

Abstract
There is a strong and growing literature showing that key aspects of brain development may be critical antecedents of adult physiology and behavior, or lead to physiological and psychiatric disorders in adulthood. Many are significantly influenced by sex-dependent factors. Neurons of the paraventricular nucleus of the hypothalamus (PVN) occupy a key position in regulating homeostatic, neuroendocrine, and behavioral functions. This brain area is a critical link for our understanding of the etiology of a number of disorders with components ranging from mood, to feeding and energy balance, and autonomic nervous system regulation. Thus based on common brain circuitry, the PVN may be a critical anatomical intersection for understanding comorbidities among depression, obesity, and cardiovascular risk. Historically, the majority of approaches to brain development examine neuronal, glial, and vascular factors independently, with notably less emphasis on vascular contributions. The realization that the PVN undergoes a unique vascular developmental process places added value on discerning the cellular and molecular mechanisms that drive its late onset angiogenesis and further implications for neuronal differentiation and function. This has ramifications in humans for understanding chronic, and sometimes fatal, comorbidities that share sex-dependent biological bases in development through functional and anatomical intersections with the hypothalamus.
Keywords: depression, cardiovascular disease, sex differences, hypothalamus, prenatal stress, comorbidity
Introduction
The comorbidity of major depressive disorder (MDD) and risk for cardiovascular disease (CVD) has a prevalence of approximately 20% [5, 25, 48, 76] and is expected be the leading cause of disability worldwide by 2020 [68, 90]. Although CVD is generally considered a “man’s disease”, since the overall rate is higher in men [59], in fact, CVD is the number one cause of death in women in the U.S., and the comorbidity of MDD and CVD is twice the rate in women than men [27, 67]. MDD has a significantly higher prevalence in women (2-fold) than men [51, 52] and MDD is an independent risk factor for the development and progression of coronary artery disease [5, 25, 50, 69]. Numerous prospective studies indicate significantly elevated risks of coronary heart disease, myocardial infarction, or cardiac death among participants with depression [74, 75, 91]. Depression predicts first cardiovascular events even among otherwise healthy people, particularly in women [91], with a risk of 1.5 to 6-fold.
Obesity is associated with MDD and CVD, although the direction of effects is controversial. Elevated body mass index (BMI) is significantly associated with anhedonia and depression, particularly in women, even controlling for other demographic variables [6, 13, 36]. Individuals with MDD typically have a higher BMI prior to onset of depressive symptoms and a history of weight fluctuation, with some evidence that these individuals, particularly women, demonstrate increased appetite, over-eating, and craving of carbohydrates, particularly in response to stress [7, 79, 82].
Although population-level studies have demonstrated the substantial sex differences in comorbidities with major public health implications worldwide, the pathways to explaining comorbidity is unclear. In part, this may be due to a lack of investigative focus in general on explaining sex differences in diseases. However, it also may be due to the fact that many investigators studying the heart and associated vascular system and/or adiposity rarely think about the brain and neuroscience perspectives and vice versa. Moreover, studies focused on CVD and MDD generally begin in adulthood.
This review argues the position that sex differences in MDD-CVD comorbidity (and associated metabolic syndrome disorders arising from conditions such as obesity) originate in part from pathogenic processes initiated in fetal development that involve shared pathophysiology between the brain, the vascular system, and the CNS control of the heart, food intake and energy balance. Fetal origins of MDD, CVD and obesity independently implicate “prenatal stress models” of hypothalamic-pituitary-adrenal (HPA) axis circuitry disruption. At the population-level, there is higher risk in women regarding shared fetal antecedents to comorbidity of MDD and poor ANS deficits (a significant CVD risk factor) [27], which implicates prenatal inflammatory and adrenal hormonal abnormalities. At the in vivo brain imaging level, fetal disruptions of HPA circuitry development are significantly associated with sex differences in adult brain activity deficits and hormonal dysregulation in MDD alone [28], that, in pilot work, were significantly associated with ANS dysregulation [41]. Previous work on the fetal programming of CVD risk alone, although not focused on sex differences, suggested adverse fetal exposures cause HPA abnormalities, elevated blood pressure and blood glucose levels, implicating glucocorticoid receptors [77, 94]. Much of the work in model animals, including our own [12, 23, 65, 101], demonstrated possible pathways in MDD involve disruption of maternal gestational glucocorticoids on nerve and angiogenic growth factors [brain derived nerve growth factor (BDNF/trkB), vascular endothelial and insulin growth factors (VEGF and IGF-1)], gonadal hormones, and gamma aminobutyric acid (GABA), on neuronal and vascular development of HPA axis regions, such as the hypothalamic paraventricular nucleus (PVN). The PVN, which is one of the most highly vascularized regions in the brain [1], is important for regulating many homeostatic, neuroendocrine, and behavioral functions and has been associated with the etiology of affective disorders, such as MDD. Furthermore, the PVN is an essential component of brain circuitries important for feeding and energy balance and serves to regulate the autonomic nervous system which is critical for appropriate cardiovascular responses. Thus, the PVN may lie at a critical crossroad for understanding comorbidities among depression, cardiovascular disease and related metabolic syndromes associated with obesity.
The neurobiological model proposed here (Figure 1, above) to explain sex differences in MDD-CVD comorbidity is that excess maternal gestational glucocorticoids (an indicator of “prenatal stress”) disrupt GABA signaling in conjunction with growth factors (VEGF, IGF-1, and BDNF) and gonadal hormones leading to sex-specific alterations in neuronal and vascular development in HPA axis brain regions (such as the PVN) that are sexually dimorphic and implicated in MDD and CVD risk through the ANS and the vasculature. Given the substantial comorbidity worldwide, a common fetal cause will have important implications for prevention or attenuation of disability in many countries, particularly in women.
Figure 1. Prenatal Stress Model of Sex-Dependent Risk for Comorbidity.
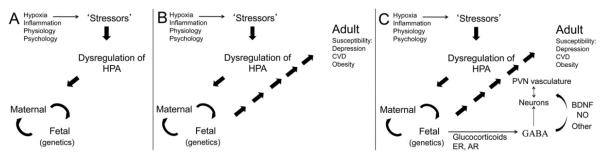
This figure shows schematically the progressive influence of external stressors to the mother on her HPA axis function (A), in addition to maternal-fetal interactions to influence adult susceptibility to disorder (B). Mechanisms that may play roles in the fetal compartment include steroid hormone receptors that could impact sex differences (estrogen and androgen receptors (ER and AR) and glucocorticoids (and their receptors). These transcription factors may impact the function of neurotransmitters (e.g., GABA), neuromodulators (Nitric oxide, NO), or growth factors (e.g., BDNF, VEGF, IGF) on neuronal, glial, or vascular compartments in the PVN.
The problem of comorbidity
It is common to consider various diseases or disorders from unitary perspectives, i.e., so-called “silo’ed investigative approaches” within specific fields of medicine. The missions of the National Institutes of Health are predicated on the unitary approach to the study of a number of disorders. For example, the National Institute of Mental Health (NIMH) focuses on depressive disorders, while the National Institute of Diabetes and Digestive and Kidney diseases (NIDDK) focuses on obesity and diabetes, and the National institute of Heart, Lung & Blood (NIHLB) focuses on cardiovascular diseases. It is likely that there are instances where these various disorders occur due to independent etiologies. However, the central theme of this review is that the greater risk, and perhaps more prevalent problem and greater expense to the healthcare system, lies in the comorbidity of one or more disorders that are found together in symptom clusters. This view of shared pathophysiology has been espoused by NIMH [44], although this view is restricted to disorders of the brain and not the comorbidites of brain disorders with general medical disorders. This review addresses the comorbidity of MDD and CVD and associated syndromes with emphasis on sex differences in the risk of each, underscoring the fact that the comorbidity of these disorders occurs more commonly in women.
In considering developmental origins of disorders that cluster together, there are at least two major etiological pathways to consider. One way is to postulate a general linear model in which one disorder helps trigger another one, resulting in several disorders in the same individual (see Figure 2a). For example, depression might arise - cause unknown, leading to overeating and obesity as downstream consequences. Obesity might then cause a cascade of medical problems leading to CVD. In a similar way, the sequence might start with obesity leading to depression, which may in turn lead to cardiovascular risk. Just as likely is that a cardiovascular problem that would lead to depression and then subsequent obesity. An alternative possibility to a linear model is a nonlinear model which is at the heart of this review. The perspective of this review is that all of these disorders have a shared biological substrate that provides a basis for an increased likelihood of several disorders arising in the same individual (see Figure 2b). Shared biological substrates could be envisioned on at least three levels: anatomical, molecular/biochemical, and genetic.
Figure 2. Sequential (A) versus Shared (B) Biological Pathways to Comorbidity.
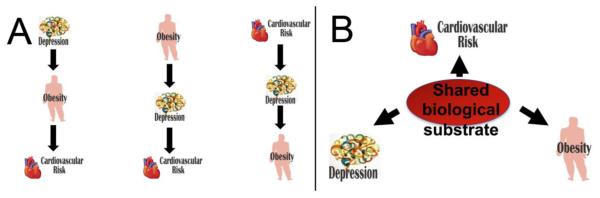
Panel A illustrates sequential pathways to comorbidity in which one disorder may lead to others. The disorders are such that any one of them may be able to trigger steps towards the others. Panel B illustrates a shared biological pathway to comorbidity in which an underlying biological problem can contribute to the onset of each of the disorders at the same time.
Anatomy as a shared biological substrate for causation
If one were to go through a list of brain structures or regions to identify those that are linked to a constellation of disorders that included depression, obesity, and cardiovascular risk, it is likely that the hypothalamus would be one of the key identified regions. It is also likely that the list would include the amygdala and/or the hippocampus. These are not disconnected regions, but rather players in a key brain circuit described many years ago by Papez [71]. However, only recently has this circuit become accessible to analysis by human in vivo MRI techniques that have demonstrated its importance in a number of disorders of the brain [31, 32, 40, 42, 62].
Of particular interest for this review, one location within the hypothalamus is notable for its role in signaling from the brain directly to the periphery. This locus, the PVN, contains neurons that control behavior, neuroendocrine and autonomic function. Specifically, neurons in regions of the PVN direct the hypothalamic-pituitary-adrenal axis (HPA) that controls much of what is considered the stress response. Other neurons in the PVN are considered preautonomic and project to brainstem and spinal cord areas that control the autonomic nervous system. Still other PVN neurons project to sites that can control behaviors, such as feeding. These features are discussed further below.
The PVN lies at the dorsal limit of the classical hypothalamus flanking the top of the third ventricle. It has been implicated in a broad array of homeostatic and behavioral functions ranging from neuroendocrine and cardiovascular functions to affective, ingestive, and defensive behaviors [reviewed 38, 85]. Depending upon the species, functional groups of PVN neurons can be defined based on cellular characteristics, position, or chemical phenotype. Each of these subdivisions has been associated with specific functions (e.g., [10]). While there is some controversy regarding the degree to which the PVN may be subdivided in humans, it seems likely that subdivisions exist based on several criteria [53, 62]. This is consistent with findings of subgroup-selective cell loss in particular human disorders (e.g., [4, 9]).
Neurons of the PVN express a number of receptors for steroid hormones and from this arises the potential for sex differences in PVN function that could be driven by circulating sex steroid hormones. Moreover, steroid hormone receptors are among the markers that indicate different zones within the PVN.
For example, cell groups reportedly contain immunoreactive estrogen receptors-α (ir-ERα), ERβ, androgen (AR) or glucocorticoid (GR) receptors [e.g., 80, 83, 84]. Immunoreactive neurotransmitters [e.g., corticotropin releasing hormone (CRH), arginine vasopressin (AVP), oxytocin (OXY) [2, 85]], and other proteins, including calbindin and nNOS [65], also characterize specific regions of the nucleus (see Figure 3). To the extent that the neurons expressing these molecules are stereotypic for particular zones in the developing PVN and controlled by key developmental factors they may be useful biomarkers for future analyses of pathology as well as normal function.
Figure 3. Molecular Complexity of PVN Neurons.

Developmental sequence for Nissl (thionin stain) architecture of the paraventricular nucleus of the hypothalamus (PVN) in mice (left) is contrasted with the complex molecular heterogeneity of PVN neurons across the nucleus (right). The molecular phenotypes of many PVN neurons are already in place by embryonic day (E)15, even though the nucleus is not yet viewable based on cell density (left). The PVN emerges from the background of cells along the third ventricle (v) as a cell-poor zone opens up around the nascent nucleus between E15 and 17. By birth at postnatal (P)0, the medial to lateral extent of the PVN on one side is about 400μm.
Defects in the healthy development of the PVN provide an anatomical basis to predict shared comorbidity for disorders related to the functions of neurons normally located in or around the PVN. A number of mechanisms may lead to potential long-term alterations in PVN function that may or may not also be sex-dependent. These include changes in gene expression, cell death, connectivity, neuronal phenotypes or positions thereof, or relationships to unique vasculature. Further, environmental stimuli in development (e.g., obstetric complications or chronic social/psychological stress) may cause changes in gene expression within neurons of the PVN, in response to a common circulating factor (such as glucocorticoids), and perhaps as a function of epigenetic marks that are placed on DNA in response to such stimuli [11, 12]. For example, we and others demonstrated that perinatal exposure of rats to the synthetic glucocorticoid, dexamethasone, change the methylation state of the BDNF gene [11, 43] and/or the levels of preproTRH [12]. In a similar fashion, levels of maternal behavior can alter the adult expression of glucocorticoid receptor [93] and estrogen receptor [16]. In many cases, such influences may be sex-dependent with responses more prevalent in females.
Disruptive events in development can also lead to apoptotic (or possibly necrotic) cell death within specific subdivisions of the PVN or the neighboring region [101]. Changes in the incidence of cell death, whether increases or decreases, can have long lasting effects on neural circuitry. For example, it has been demonstrated that prenatal exposure of rats to stress caused an increase in cell death in the fetal PVN [89] with greater levels in the female. Correspondingly, fetal exposure to dexamethasone caused increases in apoptotic cell numbers in areas that project to the PVN, such as CA1, CA3 and peri-PVN [101]. There are many connections into the PVN and the surrounding region, and increased or decreased stimulation along any of these pathways either during development or relative to later plasticity, may cause long-term changes in PVN function. This can be due to changes in inputs from intra- or extra-hypothalamic areas, or changes in synaptogenesis in the PVN itself. For example, chronic variable stress may impact synapse formation in the PVN [22] that might also be reflected as sex differences in synaptophysin containing terminals [14].
Neurogenesis and neuron migration are classic mechanisms for neural development in general. Specific stimuli may cause changes in the location of neuron phenotypes within the PVN because of altered birth or migration [65]. For example, we demonstrated that loss of GABAB receptor function by either genetic or pharmacological means (during fetal development) resulted in cells that express estrogen receptor alpha being located more laterally in the PVN in females only. Such a change (like for phenotype or apoptosis) might be useful in predicting alterations in PVN-mediated functions that underlie the development of a sex-related disorder. Interestingly, the loss of GABAB receptor functioning also disrupted the positions of cells containing immunoreactive estrogen receptor alpha in and around the ventromedial nucleus [64], but not in a sex-dependent manner. These findings suggest the importance of developmental timing and region-specificity for identification of sex-specific outcomes.
The vast majority of approaches to brain development examine neuronal, glial, and vascular factors independently, with notably less emphasis on vasculogenesis. Surprisingly, the extensive vascularity of the PVN [1, 92] occurs late in hypothalamic development. Thus, changes in vascular density or function within the PVN [23] provide another anatomical substrate to help explain shared comorbidities. The healthy development of brain vasculature proceeds through the invasion of vascular sprouts from the pial vessels towards the central ventricles. Subsequently, finer branches form via secondary angiogenic processes. Hypoxia is considered a major driving force in promoting the formation of new vessels. However, for PVN development, the region is fully vascularized at birth similar to the rest of the forebrain. By the second postnatal (P) week, a striking increase in vascularization is readily discernible [23, 66]. Whatever drives the increased vascularity does so over a period of time when the PVN receives similar blood flow as the rest of the hypothalamus and presumably sufficient oxygen. Thus, postnatal angiogenesis in the PVN (which is associated with later prenatal human development) may be driven by neural signals as suggested by the term “angioneurins” [99], but intrinsic to the unique environment and components of the PVN. Our preliminary studies in mice (Frahm, Zhang, & Tobet, unpublished observations) suggest that the mRNAs of known angiogenic factors or receptors, such as vascular endothelial growth factor (VEGF), BDNF and the BDNF receptor TrkB, have a postnatal developmental time course in the neonatal PVN in rodents (and prenatal in humans) consistent with a role in driving angiogenesis. Important for the PVN, is the fact that neural activity may impact the development of the vasculature, since GABAB receptor signaling caused a 20% decrease in vascular characteristics of length or branch points [23].
Studies of the neurovascular unit suggest that the blood brain barrier (BBB) may be a variable that may be particularly important in highly vascularized regions like the PVN. Given the several-fold greater density of blood vessels in the PVN, any disruption of BBB function will make the PVN appear selectively vulnerable compared to many other brain regions. In development, there is debate as to when the BBB ‘closes’ or begins to regulate the flow of macromolecules into and out of the brain parenchyma. In general, significantly more information is needed on the maturation and regulation of BBB function in all brain areas, including the PVN. Studies are just beginning to illuminate regulators of BBB development in the region of the PVN. Our results suggest that perinatal GABAergic [23] or glucocorticoid (Frahm and Tobet, unpublished results) treatments may influence BBB development. Evidence currently exists for both sex-dependent glucocorticoid [73] and reproductive hormone [96] influences on some aspects of BBB function, such as permeability and expression of molecular pumps.
Molecular players as shared biological substrate for causation
Hypothalamic-pituitary-adrenal axis (HPA)
The HPA axis is likely best known for its role in controlling neuroendocrine stress responses [45]. Simply put, it consists of a series of feedback loops that can be studied from an anatomical and clinical basis as well as for its molecular signaling properties. Brain regions implicated in modulating stress response circuitry provide inputs to the PVN, the neuroendocrine motor pathway for the HPA axis. The brain regions that contribute to the higher order control of HPA axis function include subregions of the amygdala, hippocampus, periaqueductal gray, medial and orbital prefrontal cortices, and anterior cingulate cortex. In brief, the regulation of the HPA axis response to a number of different stressful stimuli are gated through these various brain regions, which we demonstrated in vivo in humans using functional MRI [29, 30]. Ultimately, CRH is secreted from parvocellular PVN neurons into the hypothalamo-hypophyseal portal capillaries of the median eminence and travel to the pituitary to regulate the secretion of ACTH from pituitary corticotrophs. Although CRH has been considered the principal secretagogue driving anterior pituitary ACTH secretion, studies have shown that AVP can be co-released from neuroendocrine parvocellular CRH neurons to amplify the actions of CRH on pituitary release of ACTH [95]. Once secreted into the general circulation, ACTH drives the adrenal cortex to secrete the principal glucocorticoid hormone, cortisol in humans, and corticosterone in rodents and many other vertebrates.
By causing a rapid rise in plasma glucocorticoid concentrations, stress has long been considered to play a major role in causing adrenal hypertrophy, energy mobilization, alterations in the immune system, and gastrointestinal problems [78, 86]. In concert, rapid activation of pre-autonomic neurons in the PVN drives chromaffin cells in the adrenal medulla to rapidly release the catecholamines, epinephrine and norepinephrine, into the general circulation. These factors are also involved in the characteristic fight or flight response to stress and rapidly impact heart rate, blood pressure, and smooth muscle functions throughout the body [41]. Further, glucocorticoid release also drives adrenal medullary synthesis of the phenylethanolamine N-methyltransferase (PNMT), which is the key enzyme responsible for transforming norepinephrine to epinephrine. The combination of glucocorticoids and epinephrine secreted in the bloodstream generate diverse responses throughout the body.
In addition to the negative feedback responses at pituitary and brain levels, glucocorticoids have many other roles in brain [39, 57]. Glucocorticoid actions in brain may be impacted by sex and/or interactions with hormones of the hypothalamic-pituitary-gonadal axis [11, 21, 81]. In fact, at the human in vivo brain imaging level, gonadal hormones regulate stress response circuitry, including anterior hypothalamic regions, in healthy individuals that contribute to explaining sex differences in the brain’s response to stress [29, 30] and were disrupted in depression in tandem with gonadal hormone deficits [42] and ANS dysregulation [41]. Taken together, animal and human studies of the stress response circuitry suggest that many of the influences of glucocorticoids on the brain are based in changes in “chemical anatomy” that are relevant for considering potential comorbidities.
It is at the level of molecular players that the idea of shared biological substrates becomes an amplified concept. For example, one of the first thoughts about what prenatal stress can do is to provoke a chemical response as outlined above. These chemical responses can have wide-ranging impact in the fetal compartment (see Figure 4). In that compartment, there are strong opportunities for interactions among molecular signaling pathways in different locations. In addition to CRH, central to stress aspects of the HPA axis, there are a number of other peptides found in the PVN that have been associated with either specific or multiple human disorders and have the potential to act as the biological bases of comorbidity. For example, vasopressin and oxytocin have been implicated in anxiety and depression [70], cardiovascular risk [33, 98], and metabolic issues related to obesity [87, 97] and eating disorders [56]. Alterations in TRH have been linked to depression [100] and CVD [54] in addition to metabolic problems [72]. In support of these findings in humans, our recent animal studies show that prenatal dexamethasone exposure can alter homologous TRH neurons in the PVN of adult rats [12].
Figure 4. Impact of Maternal Conditions on Sex-Dependent Brain Development and Outcomes.

This schematic illustrates the tripartite nature of maternal impact on fetal development resulting in an adult offspring that responds differentially to specific stimuli. Sex differences arise at multiple points in the process.
Genetics as a shared biological substrate for causation
As a shared biological substrate for causation, genetics can be considered a special case of molecular/biochemical. The search for genetic associations has typically been for specific disorders, although some have begun to investigate shared genetic pathways related to comorbidities [63]. One way to consider genetic predisposition is to consider mutations that either prevent the synthesis of a functional protein or more often provide for the synthesis of a protein with suboptimal function. For example, Val66Met is an amino acid substitution in BDNF that compromises, but does not eliminate BDNF signaling [17]. This amino acid substitution has been linked to multiple psychiatric disorders [47]. However, a recent meta-analysis suggests that by examining the literature as a whole, the linkage of BDNF variants alone to MDD may be less promising than previously thought [34], and may need to be thought of in the context of epigenetic effects of environmental exposures on the regulation of BDNF, as with studies of gene-environment interactions in MDD with the serotonin-receptor transporter or other genes and early childhood adversity [15, 49].
Combining biological substrates for causation
The simplest combination of causes for comorbidity might predict a location in the brain that participates in the regulation of several components of multiple disorders and/or has some unique molecular/biochemical properties that were partially accounted for by the expression of specific genes of interest. There is a long history of research on the interaction of HPA axis dysregulation and disorders from the perspective of fetal antecedents to these disorders that includes MDD, CVD and obesity [3, 26, 28, 35]. During brain development, glucocorticoids may influence GABAergic mechanisms, as has been shown in adulthood [61, 88]. There is a significant link between GABA and morphogenetic roles in brain development. Several neurotransmitter systems have been suggested as neurotrophic factors or morphogens in various brain regions [8, 55] including, GABA, serotonin, dopamine, and endogenous opiates. Important for the current discussion, defects in GABA signaling have been found in animal models of depression [18, 19, 60] in addition to humans with MDD [24] and CVD [58]. Moreover, as we have demonstrated, the distribution of GABAergic elements may be essential for the final cytoarchitectural arrangement of cellular elements in the region of the PVN [65]. Viewed sequentially (as in Figure 1), exposure to stressors of any type during development may cause activation of the HPA axis (Figures 1a, 4). This will lead to a number of humoral signals exchanged between the maternal and fetal compartments that ultimately result in changes in brain structure and function leading to adult susceptibility to disorders. These include depression, cardiovascular risk or metabolic imbalances leading to obesity (Figures 1b, 4). Potential mechanisms of action operating in the fetal compartment include the interactions of steroid hormone receptors regulating responses to neurotransmitters (e.g., GABA) or growth factors (e.g., BDNF, VEGF, IGF) in the context of neurons, glia, or vasculature in the PVN (Figures 1c, 4).
Currently, there is reasonable evidence to implicate several key factors in the etiology of comorbid disorders. From the anatomical perspective, the PVN occupies a notable location that mediates functions at the heart of a number of disorders. It is the site in the brain where neurons involved in controlling HPA function lose negative feedback control in major depressive disorder. It may be interesting to note that other areas of focus for comorbidities include the hippocampus, amygdala, and portions of cerebral cortex that also signal transynaptically to regulate PVN activity, as demonstrated in our studies activating this circuitry in vivo [29, 30, 42]. From the molecular perspective, brain GABA and stress-related release of glucocorticoids have notable ties to fetal antecedent actions with long-term consequences for neuronal circuitry and function. From the genetic perspective, it is difficult to find strong gene linkages to particular disorders. However, there have been several studies to connect alterations in GABA signaling genes, glucocorticoid signaling related genes, and the BDNF gene to increased likelihood of several disorders. There is currently much less evidence relating changes in angiogenic genes to multiple disorders, but the number of studies is growing as investigators learn to specifically focus on such markers. Viewing the evidence through a prism that highlights sex differences will likely help clarify results that may be conflicting or insufficiently powerful because genetic sex or differences in sex steroid hormones were not considered in the model.
Acknowledgements
The ideas in this review emanated from research on a translational (human to animal) program project that was supported by the Office for Research on Women’s Health (ORWH) and National Institute of Mental Health (ORWH-NIMH P50 MH082679; Goldstein, Tobet, Handa, PIs; http://mddscor.bwh.harvard.edu). Additionally, it was in part supported by NIMH-NHLBI R01 MH074679 (Goldstein, PI). We are very grateful for the funding support and our incredible research teams at Brigham and Women’s Hospital (Clinical Neuroscience Laboratory for Sex Differences in the Brain (http://cnl-sd.bwh.harvard.edu), Colorado State University, and University of Arizona College of Medicine.
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
Reference List
- 1.Ambach G, Palkovits M. Blood supply of the rat hypothalamus. II. Nucleus paraventricularis. Acta Morphol Acad Sci Hung. 1974;22:311–20. [PubMed] [Google Scholar]
- 2.Armstrong WE, Warach S, Hatton GI, McNeill TH. Subnuclei in the rat hypothalamic paraventricular nucleus: a cytoarchitectural, horseradish peroxidase and immunocytochemical analysis. Neuroscience. 1980;5(11):1931–58. doi: 10.1016/0306-4522(80)90040-8. [DOI] [PubMed] [Google Scholar]
- 3.Asztalos E. Antenatal corticosteroids: a risk factor for the development of chronic disease. J Nutr Metab. 2012;2012:930591. doi: 10.1155/2012/930591. doi: 10.1155/2012/930591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bao AM, Hestiantoro A, Van Someren EJ, Swaab DF, Zhou JN. Colocalization of corticotropin-releasing hormone and oestrogen receptor-alpha in the paraventricular nucleus of the hypothalamus in mood disorders. Brain. 2005;128:1301–1313. doi: 10.1093/brain/awh448. [DOI] [PubMed] [Google Scholar]
- 5.Barefoot JC, Helms MJ, Mark DB, Blumenthal JA, Califf RM, Haney TL, O’Connor CM, Siegler IC, Williams RB. Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol. 1996;78:613–617. doi: 10.1016/s0002-9149(96)00380-3. [DOI] [PubMed] [Google Scholar]
- 6.Barry D, Pietrzak RH, Petry NM. Gender differences in associations between body mass index and DSM-IV mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Ann Epidemiol. 2008;18:458–66. doi: 10.1016/j.annepidem.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beauregard M, Leroux JM, Bergman S, Arzoumanian Y, Beaudoin G, Bourgouin P, Stip E. The functional neuroanatomy of major depression: an fMRI study using an emotional activation paradigm. Neuroreport. 1998;9:3253–8. doi: 10.1097/00001756-199810050-00022. [DOI] [PubMed] [Google Scholar]
- 8.Behar TN, Li YX, Tran HT, Ma W, Dunlap V, Scott C, Barker JL. GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J Neurosci. 1996;16:1808–1818. doi: 10.1523/JNEUROSCI.16-05-01808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein HG, Stanarius A, Baumann B, Henning H, Krell D, Danos P, Falkai P, Bogerts B. Nitric oxide synthase-containing neurons in the human hypothalamus: reduced number of immunoreactive cells in the paraventricular nucleus of depressive patients and schizophrenics. Neuroscience. 1998;83:867–75. doi: 10.1016/s0306-4522(97)00461-2. [DOI] [PubMed] [Google Scholar]
- 10.Biag J, Huang Y, Gou L, Hintiryan H, Askarinam A, Hahn JD, Toga AW, Dong HW. Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. J Comp Neurol. 2012;520:6–33. doi: 10.1002/cne.22698. doi: 10.1002/cne.22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carbone DL, Handa RJ. Sex and stress hormone influences on the expression and activity of brain-derived neurotrophic factor. Neuroscience. 2012 Dec 2; doi: 10.1016/j.neuroscience.2012.10.073. [Epub ahead of print] doi:pii: S0306-4522(12)01128-1.10.1016/j.neuroscience.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carbone DL, Zuloaga DG, Lacagnina AF, McGivern RF, Handa RJ. Exposure to dexamethasone during late gestation causes female-specific decreases in core body temperature and prepro-thyrotropin-releasing hormone expression in the paraventricular nucleus of the hypothalamus in rats. Physiol Behav. 2012;108:6–12. doi: 10.1016/j.physbeh.2012.07.010. doi: 10.1016/j.physbeh.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carpenter KM, Hasin DS, Allison DB, Faith MS. Relationships between obesity and DSM-IV major depressive disorder, suicide ideation, and suicide attempts: results from a general population study. Am J Public Health. 2000;90:251–7. doi: 10.2105/ajph.90.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvalho-Netto EF, Myers B, Jones K, Solomon MB, Herman JP. Sex differences in synaptic plasticity in stress-responsive brain regions following chronic variable stress. Physiol Behav. 2011;104:242–7. doi: 10.1016/j.physbeh.2011.01.024. doi: 10.1016/j.physbeh.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 16.Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ. Maternal care associated with methylation of the estrogen receptor –alph1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006;147(6):2909–2915. doi: 10.1210/en.2005-1119. [DOI] [PubMed] [Google Scholar]
- 17.Chen ZY, Bath K, McEwen B, Hempstead B, Lee F. Impact of genetic variant BDNF (Val66Met) on brain structure and function. Novartis Found Symp. 2008;289:180–8. doi: 10.1002/9780470751251.ch14. 2008. discussion 188-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cryan JF, Holmes A. The ascent of mouse: advances in modeling human depression and anxiety. Nat Rev Drug Discov. 2005;4:775–790. doi: 10.1038/nrd1825. [DOI] [PubMed] [Google Scholar]
- 19.Cryan JF, Slattery DA. GABAB receptors and depression. Current status. Adv Pharmacol. 2010;58:427–51. doi: 10.1016/S1054-3589(10)58016-5. [DOI] [PubMed] [Google Scholar]
- 20.Enhörning S, Bankir L, Bouby N, Struck J, Hedblad B, Persson M, Morgenthaler NG, Nilsson PM, Melander O. Copeptin, a marker of vasopressin, in abdominal obesity, diabetes and microalbuminuria: the prospective Malmö Diet and Cancer Study cardiovascular cohort. Int J Obes (Lond) 2012 doi: 10.1038/ijo.2012.88. Epub ahead of print. doi: 10.1038/ijo.2012.88. [DOI] [PubMed] [Google Scholar]
- 21.Fernández-Guasti A, Fiedler JL, Herrera L, Handa RJ. Sex, stress, and mood disorders: at the intersection of adrenal and gonadal hormones. Horm Metab Res. 2012;44:607–18. doi: 10.1055/s-0032-1312592. doi: 10.1055/s-0032-1312592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flak JN, Solomon MB, Jankord R, Krause EG, Herman JP. Identification of chronic stress-activated regions reveals a potential recruited circuit in rat brain. Eur J Neurosci. 2012;36:2547–55. doi: 10.1111/j.1460-9568.2012.08161.x. doi: 10.1111/j.1460-9568.2012.08161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frahm KA, Schow MJ, Tobet SA. The vasculature within the paraventricular nucleus of the hypothalamus in mice varies as a function of development, subnuclear location, and GABA signaling. Horm Metab Res. 2012;44:619–24. doi: 10.1055/s-0032-1304624. doi: 10.1055/s-0032-1304624. [DOI] [PubMed] [Google Scholar]
- 24.Gao SF, Bao AM. Corticotropin-releasing hormone, glutamate, and γ-aminobutyric acid in depression. Neuroscientist. 2011;17:124–44. doi: 10.1177/1073858410361780. [DOI] [PubMed] [Google Scholar]
- 25.Glassman AH, Shapiro PA. Depression and the course of coronary artery disease. Am J Psychiatry. 1998;155:4–11. doi: 10.1176/ajp.155.1.4. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein JM. Sex, hormones, and affective arousal dysfunction in schizophrenia. Horm Behav. 2006;50:612–622. doi: 10.1016/j.yhbeh.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein JM, Cherkerzian S, Buka S, Fitzmaurice G, Susser E, Hornig M, Gillman M, Factor-Litvak P, Sloan RP. Sex-specific impact of maternal-fetal risk factors on depression and cardiovascular risk 40 years later. J Develop Origins Health Dis. 2011a;2:353–364. doi: 10.1017/S2040174411000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein JM. Fetal Programming of Sex Differences in Stress Response Circuitry, Endocrine and ANS Deficits in Adulthood: Implications for Understanding Sex Differences in Comorbidity of Depression and CVD Risk. Presentation, 50th Annual Meeting, “Hot Topics” for American College of Neuropsychopharmacology (ACNP); Hawaii. 2011b. [Google Scholar]
- 29.Goldstein JM, Jerram M, Abbs B, Whitfield-Gabrieli S, Makris N. Sex differences in stress response circuitry activation dependent on female hormonal cycle. J Neurosci. 2010;30:431–438. doi: 10.1523/JNEUROSCI.3021-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstein JM, Jerram M, Poldrack R, Kennedy DN, Seidman LJ, Makris N. Hormonal cycle modulates arousal circuitry in women using fMRI. J Neurosci. 2005;25:9309–9316. doi: 10.1523/JNEUROSCI.2239-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstein JM, Seidman LJ, Makris N, Ahern T, O’Brien LM, Caviness VS, Kennedy DN, Faraone SV, Tsuang MT. Hypothalamic abnormalities in schizophrenia: Sex effects and genetic vulnerability. Biol Psychiatry. 2007;61:935–945. doi: 10.1016/j.biopsych.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 32.Granziera C, Hadjikhani N, Arzy S, Seeck M, Meuli R, Krueger G. In vivo magnetic resonance imaging of the structural core of the Papez circuit in humans. Neuroreport. 2011;22:227–31. doi: 10.1097/WNR.0b013e328344f75f. doi: 10.1097/WNR.0b013e328344f75f. [DOI] [PubMed] [Google Scholar]
- 33.Gutkowska J, Jankowski M. Oxytocin revisited: its role in cardiovascular regulation. J Neuroendocrinol. 2012;24:599–608. doi: 10.1111/j.1365-2826.2011.02235.x. doi: 10.1111/j.1365-2826.2011.02235.x. [DOI] [PubMed] [Google Scholar]
- 34.Gyekis JP, Yu W, Dong S, Wang H, Qian J, Kota P, Yang J. No association of genetic variants in BDNF with major depression: A meta- and gene-based analysis. Am J Med Genet B Neuropsychiatr Genet. 2013;162:61–70. doi: 10.1002/ajmg.b.32122. doi: 10.1002/ajmg.b.32122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav. 2011;59:279–89. doi: 10.1016/j.yhbeh.2010.06.007. doi: 10.1016/j.yhbeh.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 36.Heo M, Pietrobelli A, Fontaine KR, Sirey JA, Faith MS. Depressive mood and obesity in US adults: comparison and moderation by sex, age, and race. Int J Obes. 2006;30:513–9. doi: 10.1038/sj.ijo.0803122. [DOI] [PubMed] [Google Scholar]
- 37.Herman JP, Flak J, Jankord R. Chronic stress plasticity in the hypothalamic paraventricular nucleus. Prog Brain Res. 2008;170:353–64. doi: 10.1016/S0079-6123(08)00429-9. doi: 10.1016/S0079-6123(08)00429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(8):1201–13. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Herman JP, Seroogy K. Hypothalamic-pituitary-adrenal axis,_glucocorticoids, and neurologic disease. Neurol Clin. 2006;24:461–81. doi: 10.1016/j.ncl.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Holsen LM, Lawson EA, Ko E, Blum J, Makris N, Fazeli PK, Klibanksi A, Goldstein JM. Food motivation circuitry hypoactivation related to hedonic and non-hedonic aspects of hunger and satiety in women with active and weight-restored anorexia nervosa. J Psychiatry Neurosci. 2012b;37:322–32. doi: 10.1503/jpn.110156. 2012b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holsen LM, Spaeth SB, Lee J-H, Ogden LA, Klibanski A, Whitfield-Gabrieli S, Goldstein JM. Stress response circuitry hypoactivation related to hormonal dysfunction in women with major depression. J Affect Dis. 2011;131:379–87. doi: 10.1016/j.jad.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holsen LM, Lee J-H, Spaeth SB, Ogden LA, Klibanski A, Whitfield-Gabrieli S, Sloan RP, Goldstein JM. Brain hypoactivation, autonomic nervous system dysregulation, and gonadal hormones in depression: a preliminary study. Neurosci Lett. 2012a;514:57–61. doi: 10.1016/j.neulet.2012.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hossain A, Hajman K, Charitidi K, Erhardt S, Zimmermann U, Knipper M, Canlon B. Prenatal dexamethasone impairs behavior and the activation of the BDNF exon IV promoter in the paraventricular nucleus in adult offspring. Endocrinology. 2008;149:6356–65. doi: 10.1210/en.2008-0388. doi: 10.1210/en.2008-0388. [DOI] [PubMed] [Google Scholar]
- 44.Insel TR, Cuthbert BN, Garvey MA, Heinssen RK, Pine DS, Quinn KJ, Sanislow CA, Wang PS. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 45.Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann NY Acad Sci. 2008;1148:64–73. doi: 10.1196/annals.1410.012. doi: 10.1196/annals.1410.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeanneteau FD, Lambert WM, Ismaili N, Bath KG, Lee FS, Garabedian MJ, Chao MV. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc Natl Acad Sci USA. 2012;109:1305–10. doi: 10.1073/pnas.1114122109. doi: 10.1073/pnas.1114122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang X, Xu K, Hoberman J, Tian F, Marko AJ, Waheed JF, Harris CR, Marini AM, Enoch MA, Lipsky RH. BDNF variation and mood disorders: a novel functional promoter polymorphism and Val66Met are associated with anxiety but have opposing effects. Neuropsychopharmacology. 2005;30:1353–61. doi: 10.1038/sj.npp.1300703. [DOI] [PubMed] [Google Scholar]
- 48.Jones DJ, Bromberger JT, Sutton-Tyrrell K, Matthews KA. Lifetime history of depression and carotid atherosclerosis in middle-aged women. Arch Gen Psychiatry. 2003;60:153–160. doi: 10.1001/archpsyc.60.2.153. [DOI] [PubMed] [Google Scholar]
- 49.Kang H-J, Kim J-M, Stewart R, Kim S-Y, Bae K-Y, Kim S-W, Shin I-S, Shin M-G, Yoon J-S. Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog Neuro-Psychopharmacol Biol Psychiatry. 2013 doi: 10.1016/j.pnpbp.2013.01.006. (in press), http://dx.doi.org/10.1016/j.pnpbp.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 50.Kawachi I, Sparrow D, Vokonas PS, Weiss ST. Symptoms of anxiety and risk of coronary heart disease. The Normative Aging Study. Circulation. 1994;90:2225–2229. doi: 10.1161/01.cir.90.5.2225. [DOI] [PubMed] [Google Scholar]
- 51.Kendler KS, Gatz M, Gardner CO, Pedersen NL. A Swedish national twin study of lifetime major depression. Am J Psychiatry. 2006;163:109–114. doi: 10.1176/appi.ajp.163.1.109. [DOI] [PubMed] [Google Scholar]
- 52.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 53.Koutcherov Y, Mai JK, Ashwell KW, Paxinos G. Organization of the human paraventricular hypothalamic nucleus. J Comp Neurol. 2000;423:299–318. [PubMed] [Google Scholar]
- 54.Landa MS, Schuman ML, Burgueno A, Alvarez AL, Garcia SI, Pirola CJ. SiRNA-mediated silencing of the diencephalic thyrotropin-releasing hormone precursor gene decreases the arterial blood pressure in the obese agouti mice. Front Biosci. 2007;12:3431–5. doi: 10.2741/2324. [DOI] [PubMed] [Google Scholar]
- 55.Lauder JM. Neurotransmitters as growth regulatory signals: role of receptors and second messengers. TINS. 1993;16:233–240. doi: 10.1016/0166-2236(93)90162-f. [DOI] [PubMed] [Google Scholar]
- 56.Lawson EA, Holsen LM, Santin M, Meenaghan E, Eddy KT, Becker AE, Herzog DB, Goldstein JM, Klibanski A. Oxytocin secretion is associated with severity of disordered eating psychopathology and insular cortex hypoactivation in anorexia nervosa. J Clin Endocrinol Metab. 2012;97:E1898–908. doi: 10.1210/jc.2012-1702. doi: 10.1210/jc.2012-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levy BH, Tasker JG. Synaptic regulation of the hypothalamic-pituitary-adrenal axis and its modulation by glucocorticoids and stress. Front Cell Neurosci. 2012;6:24. doi: 10.3389/fncel.2012.00024. doi: 10.3389/fncel.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li DP, Pan HL. Role of gamma-aminobutyric acid (GABA)A and GABAB receptors in paraventricular nucleus in control of sympathetic vasomotor tone in hypertension. J Pharmacol Exp Ther. 2007;320:615–26. doi: 10.1124/jpet.106.109538. [DOI] [PubMed] [Google Scholar]
- 59.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 60.Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16:383–406. doi: 10.1038/mp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maggio N, Segal M. Differential corticosteroid modulation of inhibitory synaptic currents in the dorsal and ventral hippocampus. J Neurosci. 2009;29:2857–66. doi: 10.1523/JNEUROSCI.4399-08.2009. doi: 10.1523/JNEUROSCI.4399-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Makris N, Swaab DF, van der Kouwe A, Abbs B, Boriel D, Handa R, Tobet S, Goldstein JM. Volumetric Parcellation methodology of the human hypothalamus in neuroimaging: normative data and sex differences. NeuroImage. 2013;69:1–10. doi: 10.1016/j.neuroimage.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McEachin RC, Sannareddy KS, Cavalcoli JD, Karnovsky A, Vink JM, Sartor MA. Convergence of genetic influences in_comorbidity. BMC Bioinformatics. 2012 Mar 13;13(Suppl 2):S8. doi: 10.1186/1471-2105-13-S2-S8. 2012. doi: 10.1186/1471-2105-13-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McClellan KM, Calver AR, Tobet SA. GABAB receptors role in cell migration and positioning within the ventromedial nucleus of the hypothalamus. Neuroscience. 2008;151:1119–31. doi: 10.1016/j.neuroscience.2007.11.048. doi: 10.1016/j.neuroscience.2007.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McClellan KM, Stratton MS, Tobet SA. Roles for gamma-aminobutyric acid in the development of the paraventricular nucleus of the hypothalamus. J Comp Neurol. 2010;518:2710–28. doi: 10.1002/cne.22360. doi: 10.1002/cne.22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Menéndez A, Alvarez-Uria M. The development of vascularization in the postnatal rat paraventricular nucleus: a morphometric analysis. J Hirnforsch. 1987;28:325–9. [PubMed] [Google Scholar]
- 67.Moller-Leimkuhler AM. Gender differences in cardiovascular disease and comorbid depression. Dialogues Clin Neurosci. 2007;9:71–83. doi: 10.31887/DCNS.2007.9.1/ammoeller. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murray CJ, Lopez AD. Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet. 1997;349:1269–1276. doi: 10.1016/S0140-6736(96)07493-4. [DOI] [PubMed] [Google Scholar]
- 69.Musselman DL, Evans DL, Nemeroff CB. The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry. 1998;55:580–592. doi: 10.1001/archpsyc.55.7.580. [DOI] [PubMed] [Google Scholar]
- 70.Neumann ID, Landgraf R. Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci. 2012;35:649–659. doi: 10.1016/j.tins.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 71.Papez JW. A proposed mechanism of emotion. Arch Neur Psych. 1937;38:725–743. doi: 10.1176/jnp.7.1.103. [DOI] [PubMed] [Google Scholar]
- 72.Perello M, Cakir I, Cyr NE, Romero A, Stuart RC, Chiappini F, Hollenberg AN, Nillni EA. Maintenance of the thyroid axis during diet-induced obesity in rodents is controlled at the central level. Am J Physiol Endocrinol Metab. 2010;299:E976–89. doi: 10.1152/ajpendo.00448.2010. doi: 10.1152/ajpendo.00448.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petropoulos S, Gibb W, Matthews SG. Developmental expression of multidrug resistance phosphoglycoprotein (P-gp) in the mouse fetal brain and glucocorticoid regulation. Brain Res. 2010;1357:9–18. doi: 10.1016/j.brainres.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 74.Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99:2192–2217. doi: 10.1161/01.cir.99.16.2192. [DOI] [PubMed] [Google Scholar]
- 75.Rutledge T, Reis SE, Olson MB, Owens J, Kelsey SF, Pepine CJ, Mankad S, Rogers WJ, Merz CN, Sopko G, Cornell CE, Sharaf B, Matthews KA, Vaccarino V. Depression symptom severity and reported treatment history in the prediction of cardiac risk in women with suspected myocardial ischemia: The NHLBI-sponsored WISE study. Arch Gen Psychiatry. 2006;63:874–880. doi: 10.1001/archpsyc.63.8.874. [DOI] [PubMed] [Google Scholar]
- 76.Scherrer JF, Xian H, Bucholz KK, Eisen SA, Lyons MJ, Goldberg J, Tsuang M, True WR. A twin study of depression symptoms, hypertension, and heart disease in middle-aged men. Psychosom Med. 2003;65:548–557. doi: 10.1097/01.psy.0000077507.29863.cb. [DOI] [PubMed] [Google Scholar]
- 77.Seckl JR. Glucocorticoid programming of the fetus; adult phenotypes and molecular mechanisms. Mol Cell Endocrinol. 2001;185:61–71. doi: 10.1016/s0303-7207(01)00633-5. [DOI] [PubMed] [Google Scholar]
- 78.Selye H. The evolution of the stress concept. Stress and cardiovascular disease. Am J Cardiol. 1970;26:289–99. doi: 10.1016/0002-9149(70)90796-4. [DOI] [PubMed] [Google Scholar]
- 79.Sheline YI, Barch DM, Donnelly JM, et al. Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 2001;50:651–8. doi: 10.1016/s0006-3223(01)01263-x. [DOI] [PubMed] [Google Scholar]
- 80.Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990;294(1):76–95. doi: 10.1002/cne.902940107. [DOI] [PubMed] [Google Scholar]
- 81.Solomon MB, Herman JP. Sex differences in psychopathology: of gonads, adrenals and mental illness. Physiol Behav. 2009;97:250–8. doi: 10.1016/j.physbeh.2009.02.033. doi: 10.1016/j.physbeh.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Surguladze S, Brammer MJ, Keedwell P, Giampietro V, Young AW, Travis MJ, Williams SC, Phillips ML. A differential pattern of neural response toward sad versus happy facial expressions in major depressive disorder. Biol Psychiatry. 2005;57:201–9. doi: 10.1016/j.biopsych.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 83.Suzuki S, Handa RJ. Regulation of estrogen receptor-beta expression in the female rat hypothalamus: differential effects of dexamethasone and estradiol. Endocrinolog. 2004;145(8):3658–70. doi: 10.1210/en.2003-1688. [DOI] [PubMed] [Google Scholar]
- 84.Suzuki S, Handa RJ. Estrogen receptor-beta, but not estrogen receptor-alpha, is expressed in prolactin neurons of the female rat paraventricular and supraoptic nuclei: comparison with other neuropeptides. J Comp Neurol. 2005;484(1):28–42. doi: 10.1002/cne.20457. [DOI] [PubMed] [Google Scholar]
- 85.Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci. 1983;6:269–324. doi: 10.1146/annurev.ne.06.030183.001413. [DOI] [PubMed] [Google Scholar]
- 86.Szabo S, Tache Y, Somogyi A. The legacy of Hans Selye and the origins of stress research: a retrospective 75 years after his landmark brief “letter” to the editor of nature. Stress. 2012;15:472–8. doi: 10.3109/10253890.2012.710919. doi: 10.3109/10253890.2012.710919. [DOI] [PubMed] [Google Scholar]
- 87.Szczepanska-Sadowska E, Cudnoch-Jedrzejewska A, Ufnal M, Zera T. Brain and cardiovascular diseases: common neurogenic background of cardiovascular, metabolic and inflammatory diseases. J Physiol Pharmacol. 2010;61:509–21. [PubMed] [Google Scholar]
- 88.Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14:398–406. doi: 10.3109/10253890.2011.586446. doi: 10.3109/10253890.2011.586446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tobe I, Ishida Y, Tanaka M, Endoh H, Fujioka T, Nakamura S. Effects of repeated maternal stress on FOS expression in the hypothalamic paraventricular nucleus of fetal rats. Neurosci. 2005;134(2):387–395. doi: 10.1016/j.neuroscience.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 90.Ustun TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJ. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–392. doi: 10.1192/bjp.184.5.386. [DOI] [PubMed] [Google Scholar]
- 91.Vaccarino V, McClure C, Johnson BD, Sheps DS, Bittner V, Rutledge T, Shaw LJ, Sopko G, Olson MB, Krantz DS, Parashar S, Marroquin OC, Merz CN. Depression, the metabolic syndrome and cardiovascular risk. Psychosom Med. 2008;70:40–48. doi: 10.1097/PSY.0b013e31815c1b85. [DOI] [PubMed] [Google Scholar]
- 92.van den Pol AN. The magnocellular and parvocellular paraventricular nucleus of rat: intrinsic organization. J Comp Neurol. 1982;206:317–45. doi: 10.1002/cne.902060402. [DOI] [PubMed] [Google Scholar]
- 93.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Charma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 94.Welberg LA, Seckl JR. Prenatal stress, glucocorticoids and the programming of the brain. J Neuroendocrinol. 2001;13:113–128. doi: 10.1046/j.1365-2826.2001.00601.x. [DOI] [PubMed] [Google Scholar]
- 95.Whitnall MH. Subpopulations of corticotropin-releasing hormone neuroseretory cells distinguished by the presence or absence of vasopressing: confirmation of multiple corticotropin-releasing hormone antisera. Neuroscience. 1990;36(1):201–205. doi: 10.1016/0306-4522(90)90362-8. [DOI] [PubMed] [Google Scholar]
- 96.Wilson AC, Clemente L, Liu T, Bowen RL, Meethal SV, Atwood CS. Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochim Biophys Acta. 2008;1782:401–7. doi: 10.1016/j.bbadis.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 97.Wu Z, Xu Y, Zhu Y, Sutton AK, Zhao R, Lowell BB, Olson DP, Tong Q. An obligate role of oxytocin neurons in diet induced energy expenditure. PLoS One. 2012;7(9):e45167. doi: 10.1371/journal.pone.0045167. 2012. doi: 10.1371/journal.pone.0045167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yi SS, Kim HJ, Do SG, Lee YB, Ahn HJ, Hwang IK, Yoon YS. Arginine vasopressin (AVP) expressional changes in the hypothalamic paraventricular and supraoptic nuclei of stroke-prone spontaneously hypertensive rats. Anat Cell Biol. 2012;45:114–20. doi: 10.5115/acb.2012.45.2.114. doi: 10.5115/acb.2012.45.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–81. doi: 10.1038/nrn2336. doi: 10.1038/nrn2336. [DOI] [PubMed] [Google Scholar]
- 100.Zeng H, Schimpf BA, Rohde AD, Pavlova MN, Gragerov A, Bergmann JE. Thyrotropin-releasing hormone receptor 1-deficient mice display increased depression and anxiety-like behavior. Mol Endocrinol. 2007;21(11):2795–804. doi: 10.1210/me.2007-0048. [DOI] [PubMed] [Google Scholar]
- 101.Zuloaga DG, Carbone DL, Quihuis A, Hiroi R, Chong DL, Handa RJ. Perinatal dexamethasone-induced alterations in apoptosis within the hippocampus and paraventricular nucleus of the hypothalamus are influenced by age and sex. J Neurosci Res. 2012;90:1403–1412. doi: 10.1002/jnr.23026. doi: 10.1002/jnr.23026. [DOI] [PubMed] [Google Scholar]