGBD3 domain (Newman et al., 2008 and Fig. 2, middle panel). As discussed in detail below, we found that the CSB-PGBD3 fusion protein as well as solitary PGBD3 transposase do indeed bind to the (arbitrarily designated) “left end” of MER85 elements both in vitro and in vivo, but apparently do not regulate a significant number of nearby genes from these sites; rather, the CSB-PGBD3 fusion protein regulates expression of nearby genes when tethered through protein/protein interactions to c-Jun, TEAD1, CTCF1, and presumably other transcription factors bound to their cognate binding motifs (Gray et al., 2012).
Fig. 2. The CSB-PGBD3 fusion protein works together with existing transcription factors and MER85 elements to create new layers of regulation on top of established regulatory networks.

(upper panel) Genes and transcription factors in the ancestral primate genome before the CSB-PGBD3 fusion protein arose (43 Mya) and before MER85s replicated to high copy number (35 Mya). (middle panel) Anticipated result. The CSB-PGBD3 fusion protein would bind to MER85 elements through the PGBD3 domain and regulates nearby genes presumably through the N-terminal CSB domain. (lower panel) Actual results. The CSB-PGBD3 fusion protein binds to RNAPII and the c-Jun transcriptional activator through the N-terminal CSB domain, to the TEAD1 activator and CTCF silencer by as yet uncharactertized interactions, and to a subset of MER85 elements through the C-terminal PGBD3 domain (Gray et al., 2012). The binding of the fusion protein to c-Jun, TEAD1, and CTCF sites through protein/protein interactions regulates nearby genes. Colocalization of CTCF with the fusion protein suggests gene regulation through chromatin looping. TRE motifs (TPA response elements) bind AP-1 family factors such as Jun and Fos. Surprisingly, only 363 of 889 MER85s bound the fusion protein in vivo, and only 262 of the 1000 strongest fusion protein binding sites contained MER85 elements; the other 738 fusion protein binding sites are MER85-less. Overall gene regulation by MER85-bound CSB-PGBD3 fusion protein was not statistically significant (Fig. 3 and Gray et al., 2012) but this does not rule out MER85 regulation of a small number of individual genes. Adapted from Fig. 12 of Gray et al. (2012).
Taken together, the available genomic data suggest that the CSB-PGBD3 fusion protein and MER85s both arose in the common ancestor of humans and marmosets (a New World monkey). We do not know whether the CSB-PGBD3 fusion protein or MER85 elements arose first, but the transposase of the PGBD3 transposon that first inserted into CSB intron 5 must have been inactivated soon thereafter (or even beforehand) because all available CSB-PGBD3 fusion protein sequences (marmoset, rhesus macaque, baboon, orangutan, gorilla, chimpanzee, and human) share the same D352N mutation in the second of the three catalytic aspartates (Newman et al., 2008, Figs. S7 and S8; and unpublished observations) that were previously identified experimentally in the cabbage looper moth (Trichoplusia ni) piggyBac transposase (Keith et al., 2008). Any further transposition of MER85 elements would then have relied on PGBD3 transposase activity provided by what subsequently became one of the four PGBD3 pseudogenes (Newman et al., 2008).
3. The PGBD3 transposon evolved to function as a 3′ terminal exon
Mobile elements are incredibly diverse, and appear to have explored almost every imaginable replicative lifestyle in all three kingdoms of life. Yet piggyBac transposons are the only superfamily of mobile elements in which some members such as PGBD3 survive as obligate alternative 3′ exons (Newman et al., 2006). This unusual lifestyle has several advantages: (1) PGBD3 takes advantage of host gene introns as safe havens for insertion that do not interrupt resident ORFs — thus avoiding harmful insertional mutagenesis; (2) PGBD3 transcription is driven by a host gene promoter — greatly expanding the host range of PGBD3 elements; (3) PGBD3 exploits alternative splicing and the near-universality of eukaryotic mRNA splicing and polyadenylation signals in order to generate a novel N-terminal fusion protein while maintaining nearly normal levels of the host protein — thus skillfully dodging the risk of a knockout mutation; (4) judging by the ability of the T. ni piggyBac transposase to function efficiently in flies, maize, mice, humans, and many other organisms (Yusa et al., 2011), piggyBac transposase appears capable of catalyzing precise integration and excision without help from host factors — thus further expanding the host range as shown for other horizontally transferred transposons (Mitra et al., 2008; Li et al., 2012); and finally (5) the T. ni piggyBac transposase more readily tolerates N-terminal fusions than the Sleeping Beauty, hAT-like Tol2, and Mos1 Mariner transposases (Wu et al., 2006) — suggesting that piggyBac transposase naturally tolerates (or has evolved to tolerate) the N-terminal fusions required for its lifestyle as an obligate 3′ terminal exon.
4. CSB protein plays a general role in chromatin remodeling and transcription elongation
While studying the gene structure, transcription, and function of human small nuclear RNAs (U snRNAs), we discovered that loss of CSB activity causes site-specific metaphase chromosome fragility of four tandemly repeated gene families encoding small RNAs — the RNU1 locus at 1p36 containing about 30 tandem genes for U1 snRNA, the RNU2 locus at 17q21-q22 containing about 5–25 tandem genes for U2 snRNA, the RN5S locus at 1q42 containing about 200 tandem genes for 5S ribosomal RNA (rRNA), and the ancient PSU1 locus at 1q12-q22, that once encoded U1 snRNA but now consists entirely of dead or dying U1 pseudogenes (Yu et al., 2000, and references therein). These results were puzzling because (1) CSB was thought to be a DNA repair factor with an essential role in transcription-coupled nucleotide excision repair (TC-NER), yet there was no indication that these four genomic loci were subject to constitutive DNA damage or ongoing DNA repair; and (2) U1 and U2 snRNA are transcribed by RNA polymerase II like mRNAs, whereas 5S rRNA is transcribed by RNA polymerase III like tRNAs, yet loss of CSB affected all three loci equally. This forced us to consider the possibility that CSB plays a role in transcription as well as in DNA repair.
To generate clues regarding potential CSB functions in processes other than DNA repair, we resorted to an hypothesis-free approach, and compared gene expression in the classic CSB compound heterozygote CS1AN before and after rescue by stable transfection with a normal CSB cDNA expression construct (Newman et al., 2006). Surprisingly, we found that many of the CSB-regulated genes were also affected by treatments and mutations that modulate histone acetylation, DNA methylation, poly(ADP)ribosylation, and RNA polymerase II elongation — all of which affect chromatin structure. As might have been expected, however, no DNA repair signatures were observed in these undamaged cells. We therefore concluded that CSB plays a general role in maintenance and remodeling of chromatin structure, and we also suggested — based on additional signatures in the gene expression data — that CS might be “a disease of transcriptional deregulation caused by misexpression of growth suppressive, inflammatory, and proapoptotic pathways” (Newman et al., 2006).
5. The CSB-PGBD3 fusion protein induces, and coexpression of CSB represses, an innate interferon-like response in CSB-null UVSS1KO cells
The discovery of the CSB-PGBD3 fusion protein (Newman et al., 2008) cast a new light on Newman et al. (2006) where we had assumed that the CS1AN compound heterozygote, with one allele containing a nonsense mutation in exon 5 and the other a frameshift in exon 13 (Laugel et al., 2010), was a true CSB-null. In fact, CS1AN continues to express the CSB-PGBD3 fusion protein from the frameshift allele. Although this did not change our conclusion that CSB plays a general role in chromatin remodeling, it prevented us from disentangling the role (if any) of the CSB-PGBD3 fusion protein from that of CSB in gene expression.
Conservation of the CSB-PGBD3 fusion protein for 43 My suggests that it is advantageous in health when CSB is functional, but potentially harmful when functional CSB is lost or depleted in CS (Newman et al., 2008). To genetically dissect the contributions of CSB and the CSB-PGBD3 fusion protein to gene expression, we constructed four stable lines derived from the true CSB-null line, UVSS1KO, with homozygous nonsense mutations at residue 77 (Horibata et al., 2004) that prevent synthesis of the CSB-PGBD3 fusion protein (Horibata et al., 2011). Gene expression in these four stable lines — expressing either CSB protein only, CSB-PGBD3 fusion protein only, both proteins, or neither — was then characterized using Affymetrix U133A Plus 2.0 chips (Bailey et al., 2012).
Consistent with the interpretation that the fusion protein is advantageous in health but harmful in disease, we found that (1) stable expression of the CSB-PGBD3 fusion protein in CSB-null UVSS1KO cells — recreating a CS genotype — induces a constitutive innate antiviral IFN response in the complete absence of viral infection; and (2) coexpression of functional CSB with the CSB-PGBD3 fusion protein in the same cells — recreating a normal genotype — dramatically represses the IFN-like response (Bailey et al., 2012; Gray et al., 2012). Specifically, expression of the CSB-PGBD3 fusion protein alone strongly induces a host of IFN-related genes including all three subunits of the IFN-stimulated transcription factor ISGF3 (STAT1, STAT2, and IRF9); the oligoadenylate synthetases (OAS1, OAS2, OAS3, and OASL); the IFN-stimulated and inducible genes ISG15, ISG20, IFI6, IFI27, IFI44, IFI44L, IFH1, IFIT1, IFIT2, IFIT3, IFIT5, and IFITM1; the type I IFN receptor IFNAR2; and the receptor-activated JAK1 kinase. In addition, expression of CSB and the CSB-PGBD3 fusion protein together, but neither alone, upregulates the insulin growth factor binding protein IGFBP5 by 7-fold and downregulates IGFBP7 by 3-fold, consistent with the hypothesis that organismal resources are reallocated by the IGF1/insulin pathway from growth to somatic preservation in response to unrepaired DNA damage (van der Pluijm et al., 2007; Niedernhofer et al., 2006).
6. The IFN-like response induced by the CSB-PGBD3 fusion protein resembles the sustained IFN response normally maintained by unphosphorylated STATs (U-STATs)
The acute phase of a normal IFN response is effective against viruses, bacteria, and other pathogens but, like many drugs and chemotherapy regimens, is cytotoxic for the cell. Cheon and Stark (2009) and Cheon et al. (2011) have recently demonstrated that the canonical IFN response has two phases — an acute cytotoxic phase driven by STATs that are sequentially phosphorylated on tyrosine and serine (P-STATs) to meet the immediate threat, and a more moderate phase of sustained vigilance driven by unphosphorylated STATs (U-STATs) to insure that the threat has been vanquished. Ptyr-STATs are typically generated by JAK and TYK family kinases activated by IFN-stimulated IFN receptors but, as the initial IFN signal subsides, the remaining Ptyr-STATs continue to stimulate STAT transcription, initiating a positive feedback loop in which U-STATs activate their own transcription along with a more moderate subset of IFN-related genes.
Consistent with the notion that expression of the CSB-PGBD3 fusion protein in CSB-null UVSS1KO cells induces and sustains the second phase of an IFN-like response, we found that the fusion protein (1) failed to induce IFN mRNA; (2) induced 18 of the 20 genes most strongly induced in BJ fibroblasts by the Y701F-STAT1 mutant that cannot be phosphorylated on tyrosine; and (3) induced phosphorylation of STAT1 on serine but not on tyrosine, suggesting that the fusion protein induces the IFN response pathway downstream of the canonical tyrosine phosphorylation of STAT1 by IFN-activated receptor JAK/TYK kinases (Bailey et al., 2012).
Although induction of an IFN-like response by the CSB-PGBD3 fusion protein remains surprising, it is not surprising that this response resembles the second phase of a normal IFN response — a period of heightened vigilance instead of the life-and-death decisions of the initial acute response (Bailey et al., 2012). Indeed, we suggest below that the CSB-PGBD3 fusion protein may have been fixed 43 Mya and maintained ever since because it raises our threshold levels of viral and/or pathogen defense — in effect paying the smaller price of sustained vigilance to avoid the larger price of catastrophic infection.
7. CSB and the CSB-PGBD3 fusion protein induce related antiviral responses in different genetic backgrounds
The CSB compound heterozygote CS1AN expresses no functional CSB, but it continues to express the CSB-PGBD3 fusion protein from one of its two mutant CSB alleles (Horibata et al., 2011). When we rescued CS1AN by stable expression of normal CSB cDNA, gene expression analysis using the L2L Microarray Suite and Database (Newman and Weiner, 2005) revealed strong chromatin remodeling and transcription elongation signatures, but considerably weaker IFN and antiviral signatures (Newman et al., 2006). However, when we reanalyzed the same CS1AN datasets using MSigDB instead of L2L, we found that expression of CSB in the CS1AN compound heterozygote upregulated 15 IFN-related genes, 12 of which are also upregulated by expression of CSB-PGBD3 fusion protein in the CSB-null UVSS1KO line and belong to 3 related antiviral themes — viral RNA recognition (RIG-1, MDA5), regulation of protein biosynthesis (IFIT1, IFIT2) or degradation (ISG15, HERC5, RBBP6, TRIM14), and membrane-mediated antiviral activities (GBP1, IFITM1, PLSCR1, RSAD2) (Bailey et al., 2012).
The 12 proteins induced by both CSB-PGBD3 fusion protein in CSB-null UVSS1KO cells and coexpression of CSB with endogenous CSB-PGBD3 in CS1AN cells are not the only antiviral proteins induced by the fusion protein. Most dramatically, the CSB-PGBD3 fusion protein strongly induces BST2/tetherin in CSB-null UVSS1KO cells. Although unhelpfully named bone marrow stromal antigen 2 (BST2), this gene encodes the trans-membrane protein tetherin which, as the name implies, prevents budding and release of enveloped viruses by tethering nascent buds to the cell surface. Tetherin is induced 26,500-fold by expression array analysis, 143-fold by RT-PCR assay, and repressed >200-fold by coexpression of intact CSB as is the case for many other genes induced by the CSB-PGBD3 fusion protein.
Admittedly it is odd, if not paradoxical, that 12 of the same antiviral genes are induced both by expression of the CSB-PGBD3 fusion protein in a CSB-null UVSS1KO background (recreating a CS genotype) and by coexpression of CSB with endogenous CSB-PGBD3 fusion protein in the CS1AN compound heterozygote (recreating a normal genotype). Conceivably, the effects of endogenous CSB-PGBD3 fusion protein generated by the CS1AN exon 13 frameshift allele could be blunted by the 341 residue N-terminal CSB fragment expressed from the exon 5 nonsense allele (Laugel et al., 2010; Horibata et al., 2011), but then rescued by coexpression of functional CSB. Alternatively, genetic or epigenetic modifiers in the CS1AN and UVSS1KO lines may result in cell line-specific responses to stable expression of the CSB and CSB-PGBD3 fusion proteins — much as the same CS genotypes can lead to different clinical presentations.
The observation that about half of all CS patients are consangineous (Laugel et al., 2010) suggests that genetic background can significantly affect clinical outcome. For example, patient UVSS1KO has a homozygous nonsense mutation at codon 77, expresses no CSB-related polypeptides, but is only mildly UV sensitive and had not exhibited any signs of CS by age 33 (Horibata et al., 2004); whereas patient KPSX6 is homozygous for a frameshift mutation at codon 77, and also lacks any CSB-related polypeptides, but exhibited late onset (type III) CS beginning at age 45 (Hashimoto et al., 2008). Moreover, two other CS patients, CS539VI and CS548VI, have identical homozygous deletions of the CS promoter and 5′ UTR, and lack any CSB-related polypeptides, but exhibited classical early onset (type I) CS (Laugel et al., 2008). Thus the absence of CSB-related polypeptides can cause UVSS, delayed onset (type III) CS, or classical (type I) CS. The poor correlation of many CS genotypes with clinical phenotypes suggests that what is true for patients may also be true for cell lines, although perhaps not for precisely the same reasons. We can only suggest that the ability of CSB and the CSB-PGBD3 fusion protein to coregulate a subset of antiviral genes, albeit in different genetic backgrounds, may imply that the CSB-PGBD3 fusion protein was fixed in the primate lineage about 43 Mya because it modulated, augmented, diversified, or partially antagonized existing CSB functions.
8. Upregulation of the RIG-I and MDA5 viral RNA sensors by the CSB-PGBD3 fusion protein in the apparent absence of JAK/STAT signaling suggests that the fusion protein may phenocopy an RNA virus infection
Stable expression of the CSB-PGBD3 fusion protein in CSB-null UVSS1KO cells strongly upregulates the viral RNA sensors RIG-I (6- to 8-fold) and MDA5 (36-fold) that normally function in the innate, intracellular antiviral defense. The ability of the CSB-PGBD3 fusion protein to induce an IFN-like response in CSB-null UVSS1KO cells, but without inducing IFN mRNAs (assayed by microarrays) or activating JAK/STAT signaling (assayed by immunoblotting for Ptyr-STAT1), suggested that the fusion protein might phenocopy or mimic a cell autonomous RNA virus infection. Indeed, the localization of nucleic acid-sensing toll-like receptors TLR3, 7, 8, and 9 to the endoplasmic reticulum (Beutler et al., 2006) provide clear evidence that the innate immune response is designed to initiate from within the cell as well as from without. We therefore suggested that loss of the CSB chromatin remodeling protein (Newman et al., 2006), or the presence of the CSB-PGBD3 fusion protein (Newman et al., 2008) in the absence of functional CSB (Bailey et al., 2012), might disrupt chromatin remodeling by interfering with CSB-dependent functions — thereby allowing bidirectional transcription and generation of double-stranded RNA (dsRNA) that can activate the RIG-1 and/or MDA5 RNA sensors (Bailey et al., 2012). We elaborate on this working hypothesis below.
9. The CSB-PGBD3 fusion protein functions as a transcription factor when bound to chromosomal c-Jun/TRE, TEAD1, and CTCF proteins, but not when bound to MER85 elements
As shown in Fig. 2, we initially speculated that the CSB-PGBD3 fusion protein might affect gene expression by binding to MER85 elements through the C-terminal PGBD3 domain and regulating nearby promoters and enhancers through the 465 residue N-terminal CSB domain (Newman et al., 2008). What we actually found by ChIP-seq identification of genomic CSB-PGBD3 fusion protein binding sites was totally different.
Although the CSB-PGBD3 fusion protein binds to 363 of 889 genomic MER85 elements in vivo, these peaks did not correlate significantly with nearby genes that are located less than 1 Mb away and regulated by stable expression of the CSB-PGBD3 fusion protein in CSB-null UVSS1KO cells (Gray et al., 2012). Indeed, MER85s constituted only 18% of the 2,087 genomic CSB-PGBD3 fusion protein binding peaks; 44% of the fusion protein peaks were found directly and symmetrically over c-Jun/AP1/TRE, TEAD1, and CTCF motifs, and a remarkable 42% of the equally significant and symmetrical peaks were located over no recognizable sequence features whatsoever (Fig. 3). Unexpectedly, the CSB-PGBD3 fusion protein binds to AP1/TRE motifs by protein/protein interactions between c-Jun and the N-terminal CSB domain of the fusion protein (Gray et al., 2012); binding to TEAD1 and CTCF motifs is presumably mediated by the cognate factors, although we do not know whether these protein/protein interactions also involve the N-terminal CSB domain (blue) or the C-terminal PGBD3 domain (orange) of the fusion protein (Figs. 2 and 3). The remaining 42% of the CSB-PGBD3 peaks could reflect direct binding of the fusion protein to variable or gapped DNA sequence motifs that MEME cannot recognize; or indirect binding through protein/protein interactions between the N-terminal CSB-domain of the fusion protein and RNA polymerase II (Gray et al., 2012) or topoisomerase I (Horibata et al., 2011).
Fig. 3. Changes in gene expression correlate with tethering of the CSB-PGBD3 fusion protein to transcription factors bound to their cognate motifs.
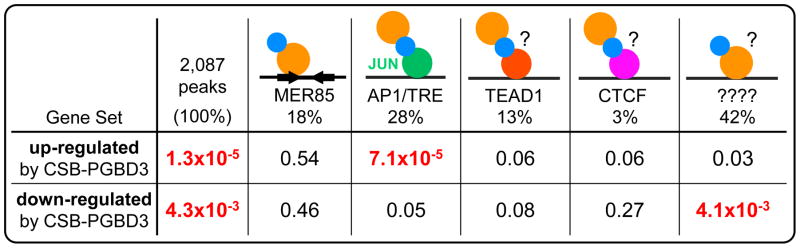
The N-terminal CSB domain (blue) and the C-terminal PGBD3 transposase domain (light orange) of the fusion protein are color coded as in Fgures 1 and 2. The CSB-PGBD3 fusion protein binds to MER85 elements through the PGBD3 domain, and to AP1/TRE motifs by interactions between bound c-Jun (dark green) and the N-terminal CSB domain of the fusion protein; similar interactions with TEAD1 (dark orange) and CTCF (magenta) may tether the CSB-PGBD3 fusion protein those motifs. The mode of binding to apparently featureless sequences is unknown. Gene sets (left) refer to genes up- or down-regulated by stable expression of the CSB-PGBD3 fusion protein in CSB-null UVSS1KO cells (Bailey et al., 2012). The most significant p-values (red font) indicate that up-regulation of nearby genes correlates with binding to AP1/TRE motifs, down-regulation with binding to unknown sequences, conformations, or modifications. TEAD1 regulates both up- and down-regulation weakly; CTCF mainly affects up-regulation. Adapted from Fig. 11 of Gray et al. (2012).
The observation that the CSB-PGBD3 fusion protein binds to c-Jun through the N-terminal CSB-domain (Gray et al., 2012) was unexpected, but suggests an intriguing scenario when combined with the discovery that the N-terminal domain of CSB autoinhibits CSB chromatin remodeling (Lake et al., 2010). Admittedly, c-Jun does not bind CSB in a coimmunoprecipitation assay from UVSS1KO cells (Gray et al., 2012, Fig. 9); however, CSB could potentially bind c-Jun after ATP-dependent relief of N-terminal repression in the context of chromatin — thereby favoring CSB remodeling near occupied c-Jun motifs. A possible implication would be that c-Jun sites (and perhaps TEAD1 and CTCF sites as well) are CSB-dependent, and are among the sites that are misregulated when the CSB-PGBD3 fusion protein is expressed in the absence of CSB.
10. Does the CSB-PGBD3 fusion protein disrupt chromatin remodeling, generating dsRNA that mimics RNA viral infection and induces an IFN response?
Using GREAT analysis (McLean et al., 2010), we found that the CSB-PGBD3 fusion protein regulates nearby genes through protein/protein interactions with c-Jun/AP1, TEAD1, and CTCF transcription factors bound to their cognate DNA motifs (Fig. 2). Although genes that are regulated in this fashion by the fusion protein correlate significantly with TGF-beta signaling (from AP1/TRE sites), IFN-gamma signaling (from AP1/TRE and TEAD1 sites), carcinogenesis (from AP1/TRE, TEAD1, and sites with no detectable motif), and IL-2 signaling (from sites with no detectable motif), the IFN-gamma correlation was no stronger than the others, and no IFN-alpha or IFN-beta correlation could be seen (Gray et al., 2012). Many other scenarios are possible, but these data suggest once again that the CSB-PGBD3 fusion protein may induce the IFN response not by regulating specific genes in the IFN signaling pathway, but by interfering with normal CSB-dependent functions in genomewide chromatin remodeling and/or transcriptional elongation.
Taken together, the absence of detectable Ptyr-STAT1 which normally drives an autocrine IFN response, the relatively modest regulation of IFN-related genes by chromatin-bound CSB-PGBD3 fusion protein, and the ability of the CSB-PGBD3 fusion protein to strongly induce the RIG-I and MDA5 RNA sensors, are all consistent with a model in which the CSB-PGBD3 fusion protein disrupts chromatin remodeling, allowing bidirectional transcription, production of double stranded RNA (dsRNA), and consequent induction of a cell autonomous innate IFN response driven by the the cytoplasmic RIG-I and MDA5 RNA sensors, or the endosomal TLR3, 7, or 8 RNA sensors (Bailey et al., 2012). On the other hand, the ability of the CSB-PGBD3 fusion protein to inhibit repair of camptothecin-induced covalent complexes between topoisomerase I and DNA suggests an alternative scenario in which the CSB-PGBD3 fusion protein inhibits repair of endogenous topoisomerase I-DNA complexes (Horibata et al., 2011) — potentially inducing an autocrine IFN response just as the camptothecin derivative Topotecan induces IFN-beta and other cytokines including TNF-alpha, IL-6, and IL-8 (Wan et al., 2012). However, the absence of detectable Ptyr-STAT1 argues against the autocrine IFN response in this scenario. Note that in either scenario, the CSB-PGBD3 fusion protein could be regarded as a “recessive negative” because, unlike a “dominant negative,” the fusion protein is largely recessive to functional CSB (Horibata et al., 2011; Bailey et al., 2012) consistent with CS genetics (Nance and Berry, 1992; Laugel et al., 2010).
11. The N-terminal CSB and C-terminal PGBD3 domains of the CSB-PGBD3 fusion protein can independently affect gene expression
We had expected that gene regulation by the CSB-PGBD3 fusion protein would require both the N-terminal CSB domain (functioning as the “effector” end of the molecule) and the C-terminal PGBD3 transposase domain (tethering the effector end to chromosomal MER85 elements) (Fig. 2). However, when CSB-null UVSSS1KO cells were stably transfected with the artificial chimeras CSB-LacI or eGFP-PGBD3, using the natural CSB-PGBD3 fusion protein as a positive control, we found that the N-terminal domain of CSB alone (in the CSB-LacI chimera) induced many of the same IFN-related genes as the fusion protein, while PGBD3 alone (in the eGFP-PGBD3 chimera) induced some of the same genes as the N-terminal CSB domain and the fusion protein (Gray et al., 2012; Fig. 11). These results were also consistent with our ChIP-seq evidence that the CSB-PGBD3 fusion protein does not regulate IFN-related genes by binding to nearby MER85 elements (Fig. 3, and Gray et al., 2012). The simplest, albeit provisional, interpretations of these data are that (1) the PGBD3 domain of the CSB-PGBD3 fusion protein may serve primarily to stabilize, localize, or deliver the N-terminal CSB domain to the cell; and (2) the PGBD3 domain may independently regulate some genes — perhaps by DNA wrapping or bending as seen for other DNA transposases (Montaño et al., 2012). However, the ability of the N-terminal CSB and C-terminal PGBD3 domains of the CSB-PGBD3 fusion protein to induce some of the same IFN-related genes currently defies explanation.
12. What do we know about the function of the N-terminal CSB domain?
The N-terminal CSB domain of the CSB-PGBD3 fusion protein has emerged as a central player that may be largely responsible for induction of the IFN response. Comparatively little is known about the role of the N-terminal domain in normal CSB functions, but it is clear that this domain can exert both positive and negative effects: (1) Intact CSB is required for efficient transcription of ribosomal RNA in vivo (Bradsher et al., 2002), and the stable N-terminal CSB fragment found in the compound heterozygote CS1AN interferes with rRNA transcription in vitro (Lebedev et al., 2008). Admittedly the N-terminal CSB domain of the CSB-PGBD3 fusion protein is somewhat larger than the stable CS1AN fragment (465 versus 341 residues), but the two may behave similarly. Thus loss of CSB, or the continued presence of the fusion protein in the absence of CSB, could cause the cachexia typical of CS (Nance and Berry, 1992) by impairing rRNA transcription as suggested by Bradsher et al. (2002). (2) UV-induced association of CSB with chromatin requires ATP-dependent relief of N-terminal autorepression (Lake, 2010), suggesting that the N-terminus regulates CSB chromatin remodeling activity. (3) Surprisingly, both the CSB-PGBD3 fusion protein as well as the stable N-terminal CSB fragments found in CS1AN (341 residues) and CS3PV (452 residues) inhibit repair of covalent complexes between DNA topoisomerase I and DNA (Horibata et al., 2011). Thus stable N-terminal CSB fragments and the CSB-PGBD3 fusion protein may be partially redundant in CS individuals who express both. (4) Using host cell reactivation (HCR) assays, we found that the CSB-PGBD3 fusion protein inhibits TC-NER of oxidative damage only slightly, but strongly synergizes with CSB to stimulate UV repair by 200–250% (Bailey et al., 2012; Fig. 2B). As HCR assays are difficult to interpret mechanistically, the safest conclusion may be that the fusion protein can, as might be expected for a protein that shares an N-terminal CSB domain of 465 residues, either help or hinder CSB-dependent TC-NER. And finally (5) given the role of acidic regions as transcriptional activators with the potential to modulate chromatin structure (Neumann et al., 2012), it is tempting to speculate that the acidic N-terminal domain of CSB in the CSB-PGBD3 fusion protein might facilitate PGBD3 transposition by opening the chromatin structure of donor elements or target sites. On the other hand, although the N-terminal domain of CSB is conserved, deletion of the 39 residue acidic region does not affect transcription-coupled repair (TC-NER) or general genome repair (GGR) of UV-induced crosslinks and bulky adducts (Sunesen et al., 2000).
13. Does the CSB-PGBD3 fusion protein confer increased resistance to infectious disease?
We speculated that the ability of the CSB-PGBD3 fusion protein to induce a constitutive IFN response may have increased basal resistance to viral, and perhaps even bacterial, fungal, protozoan, or multicellular parasitic infections (Bailey et al., 2012). However, this speculation is not easily tested because (1) the potential infectious agents are extraordinarily diverse and constantly changing over time and place (Hayward et al., 2011); (2) physiological traits like pathogen resistance are extremely difficult to quantify in natural populations (Kingsolver et al., 2001) and are surely multigenic (Ferris et al., 1983); (3) although evolutionary biologists agree that immunity is metabolically costly, and that too weak or too strong an immune response to any particular pathogen could be harmful or lethal (Colditz, 2008), very few evolutionary biologists have dared to tackle the role of natural selection in shaping the subtlety, flexibility, redundancy, and complexity of the innate and adaptive immune responses — a challenge that will ultimately require a systems biology approach (Tan et al., 2012); and finally (4) although the mouse might appear to be an unpromising experimental model because it lacks both an endogenous CSB-PGBD3 fusion protein as well as MER85 elements, it could serve as a true null background for examining the regulatory effects of the human CSB-PGBD3 fusion protein, or a murinized version of the fusion protein in which the human N-terminal CSB domain is replaced by the highly homologous mouse CSB sequence.
The elevated IFN response we propose here could be seen as Nature’s own form of IFN therapy. Oral or systemic IFN treatment has been shown to help combat viral infection, and combination therapy with pegylated interferon and ribavirin is the standard of care for treating chronic hepatitis C (HCV) infections (Beilharz et al., 2007); Wyles, 2012). Viral proteins can also suppress IFN signaling or expression, the best-studied examples being the HCV NS5A protease (Kumthip et al., 2012) and NS2 protease (Kaukinen et al., 2013). Thus IFN treatment may contribute to the success of HCV combination therapy precisely because the virus suppresses the normal cellular IFN response. An elevated IFN response — perhaps when CSB is depleted by stress or DNA damage — could buy time for the innate or adaptive immune system to fight the early stages of viral infection, or to compensate for viral suppression of the IFN response later in infection.
14. A role for the CSB-PGBD3 fusion protein in CS is not inconsistent with the genetics of Cockayne syndrome
Our hypothesis that expression of the CSB-PGBD3 fusion protein in the absence of fully functional CSB may contribute to CS disease is not inconsistent with CS genetics. CSB defects are responsible for 70% of CS disease, CSA defects for 20%, and XPB, XPD, and XPG defects for the remaining 10% (Laugel et al., 2010). Over 90% of all CS patients with known CSB mutations are capable of expressing the CSB-PGBD3 fusion protein from at least one allele in the absence of functional CSB (Laugel et al., 2010); and the remaining 30% of CS patients with mutations in CSA, XPB, XPD, and XPG all express the fusion protein but under conditions where CSB cannot function fully in TC-NER. Thus about 90% of all CS patients continue to express the CSB-PGBD3 fusion protein under conditions that potentially sequester CSB in inactive TC-NER complexes or otherwise affect CSB function. As we have argued elsewhere (Bailey et al., 2012), apparent exceptions may reflect the modifying effects of variants or mutations in other DNA repair genes (Karahalil, Bohr, and Wilson, 2012); the modifying effects of genetic background (or possibly environment) in CS, UV Sensitive syndrome (UVSS), and DeSanctis-Cacchione syndrome patients with similar or identical mutations (Horibata et al., 2004; Laugel et al., 2008; Colella et al., 2000); or the observation that nearly half of all CS patients are consanguineous or descended from small founder populations (Laugel et al., 2010). In fact, it may be useful to regard the CSB-PGBD3 fusion protein as a part of the genetic background that could be responsible for the unusual clinical heterogeneity of CS disease (Nance and Berry, 1992).
15. Summary
The fixation of the CSB-PGBD3 fusion protein in the common ancestor of humans and marmosets, and subsequent conservation of the fusion protein for about 43 Mya, strongly suggest that the fusion protein is advantageous in healthy individuals. The large size of the N-terminal CSB domain (52 kDa) that is shared by CSB and the CSB-PGBD3 fusion protein also suggests (but does not guarantee) that the fusion protein will mimic, modulate, or functionally interact with CSB; and any protein that affects CSB function in health could, in principle, affect loss of CSB function in CS disease. The ability of the CSB-PGBD3 fusion protein to induce a constitutive IFN response in CSB-null UVSS1KO cells suggests that the fusion protein may have increased levels of immune defense against viruses and possibly bacterial or multicellular pathogens. Although the CSB-PGBD3 fusion protein binds to dispersed MER85 elements through the C-terminal piggyBac transposase domain, it regulates nearby genes primarily when tethered to transcription factors such as the activators c-Jun and TEAD1, and the silencer CTCF, apparently through the N-terminal CSB domain. However, the genes that are regulated by chromatin-bound CSB-PGBD3 fusion protein exhibit only a weak IFN-gamma signature. We therefore speculate that the fusion protein induces the IFN response not by upregulating specific genes, but by interacting with CSB-dependent chromatin remodeling functions — perhaps leading to bidirectional transcription, generation of dsRNA, and engagement of intracellular RNA sensors such as endosomal TLR3, 7, and 8, or cytoplasmic RIG-I and MDA5.
Brooks et al. (2008) were the first to notice that three different childhood neuropathies — Cockayne syndrome, trichiothiodystrophy (TTD), and Aicardi-Goutières syndrome (AGS) — exhibit potentially related forms of cerebral calcification and abnormal myelination (Brooks et al., 2008; but see Livingston et al., 2012). AGS is caused by a constitutive type I interferon response or “interferonopathy” resulting from defects in any of 5 genes (Crow, 2011; Rice et al., 2012: Stetson, 2012), whereas functional CSB protein and the CSB-PGBD3 fusion protein can both induce interferon-related viral defense genes depending on genetic background (Section 7). We are intrigued by the similarities between CS and AGS, and further speculate that an inappropriate interferon response may contribute to the heterogeneous clinical presentation of CS.
Highlights.
Cockayne syndrome (CS) is a devastating progeria usually caused by CSB mutations.
A piggyBac transposon inserted into the primate CSB gene over 43 million years ago.
The CSB gene now encodes both the CSB protein and a CSB-piggyBac fusion protein.
The CSB-PGBD3 fusion protein induces an interferon response in CSB-null cells.
We speculate that an inappropriate interferon response contributes to CS disease.
Acknowledgments
We thank P. J. Brooks for encouragement, and for first pointing out the potential similarities between Aicardi-Goutières and Cockayne syndrome (Brooks et al., 2008). The work described in this review was supported by NIH awards R01 GM41624 (AMW) and the Cell and Molecular Biology Training Program T32 GM007270 (LTG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anindya R, Mari PO, Kristensen U, Kool H, Giglia-Mari G, Mullenders LH, Fousteri M, Vermeulen W, Egly JM, Svejstrup JQ. A ubiquitin-binding domain in Cockayne syndrome B required for transcription-coupled nucleotide excision repair. Mol Cell. 2010;38:637–648. doi: 10.1016/j.molcel.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AD, Gray LT, Pavelitz T, Newman JC, Horibata K, Tanaka K, Weiner AM. The conserved Cockayne syndrome B-piggyBac fusion protein (CSB-PGBD3) affects DNA repair and induces both interferon-like and innate antiviral responses in CSB-null cells. DNA Repair (Amst) 2012;11:488–501. doi: 10.1016/j.dnarep.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz MW, Cummins JM, Bennett AL. Protection from lethal influenza virus challenge by oral type 1 interferon. Biochem Biophys Res Commun. 2007;355:740–744. doi: 10.1016/j.bbrc.2007.02.019. [DOI] [PubMed] [Google Scholar]
- Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- Bradsher J, Auriol J, Proietti de Santis L, Iben S, Vonesch JL, Grummt I, Egly JM. CSB is a component of RNA pol I transcription. Mol Cell. 2002;10:819–829. doi: 10.1016/s1097-2765(02)00678-0. [DOI] [PubMed] [Google Scholar]
- Brooks PJ, Cheng TF, Cooper L. Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair (Amst) 2008;7:834–848. doi: 10.1016/j.dnarep.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci U S A. 2009;106:9373–9378. doi: 10.1073/pnas.0903487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon H, Yang J, Stark GR. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J Interferon Cytokine Res. 2011;31:33–40. doi: 10.1089/jir.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colditz IG. Six costs of immunity to gastrointestinal nematode infections. Parasite Immunol. 2008;30:63–70. doi: 10.1111/j.1365-3024.2007.00964.x. [DOI] [PubMed] [Google Scholar]
- Colella S, Nardo T, Mallery D, Borrone C, Ricci R, Ruffa G, Lehmann AR, Stefanini M. Alterations in the CSB gene in three Italian patients with the severe form of Cockayne syndrome (CS) but without clinical photosensitivity. Hum Mol Genet. 1999;8:935–941. doi: 10.1093/hmg/8.5.935. [DOI] [PubMed] [Google Scholar]
- Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238:91–98. doi: 10.1111/j.1749-6632.2011.06220.x. [DOI] [PubMed] [Google Scholar]
- Ferris SD, Sage RD, Huang CM, Nielsen JT, Ritte U, Wilson AC. Flow of mitochondrial DNA across a species boundary. Proc Natl Acad Sci U S A. 1983;80:2290–2294. doi: 10.1073/pnas.80.8.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray LT, Weiner AM. Ubiquitin recognition by the Cockayne syndrome group B protein: binding will set you free. Mol Cell. 2010;38:621–622. doi: 10.1016/j.molcel.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Gray LT, Fong KK, Pavelitz T, Weiner AM. Tethering of the Conserved piggyBac Transposase Fusion Protein CSB-PGBD3 to Chromosomal AP-1 Proteins Regulates Expression of Nearby Genes in Humans. PLoS Genet. 2012;8:e1002972. doi: 10.1371/journal.pgen.1002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S, Suga T, Kudo E, Ihn H, Uchino M, Tateishi S. Adult-onset neurological degeneration in a patient with Cockayne syndrome and a null mutation in the CSB gene. J Invest Dermatol. 2008;128:1597–1599. doi: 10.1038/sj.jid.5701210. [DOI] [PubMed] [Google Scholar]
- Hayward AD, Wilson AJ, Pilkington JG, Clutton-Brock TH, Pemberton JM, Kruuk LE. Natural selection on a measure of parasite resistance varies across ages and environmental conditions in a wild mammal. J Evol Biol. 2011;24:1664–1676. doi: 10.1111/j.1420-9101.2011.02300.x. [DOI] [PubMed] [Google Scholar]
- Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101:15410–15415. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horibata K, Saijo M, Bay MN, Lan L, Kuraoka I, Brooks PJ, Honma M, Nohmi T, Yasui A, Tanaka K. Mutant Cockayne syndrome group B protein inhibits repair of DNA topoisomerase I-DNA covalent complex. Genes Cells. 2011;16:101–114. doi: 10.1111/j.1365-2443.2010.01467.x. [DOI] [PubMed] [Google Scholar]
- Karahalil B, Bohr V, Wilson D., 3rd Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk. Hum Exp Toxicol. 2012;31:981–1005. doi: 10.1177/0960327112444476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukinen P, Sillanpaa M, Nousiainen L, Melen K, Julkunen I. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J Med Virol. 2013;85:71–82. doi: 10.1002/jmv.23442. [DOI] [PubMed] [Google Scholar]
- Keith JH, Schaeper CA, Fraser TS, Fraser MJ., Jr Mutational analysis of highly conserved aspartate residues essential to the catalytic core of the piggyBac transposase. BMC Mol Biol. 2008;9:73. doi: 10.1186/1471-2199-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsolver JG, Hoekstra HE, Hoekstra JM, Berrigan D, Vignieri SN, Hill CE, Hoang A, Gibert P, Beerli P. The strength of phenotypic selection in natural populations. Am Nat. 2001;157:245–261. doi: 10.1086/319193. [DOI] [PubMed] [Google Scholar]
- Kumthip K, Chusri P, Jilg N, Zhao L, Fusco DN, Zhao H, Goto K, Cheng D, Schaefer EA, Zhang L, Pantip C, Thongsawat S, O’Brien A, Peng LF, Maneekarn N, Chung RT, Lin W. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J Virol. 2012;86:8581–8591. doi: 10.1128/JVI.00533-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake RJ, Geyko A, Hemashettar G, Zhao Y, Fan HY. UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol Cell. 2010;37:235–246. doi: 10.1016/j.molcel.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Laugel V, Dalloz C, Stary A, Cormier-Daire V, Desguerre I, Renouil M, Fourmaintraux A, Velez-Cruz R, Egly JM, Sarasin A, Dollfus H. Deletion of 5′ sequences of the CSB gene provides insight into the pathophysiology of Cockayne syndrome. Eur J Hum Genet. 2008a;16:320–327. doi: 10.1038/sj.ejhg.5201991. [DOI] [PubMed] [Google Scholar]
- Laugel V, Dalloz C, Tobias ES, Tolmie JL, Martin-Coignard D, Drouin-Garraud V, Valayannopoulos V, Sarasin A, Dollfus H. Cerebro-oculo-facio-skeletal syndrome: three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J Med Genet. 2008b;45:564–571. doi: 10.1136/jmg.2007.057141. [DOI] [PubMed] [Google Scholar]
- Laugel V, Dalloz C, Durand M, Sauvanaud F, Kristensen U, Vincent MC, Pasquier L, Odent S, Cormier-Daire V, Gener B, Tobias ES, Tolmie JL, Martin-Coignard D, Drouin-Garraud V, Heron D, Journel H, Raffo E, Vigneron J, Lyonnet S, Murday V, Gubser-Mercati D, Funalot B, Brueton L, Sanchez Del Pozo J, Munoz E, Gennery AR, Salih M, Noruzinia M, Prescott K, Ramos L, Stark Z, Fieggen K, Chabrol B, Sarda P, Edery P, Bloch-Zupan A, Fawcett H, Pham D, Egly JM, Lehmann AR, Sarasin A, Dollfus H. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum Mutat. 2010;31:113–126. doi: 10.1002/humu.21154. [DOI] [PubMed] [Google Scholar]
- Lebedev A, Scharffetter-Kochanek K, Iben S. Truncated Cockayne syndrome B protein represses elongation by RNA polymerase I. J Mol Biol. 2008;382:266–274. doi: 10.1016/j.jmb.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Li X, Ewis H, Hice RH, Malani N, Parker N, Zhou L, Feschotte C, Bushman FD, Atkinson PW, Craig NL. A resurrected mammalian hAT transposable element and a closely related insect element are highly active in human cell culture. Proc Natl Acad Sci U S A. 2012;109 doi: 10.1073/pnas.1121543109. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Stivaros S, MSVDK, Crow YJ. Recognizable phenotypes associated with intracranial calcification. Dev Med Child Neurol. 2012;55:46–57. doi: 10.1111/j.1469-8749.2012.04437.x. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, Fain-Thornton J, Craig NL. piggyBac can bypass DNA synthesis during cut and paste transposition. Embo J. 2008;27:1097–1109. doi: 10.1038/emboj.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano SP, Pigli YZ, Rice PA. The Mu transpososome structure sheds light on DDE recombinase evolution. Nature. 2012;491:413–417. doi: 10.1038/nature11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- Neumann FR, Dion V, Gehlen LR, Tsai-Pflugfelder M, Schmid R, Taddei A, Gasser SM. Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev. 2012;26:369–383. doi: 10.1101/gad.176156.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, Weiner AM. L2L: A simple tool for discovering the hidden signficance in microarray expression data. Genome Biol. 2005;6:R81. doi: 10.1186/gb-2005-6-9-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103:9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, Bailey AD, Fan HY, Pavelitz T, Weiner AM. An abundant evolutionarily conserved CSB-PiggyBac fusion protein expressed in Cockayne syndrome. PLoS Genet. 2008;4:e1000031. doi: 10.1371/journal.pgen.1000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- Niwa M, Rose SD, Berget SM. In vitro polyadenylation is stimulated by the presence of an upstream intron. Genes Dev. 1990;4:1552–1559. doi: 10.1101/gad.4.9.1552. [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, Del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB. Endogenous retroelements and autoimmune disease. Curr Opin Immunol. 2012;24:692–697. doi: 10.1016/j.coi.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunesen M, Selzer RR, Brosh RM, Jr, Balajee AS, Stevnsner T, Bohr VA. Molecular characterization of an acidic region deletion mutant of Cockayne syndrome group B protein. Nucleic Acids Res. 2000;28:3151–3159. doi: 10.1093/nar/28.16.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Pan R, Qiao L, Zou X, Pan Z. Modeling and dynamical analysis of virus-triggered innate immune signaling pathways. PLoS One. 2012;7:e48114. doi: 10.1371/journal.pone.0048114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pluijm I, Garinis GA, Brandt RM, Gorgels TG, Wijnhoven SW, Diderich KE, de Wit J, Mitchell JR, van Oostrom C, Beems R, Niedernhofer LJ, Velasco S, Friedberg EC, Tanaka K, van Steeg H, Hoeijmakers JH, van der Horst GT. Impaired genome maintenance suppresses the growth hormone--insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2007;5:e2. doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One. 2012;7:e32542. doi: 10.1371/journal.pone.0032542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S, Kaminski JM. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A. 2006;103:15008–15013. doi: 10.1073/pnas.0606979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyles DL. Beyond telaprevir and boceprevir: resistance and new agents for hepatitis C virus infection. Top Antivir Med. 2012;20:139–145. [PMC free article] [PubMed] [Google Scholar]
- Yu A, Fan HY, Liao D, Bailey AD, Weiner AM. Activation of p53 or loss of the Cockayne syndrome group B repair protein causes metaphase fragility of human U1, U2, and 5S genes. Mol Cell. 2000;5:801–810. doi: 10.1016/s1097-2765(00)80320-2. [DOI] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]