

Abstract
Summary
The members of the immunoglobulin superfamily (IgSF) control innate and adaptive immunity, and are prime targets for the treatment of autoimmune diseases, infectious diseases and malignancies. We describe a computational method, termed the Brotherhood Algorithm, which utilizes intermediate sequence information to classify proteins into functionally related families. This approach identifies previously unrecognized functional relationships within the IgSF and predicts the existence of new receptor-ligand interactions. As a specific example, we describe new members of the nectin/nectin-like family of cell adhesion and signaling proteins, as well as new receptor-ligand interactions within this family. Guided by the Brotherhood approach, we present the high resolution structural characterization of a previously undescribed homophilic interaction involving the class-I MHC-restricted T-cell-associated molecule (CRTAM) – a newly defined nectin-like family member. The Brotherhood Algorithm is likely to have significant impact on structural immunology by identifying those proteins and complexes for which structural characterization will be particularly informative.
Keywords: Immune regulatory proteins, Brotherhood algorithm, Class-I MHC-restricted T-cell-associated molecule, CRTAM, Immunoglobulin Superfamily, functional classification
Introduction
The immunoglobulin superfamily (IgSF) includes hundreds of structurally similar cell-surface and secreted proteins that support a wide range of recognition and adhesive processes required for complex morphogenetic and developmental pathways, and for the protective advantages afforded by innate and adaptive immune responses. The comprehensive identification of these proteins and the complexes they form, along with molecular-level mechanistic understanding, is essential for defining the repertoire of physiological and pathological immune responses. These proteins represent important targets for immune-based therapeutics for the treatment of infectious diseases, cancer, and autoimmune diseases.
Based on the mechanistic and therapeutic importance of these molecules, a systematic structural analysis of the entire ensemble of cell surface immune regulatory proteins and their cognate complexes remains one of the major goals of structural immunology. Indeed large-scale efforts are now beginning to focus on this task, including the SPINE2 Program supported by the European Commission (www.spine2.eu) and the Immune Function Network supported by National Institutes of Health Protein Structure Initiative (http://sbkb.org/kb/centers.jsp?pageshow=20). However, given that the number of targets is estimated in the thousands (considering all full-length proteins, protein domains and protein complexes involved in the immune response), it remains impractical for one laboratory, or even a substantial consortium of laboratories, to structurally and functionally characterize all targets. These goals are further complicated by the fact that many of the biologically important receptor-ligand interactions remain unknown. Therefore, there exists a need to develop complementary strategies to identify, select and prioritize protein targets for experimental analysis.
Clustering of macromolecules into groups or families on the basis of sequence similarity frequently permits the prediction of at least some aspects of function and mechanism. Beyond simple assignment of putative function based on sequence similarity to an already annotated protein (i.e., annotation transfer), clustering can generate specific hypotheses that drive the identification of proteins for which direct structural and functional analyses are most likely to yield novel insights. Computational methods for clustering typically rely on the assumption that proteins with similar sequences are evolutionarily related and share similar structural features (Rost, 1997). CD-HIT (Li and Godzik, 2006) and BLASTCLUST (Dondoshansky, 2002) are widely used methods that cluster homologous proteins on the basis of explicit pairwise sequence comparisons. Other methods such as SCI-PHY (Brown et al., 2007) utilize multiple sequence alignments and phylogenomic inferences to functionally classify Superfamilies. In contrast to these approaches, which directly compare sequences or their profiles, are methods that exploit intermediate (i.e., transitive) sequences. These methods assume that evolutionary relationships detected by sequence similarity are transitive. For example, if the sequences of proteins a and b are similar and the sequences of proteins b and c are similar, then proteins a and c are considered to be evolutionarily related, even if direct pairwise similarity between a and c cannot be established (Gerstein, 1998; John and Sali, 2004; Park et al., 1997; Pegg and Babbitt, 1999; Salamov et al., 1999). While all of these computational methods have provided considerable insight into sequence and structural relationships, there is a continued need for the development of computational approaches that yield enhanced functional insight. The successes of existing methods in defining protein function is limited, as they are prone to false positive errors and therefore require relatively high similarity between the compared sequences. This requirement may leave many functionally related proteins unclassified (i.e., false negatives) (Gerlt and Babbitt, 2000; Jeong and Chen, 2001; Rost, 1997; Schnoes et al., 2009). These complications are of particular relevance to large and functionally diverse superfamilies, such as the IgSF, which can exhibit low sequence identity (i.e., <15%) among its members.
Here, we describe a new intermediate sequence search method, termed the “Brotherhood” method, which relies solely on sequence data to classify proteins into functional families. Using the Brotherhood method, we generated a global similarity network map of the complete set of human extracellular and integral membrane proteins within the IgSF, which provides an overview of families and ungrouped proteins (i.e., singletons). This mapping results in hypotheses regarding structural and functional similarities both within and between protein families and immediately allows for the prioritization of targets for structural, biochemical and functional analyses. The nectin/nectin-like family serves as a case study to highlight the potential of the Brotherhood method to expand established functional families by the inclusion of previously unassigned proteins, as well as the potential to de-orphan receptors and ligands by identifying new receptor-ligand interactions. We also report the 2.3 Å resolution crystal structure of the Class I-restricted T-cell-associated molecule (CRTAM), which the Brotherhood method suggests is evolutionarily and functionally related to the nectin-like proteins. CRTAM is a costimulatory protein that binds nectin-like 2 (nec-l2) and has been implicated in promoting NK-cell cytotoxicity, the secretion of cytokines (e.g., interferon-γ and IL-22) in CD8+ and CD4+ T cells (Boles et al., 2005), and late-stage polarization in T cells (Yeh et al., 2008). Consistent with our computational analysis, the crystal structure of CRTAM revealed an antiparallel homodimer with high structural similarity to nectin-like 1 (nec-l1) and nectin-like 3 (nec-l3) from the nectin-like subfamily, thereby supporting its placement within this subfamily and validating the utility of the Brotherhood method. This structure suggests that CRTAM forms a previously unappreciated homophilic trans-interaction involved in modulating immune function. Finally, the computational classification of the IgSF into evolutionarily related families immediately identifies proteins predicted to possess unique structural and functional features. The family classification obtained from this study is currently used to guide target selection for structural and functional studies at the New York Structural Genomics Consortium and the Immune Function Network (http://www.nysgrc.org/ and http://www.sbkb.org/kb/centers.jsp?pageshow=20).
Results
The Brotherhood Algorithm
The method examines the relationship between two query proteins by determining the number of intermediate sequences shared by the two proteins relative to the total number of evolutionarily related sequences for each of the two proteins (Fig. 1A). This overlap fraction (i.e., number of blast hits shared by two sequences normalized by the total number of blast hits for each sequence) represents a powerful metric for defining functional relatedness. We generated a family classification of 561 human IgSF proteins by the Brotherhood method (Fig. 1A) with an overlap threshold set at a minimum of 45%. These results were compared with three popular methods: 1) CD-HIT (Li and Godzik, 2006) with a range of sequence identity thresholds, 2) SCI-PHY (Brown et al., 2007), and 3) all-to-all pairwise BLAST comparisons (Atkinson et al., 2009) using a range of e-value thresholds. The all-to-all BLAST comparison performed similarly to CD-HIT, therefore we present a detailed comparison of the performance of the Brotherhood method with CD-HIT and SCI-PHY.
Figure 1.

A graphical presentation of functional families within the IgSF using three clustering strategies. Each member of the IgSF is represented by a circle. Members of 14 hand-curated families are represented with colored circles. A) Schematic representation of the Brotherhood method. The groups of evolutionarily related proteins are generated for each protein using Blast. Proteins are matched if their Blast-groups intersect (gray area) and do not differ (white area) above a certain threshold. B) Clusters generated using Brotherhood method, with an overlap threshold of 0.45. C) Clusters generated by SCI-PHY. D) Clusters generated by CD-HIT with a 30% sequence identity threshold.
To assess the ability of each method to cluster functionally related proteins, we utilized 14 known and well-curated families within the IgSF. The Brotherhood method generated 17 clusters and four singletons, with 11 of the 14 test families remaining intact (Fig. 1B). The SCI-PHY method generated 22 clusters and 27 singletons with only three of the 14 test families remaining intact (Fig. 1C). The CD-HIT30 method resulted in 20 clusters and 26 singletons with four of the test families remaining intact (Fig. 1D). At the thresholds employed, none of the methods resulted in clusters that mixed members of the original 14 families. Decreasing the sequence identity threshold for CD-HIT below 30% reduces the numbers of clusters, ungrouped family members, and false negatives; however, this is accompanied by a significant increase in false positives, (i.e. clustering together proteins from different families; Fig. 1, see also Fig. S1).
An examination of the distribution of pairwise protein sequence comparison scores among proteins that belong to the same (“intra-family”) and different test families (“inter-family”) clearly demonstrates that direct pairwise comparison cannot fully distinguish between true and false matches (Fig. 2A). In contrast, the distribution of intra-family and inter-family Brotherhood pairwise scores shows that an overlap threshold of ~0.45 is able to completely discriminate true and false matches for all 14 test families (Fig. 2B). The only exceptions are the CD2 and CD58 proteins presumed to belong to the SLAM family; it is notable that at least one previous report does not consider CD2 and CD58 to be members of the SLAM family (Engel et al., 2003). In another example, using pairwise BLAST comparisons, the CD28-CTLA-4-ICOS family can be constructed by connecting CD28 to CTLA-4 with a log(e-value) of −11.3 and CD28 with ICOS with a log(e-value) of −10.4 (Fig. 2A). However, the assumption of functional relationships in this score range introduces 167 false positive (inter-family) connections into the functional similarity network. In contrast, the Brotherhood method connects CD28 to both CTLA-4 and ICOS with overlap scores of 90% and 55%, respectively; these scores are significantly above the range of false positives (Fig. 2B).
Figure 2.
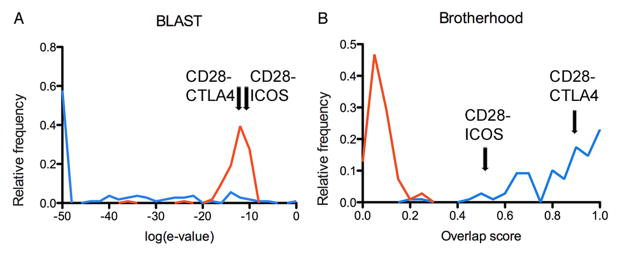
Comparing the performances of two methods (a) Blast pairwise comparison and (b) Brotherhood method, to discriminate between correct (true positives, in blue) and incorrect (false positives, in red) family classifications, respectively. The normalized distributions of scores are shown on each plot for 106 pairs of true and false positive cases. The true positive cases were obtained from the hand curated set of IgSF proteins, by comparing pairs of proteins within the same functional family, while the 106 false positives were selected as the highest scoring cases out of 5861 comparisons where proteins from different families were incorrectly clustered. The greater separation of true and false positive scores in the Brotherhood approach reflects a higher accuracy for functional classification. Arrows mark the two top scores (CD28-CTLA-4 and CD28-ICOS) that correspond to the construction of the CD28 family by both methods; these validated relationships are well defined by the Brotherhood approach, but are buried beneath the false positive signal generated by BLAST.
As demonstrated in past applications, the use of intermediate sequences was expected to increase sensitivity in detecting remote family members (Gerstein, 1998; John and Sali, 2004; Park et al., 1997; Pegg and Babbitt, 1999; Salamov et al., 1999); however, intermediate sequence analysis can also reduce specificity. In practice, we observed that almost all IgSF members of a given family are also connected through intermediate sequences to non-family related IgSF proteins. For example, the protein Sialoadhesin (sialic acid-binding Ig-like lectin 1), one of 15 members of the SIGLEC family, can be linked to Vascular endothelial growth factor receptor 2 (VEGFR2), a member of the PDGFR family, through 62 intermediate sequences. Therefore, it is critical to properly control the signal-to-noise ratio used in the intermediate sequence analysis. The Brotherhood method reduces this “noise” by requiring a certain ratio of detected intermediate sequences compared to the total number of related sequences. In the example of Sialoadhesin and VEGFR2, of 250 significant hits returned by a BLAST search for each protein, only 62 are shared, resulting in an overlap score of about 25%. This overlap score is significantly smaller than the threshold required to recapitulate the hand-curated functional families. In notable contrast, within the SIGLEC and PDGFR families the overlap scores range from 51% to 99% and 73% to 91%, respectively, resulting in the clear and accurate recapitulation of these hand-curated families.
Validating a new functional assignment
Brotherhood analysis of the entire ensemble of human secreted and integral membrane proteins in the IgSF yields 63 ungrouped proteins (singletons), compared to 117 and 129 singletons generated using CD-HIT30 and SCI-PHY, respectively (Fig. 1). The observation of fewer singletons and more highly populated families suggests that the Brotherhood method can identify new family members that escape detection with the established CD-HIT30 and SCI-PHY methods. For example, analysis of the nectin/nectin-like cluster highlights the potential of the Brotherhood algorithm to identify new family members and to provide new functional insights, including the prediction of unappreciated receptor:ligand interactions. The nectin/nectin-like family is composed of nine cell adhesion proteins with a common ectodomain architecture consisting of three Ig-like domains: two membrane-proximal Ig-C2 domains and a membrane-distal Ig-V domain that is responsible for Ca2+-independent adhesion through homophilic or heterophilic trans-interactions with other members of the family (Fig. 1, and see also Fig. S2). This family can be further classified into two subgroups that consist of four nectin and five nectin-like proteins based on their ability to directly bind afadin, a protein that physically links the nectins to the actin cytoskeleton (Takai et al., 2008). CD-HIT30, BLAST and SCI-PHY were unsuccessful at clustering all nine members of the nectin/nectin-like family or separating them into the two subgroups (Figs. 1C and 1D). In contrast, the Brotherhood method successfully clustered all nine nectin/nectin-like proteins into a single cluster with the two subgroups (nectin and nectin-like) clearly segregated (Fig. 1B). Contrary to its assigned name, the Brotherhood method clustered the nectin-like 5 (nec-l5) ectodomain with the nectin proteins rather than with the nectin-like proteins. This assignment is supported by the facts that the nec-l5 ectodomain sequence and its gene structure are more similar to the ectodomains of the nectins proteins than the nectin-like proteins (Fig. 1 and see also Fig. S3 and S4)
Most interestingly, the Brotherhood method indicated that five additional IgSF proteins were associated with the nectin/nectin-like family, including CRTAM, CD226, CD96, CD200 and TIGIT (T-cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motif), suggesting either five false positive assignments or an expansion of this cluster into a larger 14 member family (Fig. 1B and see also Fig. S1). All five of these proteins were classified as singletons using the CD-HIT30 and SCI-PHY IgSF networks. An extensive literature search, at the time of this analysis, revealed that with the exception of CD200 and TIGIT, the remaining 12 proteins had been previously reported to possess binding partners that reside within this nectin/nectin-like cluster (Takai et al., 2008) (Fig. 3). Notably, Yu et al. reported a functional relationship between TIGIT and the nectin/nectin-like family by experimentally screening a library of approximately 1000 purified cell surface proteins as Ig-fusion constructs and demonstrated that TIGIT directly binds to nec-l5, nectin-3, and to a lesser extent to nectin-2 (Yu et al., 2009). Thus, four of the five newly assigned proteins were found to recognize ligands that are similar to the ligands of the nine previously known members of the nectin/nectin-like family. This observation suggests that all members of the expanded family utilize similar binding mechanisms to recognize related binding partners within the nectin/nectin-like family and thus share significant evolutionary and functional relationships.
Figure 3.
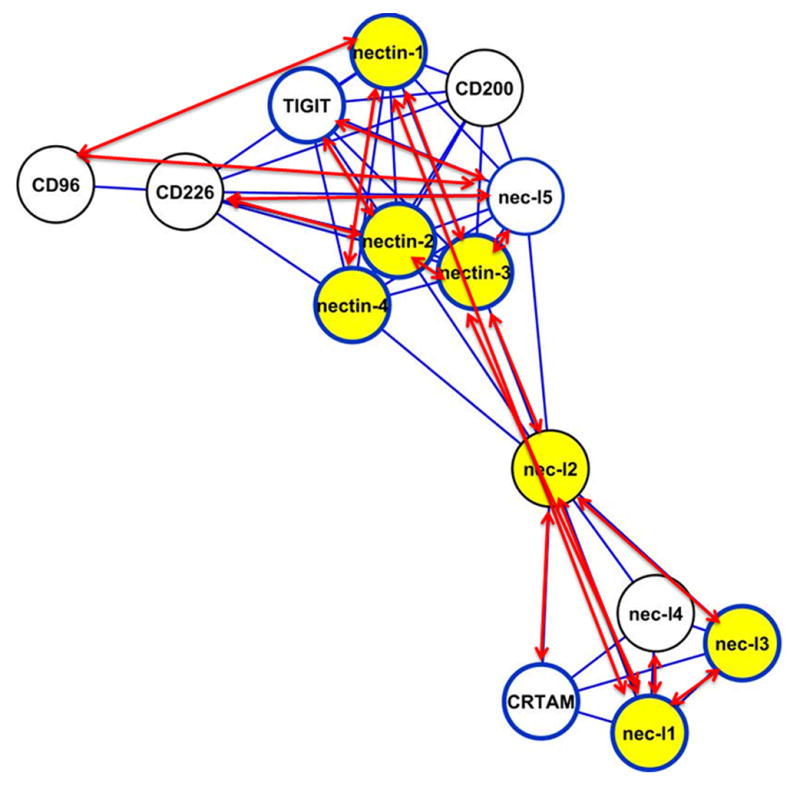
The Brotherhood-defined nectin/nectin-like cluster with the mapping of experimental information. Red two-headed arrows correspond to known receptor-ligand heterophilic interactions; yellow filled circles represent known receptor-ligand homophilic interactions; circles with blue outlines represent proteins with a known three dimensional structure.
CRTAM exhibits a homophilic interaction with a mode of dimerization similar to that exhibited by nectin-like proteins
When examined by size exclusion chromatography (SEC), the elution profile of the CRTAM Ig-V domain exhibits peaks consistent with dimeric (~28kD) and monomeric (~14kD) species (Fig. 3 and see also Fig. S5). Sedimentation equilibrium analysis demonstrated that CRTAM self-associates in monomer-dimer equilibrium with an equilibrium dissociation constant (Kd) of ~10 μM (Fig. 3 and see also Fig. S6).
The crystal structure of the CRTAM N-terminal Ig-V domain was determined by molecular replacement and refined to a resolution of 2.3 Å (Table 1). The CRTAM structure exhibits the expected Ig-V domain fold composed of nine anti-parallel β-strands organized into a two-layered β-sheet assembly, with the A, G, F, C, C′, C″ strands forming the front sheet and the B, E, and D strands forming the back sheet. The asymmetric unit contains four independent CRTAM Ig-V domains related by a 4-fold non-crystallographic symmetry (NCS) axis oriented approximately parallel to the crystallographic c-axis. Each of the four molecules forms a tight antiparallel dimer with a CRTAM Ig-V domain from an adjacent asymmetric unit related by a crystallographic two-fold axis. The antiparallel dimer buries approximately 700 Å2 of solvent accessible surface area per protomer (1400 Å2 total), while the average interface between pairs of molecules in the NCS tetramer is 343 Å2 per protomer. Based on the evaluation of energetic considerations and physicochemical properties by the PISA server (Krissinel and Henrick, 2007), the antiparallel dimer is predicted to be the only stable assembly in solution.
Table 1.
Data collection and refinement statistics
| Data collection | |
|---|---|
| PDB code | 3RBG |
| Space group | C2221 |
| Unit cell length (Å) | a = 116.020, b = 116.291, c = 79.018 |
| Unit cell angles (deg) | α = 90.0, β = 90.0, γ = 90.0 |
| Wavelength (Å) | 1.081 |
| Resolution range (Å) | 2.32 – 40.0 (2.32 – 2.36) |
| Unique reflections (N) | 23,676 (1168) |
| Redundancy | 5.5 (5.6) |
| Completeness | 98.9 (99.8) |
| Rmergea | 0.076 (0.462) |
| <I/σ> | 21.7 (4.9) |
| Refinement | |
| Resolution range (Å) | 2.32 – 28.47 (2.32 – 2.36) |
| Rworkb | 0.198 (0.278) |
| Rfree | 0.234 (0.327) |
| Average B factor (Å2) | 30.1 |
| rms bond (Å) | 0.013 |
| rms angle (deg) | 1.453 |
| Residues in most favored region (%) | 96.21 |
| Residues in additionally allowed region (%) | 3.52 |
| Residues in generously allowed region (%) | 0 |
| Residues in unfavorable region (%) | 0.27 |
Rmerge = Σ|Ih−〈 Ih 〉|/ΣIh, where h is the reflection index and Ih is the average intensity over symmetry equivalents
Rwork = Σ|Fc − Fo|/ΣFo
Rfree calculated as Rwork on a subset (5%) of the reflection data that is not included in the refinement calculation.
Values in parenthesis correspond to the highest resolution bin.
The antiparallel dimer interface is similar in general organization to other IgSF interfaces and is formed by 17 residues from each CRTAM Ig-V contributed by the C, C′, C″ and F strands, and the C-C′, C′-C″, C″-D, and F-G loops (Fig. 4). The antiparallel dimer appears to be stabilized by a network of four hydrogen bonds, involving three residues from each molecule (Gln49, Thr57, and Tyr101), with side chain-side chain interactions between Gln49 and Thr57 and a side chain-backbone interaction with Tyr101. This network is surrounded by a set of hydrophobic interactions involving Phe56, Leu60, Val65, Leu66, Leu99 and Val105 (Fig. 4B). Due to the two-fold symmetry of the homodimer, interface residues from one of the molecules are also present at the symmetry-related region of the other molecule.
Figure 4.

CRTAM structure. A) Ig-V domains in the CRTAM homodimer are organized in an antiparallel manner. Ig-V domains are in blue and grey, with their interface residues colored in red and green, respectively. Strands of the Ig-V domains are labeled according to convention and stick models illustrate the interface residues. B) Blow-up of the interface. Dashed lines represent hydrogen bonds.
The sequence of the full CRTAM ectodomain shares less than 30% sequence identity with any of the ectodomains of nectin/nectin-like family members, while the N-terminal Ig-V domain itself shares approximately 35% sequence identity with the Ig-V domains of the nectin-like proteins (nec-l1 – nec-l4). Structurally, the antiparallel CRTAM dimer is similar to the nec-l1 and nec-l3 homophilic dimers (PDB codes 1Z9M and 3M45, respectively (Dong et al., 2006; Fogel et al., 2010)) with RMSDs of about 1.9 Å for 192 structurally equivalent residues. CRTAM, nec-l1 and nec-l3 have comparable interfaces, burying 706.3 Å2, 708.8 Å2, and 689.1 Å2 of solvent accessible surface area per protomer, respectively. Of the 17 residues at the CRTAM interface, 16 are in analogous positions at the nec-l1 or nec-l3 interfaces and six of these residues are invariant in all three proteins (Ser47, Gln49, Thr57, Leu66, Val105 and Thr107) (Fig. 5). One residue, Asn45, is conserved in nec-l3 but not in nec-l1, and five additional residues have similar physicochemical properties in all three proteins (Leu60, Val65, Lys67, Tyr101 and Ser102). Two of the four hydrogen bonds at the core of the CRTAM dimer interface and the residues that form them (Gln49 and Thr57) are conserved in the interfaces of both nec-l1 and nec-l3.
Figure 5.

Structural comparison of CRTAM and nectin-like proteins. A) The ribbon diagram of the structural superposition of CRTAM (in blue) with nec-l3 (in orange). N45 of CRTAM and the N-acetylglucosamine residues on N60 of nec-l3 are displayed as spheres. B) Structure-based alignment of CRTAM, necl-1 and nec-l3 sequences. Invariant and conserved alignment positions are highlighted in red background and red letters, respectively. The secondary structure corresponding to CRTAM and necl-3 are shown above and below the alignment, respectively, where arrows represent strands. Black circles mark residues forming the CRTAM Ig-V homodimer interface. The vertical black arrow marks the sequence positions of N45 and N60 of CRTAM and nec-l3 respectively.
The similarity of CRTAM to the nectin-like subfamily contrasts that with the nectin subfamily. For example, structural superposition of the homophilic Ig-V dimers of CRTAM and nectin-1 results in a RMSD of 4.2 Å for 186 aligned Cα positions, which is more than twice the RMSD found in the comparison between CRTAM and the nectin-like homodimers (Fig. 5, and see also Table S1.). In addition a structure-based multiple sequence alignment of CRTAM with the nectin/nectin-like family identified only one CRTAM interfacial residue conserved between CRTAM and the nectin proteins, as well as TIGIT, CD226, CD96 and CD200, in contrast to the 11 conserved interface residues between CRTAM, necl1 and necl3 (Fig. 5, and see also Fig. S3).
CRTAM gene structure
The exon/intron organization (exon length and phasing) of the gene segment encoding the ectodomain of CRTAM exhibits the same pattern present in all of the genes of the nectin-like subgroup members and differs from that of the nectin subgroup members (Fig. 3, and see also Fig. S4). The CRTAM gene is composed of 10 exons, all of which are associated with phase-one introns (i.e., the codon is interrupted after the first nucleotide). The first exon of CRTAM encodes for the first 15 amino acids and includes the secretion signal sequence. The second and third exons encode the CRTAM Ig-V domain and the fourth and fifth exons encode the CRTAM Ig-C2 domain. The nectin-like genes are also split within and between the Ig-V and the first Ig-C2 domains at similar positions and with the same intron phasing (Fig. 3, and see also Fig. S4). In contrast, the nectins have a distinct exon/intron pattern in which neither the Ig-V nor the first Ig-C2 coding regions are interrupted (Fig. 3, and see also Fig. S4).
Discussion
We introduced the Brotherhood Algorithm, a computational method for functional classification of proteins and utilized it to identify functional families of IgSF members in the human proteome. A better understanding of the molecular mechanisms underlying receptor-ligand recognition in the IgSF will provide insights into immune function and may result in new therapeutic strategies. However, as is the case with other large protein superfamilies, the size of the IgSF makes it impractical to subject all individual members and their complexes to experimental analysis.
In the absence of experimental information, protein family classification is typically based on amino acid sequence comparisons. However, the application of traditional sequence comparison methods results in the formation of incomplete families, with greater than 20% of the IgSF proteins remaining unclustered (i.e., singletons). Reducing the sequence identity threshold to allow for the clustering of more proteins results in the rapid accumulation of false positives. We demonstrated that an intermediate sequence search algorithm, the Brotherhood method, was able to cluster proteins sharing low sequence identity and reduce the number of unclustered IgSF proteins by almost half. The sensitivity of the Brotherhood method is achieved by considering many weakly (i.e., high Blast evalue) linked intermediate sequences; however, we observed that almost all families were interconnected through intermediate sequences. This observation was expected as all proteins in this study share the immunoglobulin fold. To avoid high numbers of false positives, Brotherhood specificity is achieved by requiring that the number of intermdiate sequences found is proportional to the overall number of homologs (i.e., significant BLAST hits) found for each query. For the IgSF an overlap of 45% between the number of intermediate sequences and all homologous sequences was choosen empiricially to recapitulate a set of hand-curated families. As with other approaches, the appropriate value of the threshold is likely to differ for different superfamilies. The precise value of the overlap is not critical, as the goal is to generate high quality hypotheses regarding functional relationships for direct experimental validation. Small numbers of incorrect suppositions can be tolerated if they are outweighed by the number of correct conjectures. Based on the current work, the Brotherhood approach appears to outperform other existing methods in this respect.
The utility of the Brotherhood method was demonstrated using the nectin/nectin-like family as a specific example (see Experimental Procedures and Supplemental Information for additional examples of large families defined by the Brotherhood method). A recent report identified CRTAM as a distant relative to the nectin/nectin-like family (Patino-Lopez et al., 2006); however, this similarity was inferred on the basis of low sequence identity and a weak BLAST similarity score between CRTAM and the nectin-like proteins. Our all-to-all BLAST comparison demonstrates that at this level of sequence similarity one cannot distinguish between true and false positive connections. For example, Kin of IRRE-like protein (KIRR) 1 and KIRR 2, which are decidedly not part of the nectin-like family, have a lower (i.e., more significant) e-value to nectin-like proteins than does CRTAM. Nevertheless, the Brotherhood method strongly supports inclusion of CRTAM in the nectin-like subfamily with overlap scores with the four nectin-like proteins ranging between 54% and 62%, which are well above our threshold of 45%. The overlap scores of KIRR 1 and KIRR 2 with the nectin-like proteins are in the range of 11–40%. CRTAM itself exhibits overlap scores with KIRR1 and KIRR2 of only 3% and 0.5%, respectively.
Equilibrium sedimentation analysis demonstrated that in solution the CRTAM Ig-V domain exists in dynamic equilibrium between dimer and monomer, with a Kd of approximately 10 μM. The affinity of the CRTAM homophilic interaction is comparable to those of other physiologically relevant homophilic and heterophilic interactions involving IgSF members, which typically exhibit three-dimensional Kds in the ~0.1 to 100 μM range (Davis et al., 2003; van der Merwe and Davis, 2003). For instance, the heterophilic CD2:CD58, 2B4:CD48 and KIR:MHC-I complexes are characterized by Kds of ~10 μM, and the CD28:CD86 complex is characterized by a Kd of 20 μM (Davis et al., 2003; van der Merwe and Davis, 2003). Within the nectin/nectin-like family, the CRTAM homophilic association is weaker than the homophilic interaction of nectin-2 (Kd of 0.4 μM); stronger than the homophilic interactions of nectin-3 and nectin-4 (Kds of 228 and 153 μM, respectively); and similar to the homophilic association of nectin-1 (Kd of 17.5 μM) (Harrison et al., 2012). It is important to underscore that while three-dimension affinities measured in solution provide important mechanistic insights, they cannot fully recapitulate the physiological constraints relevant to the in vivo functions of cell surface molecules, which critically depend on surface density and entropic contributions (Wu et al., 2011). These interactions are further dependent on the correct spatial, temporal and cell-specific expression of the interacting receptor:ligand pairs.
Despite the low sequence identity between CRTAM and the nectin-like proteins (less than 35%), the CRTAM antiparallel homodimer exhibits high structural similarity to nec-l1 and nec-l3 homodimers, including low RMSDs and interfaces involving residues with similar physicochemical properties at structurally equivalent positions. In contrast, the CRTAM dimer exhibited high RMSDs (more than double) when compared to other dimers within the IgSF (Fig.5, and see also Table S1) and the CRTAM interface residues were not conserved outside of the nectin-like subgroup. In addition to this structural evidence, the evolutionary and functional relationships between CRTAM and the nectin-like proteins are supported by their shared gene structure (Fig. 3, and see also Fig. S4).
The crystal structure of CRTAM has potential functional implications, as the structures of nec-l1 and nec-l3 were suggested to represent the organization of these proteins in homophilic trans-interactions (Dong et al., 2006; Fogel et al., 2010). Based on structural similarity of these three proteins, CRTAM may form a previously uncharacterized cell-to-cell homophilic trans-interaction mediated by its Ig-V domain, which is similar in detailed organization to the nec-l1 and nec-l3 homophilic dimers. This CRTAM:CRTAM interaction may play a role in cell–to-cell signaling involving T cells, B cells and NK cells. Furthermore, this putative CRTAM:CRTAM homophilic dimer could effectively compete with the CRTAM:nec-l2 heterophilic interaction and thus serve as a mechanism to regulate this heterophilic interaction involved in a range of innate and adaptive immune processes.
While several proteins of the nectin/nectin-like family, as well as other members of the IgSF, form homophilic trans-interactions mediated by the front sheet of their Ig-V domains, this is not a general feature of the Ig-V fold. For example, members of the CD28 receptor family (e.g., CD28, CTLA-4, ICOS), PD1, BTLA and members of the B7 ligand family (e.g., B7-1, B7-2, ICOS-ligand, PD-L1, PD-L2, B7-H3, B7-H4) do not appear to participate in homophilic trans-interactions. Instead, many of these molecules function via the formation of heterophilic trans-associations (e.g, CD28:B7-1, CD28:B7-2, CTLA-4:B7-1; CTLA-4:B7-2, ICOS:ICOS-ligand, PD-1:PD-L1, PD-1:PD-L2). Notably, physiologically relevant homophilic cis-interactions are also known for some these molecules (i.e., CD28, CTLA-4, ICOS and B7-1), which involve unusual side-to-side (e.g., CD28, CTLA-4, ICOS) or back sheet-to-back sheet interactions (e.g., B7-1). Thus, the suggestion that CRTAM forms a homophilic trans-interaction is not a trivial prediction.
The human CRTAM Ig-V domain was expressed in E. coli and is therefore not glycosylated; however, three potential glycosylation sites are present at N21, N45, and N85. Of these, only N45 is predicted to be near the observed dimer interface. Nec-l3 is the only protein in the nectin-like sub-family that possesses a glycan-modified asparagine (N60 in nec-l3) at a position structurally equivalent to N45 in CRTAM (Fig. 5) (Fogel et al., 2010). In the nec-l3 structure, N-acetylglucosamine on N60 makes contacts with interface residues; however, only part of the naturally occurring glycan is present due to enzymatic treatment during sample preparation. Structural considerations suggested that intact N-linked glycan would interfere with trans-adhesion and cell-based assays indeed demonstrate that unmodified glycosylation of N60 reduces the nec-l3 adhesive trans-interaction (Fogel et al., 2010). This behavior suggests the possibility of similar glycosylation-dependent modulation of CRTAM binding activity. However, while most nec-l3 orthologs contain this potential glycosylation, N45 is not conserved in many CRTAM orthologs. Notably, in the closely related Gorilla ortholog, N45 is not conserved, supporting a less than universal role for this glycan modification in CRTAM function (Fig. 5, and see also Fig S7.).
The global IgSF protein similarity network generated with the Brotherhood method offers significant opportunities to pursue hypothesis-driven structural biology by highlighting sequences predicted to possess structural and functional similarity, as well as sequences with unique structural features.
These considerations are of particular relevance to large-scale structural genomics efforts which seek to systematically define the reperoire of interactions responsible for complex cellular communication, including the human immune response. The results of the Brotherhood method thus allow for the identification and prioritization of those proteins for which a detailed structual analysis is most likely to yield new functional and mechanistic insights. For example, the singletons, which do not cluster with other member of the IgSF, immediately represent interesting targets because of their promise to reveal unique structure and function. We previously examined one of these singletons, termed VISTA (V-domain Ig suppressor of T-cell activation), and discovered that it has a unique distribution of cysteine residues compared to all IgSF proteins. This pattern suggests distinctive structural features in the form of new inter and/or intra molecular disulfide bonds that support VISTA function (Wang et al., 2011).
Perhaps most importantly, the IgSF similarity network affords the opportunity to identify novel candidate receptor-ligand pairs that can be readily subjected to experimental verification. For example, based on the Brotherhood-generated functional clustering and existing biochemical properties, we hypothesized that all 14 members of the extended nectin/nectin-like family, including TIGIT, would recognize a ligand within this family. Indeed, subsequent to our initial analysis, TIGIT was demonstrated to bind nec-l5, nectin-2 and nectin-3 (Yu et al., 2009). These efforts required direct binding experiments involving a library of over 1000 soluble proteins, which is outside the capabilities of a typical academic laboratory. This challenge becomes even greater when the entire secretome is considered, making exhaustive experimental interrogation impractical. In contrast, the application of the Brotherhood method led to a hypothesis that significantly narrowed the potential TIGIT binding partners to the 14 members of the extended nectin/nectin-family, which is fully tractable. This informatics-guided approach would have resulted in significant reductions in both time and resources.
The current study focused on the 561 human members of the IgSF, which represents a small subset of the entire human secretome. The Brotherhood method can easily be generalized to all extracellular proteins, or any other protein group, making it an effective tool for identifying and prioritizing proteins in standard laboratory and structural genomics settings. The Brotherhood method can also be readily applied to proteins from different genomes, including those of pathogens, so as to expand family definitions and define pan-genomic functional and evolutionary relationships.
Experimental Procedures
Selection of hand-curated families for this study
Following are the 14 families, representing a total of 246 IgSF proteins that were used to benchmark the Brotherhood approach against other methods (proteins are denoted by their Uniprot names). For each family we indicate newly predicted members as “Brotherhood additions” (a total of 9):
CD28 family (3 members): CD28, CTLA-4, ICOS (Chattopadhyay et al., 2009).
Pregnancy specific glycoproteins (PSG) and Carcinoembryonic antigen-related cell adhesion molecules (CEAM) (23 members): PSG1, PSG2, PSG3, PSG4, PSG5, PSG6, PSG7, PSG8, PSG9, PSG10, PSG11, CEAM1, CEAM3, CEAM4, CEAM5, CEAM6, CEAM7, CEAM8, CEA16, CEA18. CEA19, CEA20, CEA21 (Bairoch et al., 2005; Streydio et al., 1988). Brotherhood addition: Hepatocyte cell adhesion molecule (HECAM).
T-cell immunoglobulin and mucin domain-containing protein (TIM) (3 members): TIM1, TIM3, TIM4 (Chattopadhyay et al., 2009).
Signaling lymphocytic activation molecule (SLAM) (11 members): SLAF1, SLAF5, SLAF6, SLAF7, SLAF8, SLAF9, CD244, CD48, LY9, CD2, CD58. (Chattopadhyay et al., 2009).
-
Nectin and Nectin-like (9 members): nec-l1, nec-l2, nec-l3, nec-l4, nectin-1, nectin-2, nectin-3, nectin-4, nec-l5 (Takai et al., 2008).
Brotherhood additions: CRTAM, TIGIT, CD226, CD96, and CD200.
Sialic acid binding Ig-like lectin family (SIGLEC) (15 members): CD22, CD33, MAG, SIGL5, SIGL6, SIGL7, SIGL8, SIGL9, SIG10, SIG11, SIG12, SIG14, SIG15, SIG16, SN (Bairoch et al., 2005; Pillai et al., 2012).
-
JAM/CXR (Junctional adhesion molecule/cortical thymocyte marker in Xenopus) (10 members): ACAM, CXAR, ESAM, GPA33, IGS11, JAM1, JAM2, JAM3, VSIG1, VSIG2 (Bazzoni, 2003; Eguchi et al., 2005; Scanlan et al., 2006).
Brotherhood addition: VSIG8.
-
B7-butyrophlin family (20 members) (Bairoch et al., 2005; Carreno and Collins, 2002):
B7: CD80, CD86, ICOSL, PD1L1, PD1L2, CD276 (B7H3), VTCN1 (B7H4),
Butyrophilin: BT1A1, BT2A1, BT2A2, BT2A3, BT3A1, BT3A2, BT3A3, BTNL2, BTNL3, BTNL8, BTNL9,
-
Others: ERMAP, MOG,
Brotherhood addition: Human endogenous retrovirus-H long terminal repeat-associating protein 2 (HHLA2).
Semaphorin (14 members): SEM3A, SEM3B, SEM3C, SEM3D, SEM3E, SEM3F, SEM3G, SEM4A, SEM4B, SEM4C, SEM4D, SEM4F, SEM4G, SEM7A (Yazdani and Terman, 2006).
-
CSF/PDGFR family (8 members): CSF1R, FLT3, KIT, PGFRA, PGFRB, VGFR1, VGFR2, VGFR3 (Bairoch et al., 2005).
Brotherhood addition: Platelet-derived growth factor receptor-like protein
-
MHC-I (73 members) (Bairoch et al., 2005):
HLA-A: 1A01, 1A02, 1A03, 1A11, 1A23, 1A24, 1A25, 1A26, 1A29, 1A30, 1A31, 1A32, 1A33, 1A34, 1A36, 1A43, 1A66, 1A68, 1A69, 1A74, 1A80.
HLA-B 1B07, 1B08, 1B13, 1B14, 1B15, 1B18, 1B27, 1B35, 1B37, 1B38, 1B39, 1B40, 1B41, 1B42, 1B44, 1B45, 1B46, 1B47, 1B48, 1B49, 1B50, 1B51, 1B52, 1B53, 1B54, 1B55, 1B56, 1B57, 1B58, 1B59, 1B67, 1B73, 1B78, 1B81, 1B82.
HLA-C: 1C01, 1C02, 1C03, 1C04, 1C05, 1C06, 1C07, 1C08, 1C12, 1C14, 1C15, 1C16, 1C17, 1C18.
Non-classical: HLA-E, HLA-F, HLA-G.
-
MHC-II (31 members) (Bairoch et al., 2005):
HLA-DP: DPA1, HB2S, DPB1.
HLA-DQ: 2DA1, 2DA2, HA21, HA27, HB25, HB24, HB23, HB22, HB21, DQB2.
HLA-DR: 2DRA, HB2C, HB2B, DRB5, DRB4, DRB3, 2B1F, 2B1B, 2B1A, 2B19, 2B18, 2B17, 2B14, 2B11.
HLA-DO: 2DOA, 2DOB.
HLA-DM: 2DMA, 2DMB.
Killer cell immunoglobulin-like receptor (15 members): KI2L1, KI2L2, KI2L3, KI2L4, KI2LA, KI2LB, KI2S1, KI2S2, KI3L1, KI3L2, KI3L3, KI3S1, KI2S3, KI2S4, KI2S5 (Marsh et al., 2003).
Leukocyte-associated immunoglobulin-like receptor family (LIRA) (11 members): LIRA1, LIRA2, LIRA3, LIRA4, LIRA5, LIRA6, LIRB1, LIRB2, LIRB3, LIRB4, LIRB5 (Brown et al., 2004)
The first 10 families, with a total of 116 IgSF proteins were used for the inter-intra family relationships analysis in Fig. 2; the sequentially very similar and highly redundant last four families were excluded from this analysis.
The IgSF data set
Human secreted and integral membrane IgSF proteins were collected from Uniprot/SWISS-PROT database (Bairoch et al., 2005) on the basis of both curated and InterPro (Hunter et al., 2009) annotations. In order to identify IgSF proteins, we searched for certain regular expressions in the UNIPROT/SWISS-PROT flat-file (version date: February 9th 2010). Within the comment lines of the flat-file (“CC” lines) we searched for one of two expressions: “SIMILARITY: Contains … Ig-like…” or “SIMILARITY: Belongs to the immunoglobulin superfamily”. In the Database cross-Reference (“DR”) lines, we searched for one of the InterPro IgSF Ids (i.e., IPR003006, IPR003596, IPR003597, IPR003598, IPR003599, IPR013098, IPR013106, IPR013151, IPR013162, IPR007110, IPR013783, IPR013270, IPR008424, and IPR010457). Finally, we selected those proteins that had a description in their Feature Table (FT) lines of an Ig domain. Immunoglobulin (antibodies) and T-cell receptors were excluded from our dataset.
The Brotherhood Algorithm
The method compares the relationship between two query proteins by analyzing the fraction of intermediate (shared) proteins relative to all evolutionarily related proteins. First, an evolutionarily related group of proteins is constructed for each IgSF query using BLAST against the NCBI NR proteins database (Wheeler et al., 2008). Then a list is generated from the evolutionarily related group of the significant BLAST hits using an e-value cutoff of 0.001. In the second step, the evolutionary relationship between members of the IgSF is tested by comparing the overlap of the intersection of their Blast-derived groups with the overall size of the smaller Blast-group (Fig. 1A). A pair of query proteins is deemed “related” if the overlap is more than 45%. The overlap threshold of 45% was chosen empirically (Fig. 2).
Network of protein functional relationships
Networks were generated with Cytoscape (Shannon et al., 2003) using the Organic layout (Shannon et al., 2003). Nodes and edges represent proteins and evolutionary relationships between proteins, respectively. Protein evolutionary relationships were evaluated with BLAST e-values (Altschul et al., 1997), Brotherhood method, CD-HIT (Li and Godzik, 2006), and PSI-PHY (Brown et al., 2007). The BLAST network was constructed as described in Atkinson et al. (Atkinson et al., 2009). CD-HIT (Li and Godzik, 2006) was run locally with the following parameters in hierarchical order: first we clustered at 60% pairwise sequence identity and word size of four, we then clustered at 40% sequence identity using a word size of three, and finally we used the psi-cd-hit script from the CD-HIT suite to cluster at 30% sequence identity.
Molecular Cloning of CRTAM
The extracellular Ig-V domain of human CRTAM (residues 18–117) was cloned into the pET28a expression vector, expressed in E. coli and refolded from inclusion bodies as described by (Zhang et al., 2002) with minor modifications. The refolding buffer was composed of 200 nM Tris-HCl pH 8.0, 10 mM EDTA, 0.5 M L-Arginine, 6.5 mM cysteamine, and 3.7 mM cystamine. CRTAM refolded at at 4°C was subjected to gel filtration chromatography on superdex G-75.
Analytical ultracentrifugation sedimentation equilibrium experiments
Sedimentation equilibrium experiments were performed at 20°C using a Beckman XL-I analytical ultracentrifuge, six-sector cells, and an AN-60Ti rotor. Protein buffer was composed of 20 mM Tris-Hcl pH 8.2, 150 mM NaCl, and 1 mM EDTA. Absorption scans collected at 280 nm for three different protein concentrations (38 μM, 21 μM, and 5 μM) at rotor speeds of 20,000 or 25,000 rpm were globally analyzed using HeteroAnalysis version 1.1.44 (Cole, 2004). Equilibrium was confirmed by comparing scans taken at 22 and 24 hr at the indicated speed. Protein concentration was estimated from the extinction coefficient of 18575 M−1cm−1 determined using the ProtParam webserver (Gasteiger E., 2005). Buffer density and partial specific volume (0.7365) were calculated using SEDNTERP version 1.01 (Laue, 1992).
X-ray crystallization experiments
Diffraction quality crystals of CRTAM were obtained at 4°C with the sitting drop vapor diffusion method by mixing 0.3 μl of protein (12 mg/mL in 100 mM Tris-Hcl pH 8.2, 150 mM NaCl, 1 mM EDTA) with 0.3 μl of reservoir solution (0.49 M monobasic sodium phosphate monohydrate, 0.91 M dibasic potassium phosphate, pH 6.9) and allowing equilibration over 70 μl of reservoir solution. Prior to data collection, crystals were cryo-protected with mother liquor supplemented with 1:1 2M LiSO4 and flash-cooled in liquid nitrogen. Data extending to 2.3Å resolution were collected at a wavelength of 1.081Å at beam line X29A of the National Synchrotron Light Source using an ADSC Quantum-315 CCD detector. Diffraction data were indexed, integrated and scaled with HKL2000 (Otwinowski and Minor, 1997). Diffraction data from these crystals were initially processed in tetragonal point groups 4 and 422 with Rmerge of 9.3% and 8.3%, respectively. These data were used for molecular replacement with PHASER 1.1(McCoy et al., 2007) and the monomer of nec-l1 (34% identical; PDB Code 1Z9M (Dong et al., 2006)), truncated with CHAINSAW (CCP4-suite (Stein, 2008)) as the search model. After several rounds of modeling with COOT (Emsley and Cowtan, 2004) and refinement with REFMAC5 (Murshudov et al., 1997), the CRTAM model appeared complete, but Rfree remained around 37% indicating an anomaly in the diffraction data. Statistical tests for twinning, as implemented in CCP4-suite were not effective in confirming twinned. However, the observed similarity in the length of a and b axes in orthorhombic/monoclinic space groups, the packing of CRTAM-dimers parallel to ab-plane with four-fold symmetry, together with the difficulty to refine the structure in any tetragonal space groups led us to consider pseudo-merohedral twinning mimicking higher lattice symmetry. This existing model placed in C2221 cell and refined using the twin-law operator k, h, -l, converged with Rwork and Rfree of 0.199 and 0.233, respectively. The twin refinement substantially improved the quality of the electron density allowing us to model several residues with higher confidence. The final model contains four molecules of the CRTAM Ig-V domain, four phosphate ions and 120 water molecules. Non-native residues introduced at the C-terminus during the cloning process were disordered and not modeled. Data collection, phasing, and refinement statistics are presented in Table 1.
Supplementary Material
Highlights.
An intermediate sequence based clustering algorithm, Brotherhood, is introduced
Brotherhood method is used to assign functional families within the human IgSF
Newly annotated nectin-like family member, CRTAM structure is solved
Previously undescribed homophilic trans interaction is observed for CRTAM
Acknowledgments
We are grateful to Drs. Eduardo Fajardo, Rafael Toro, Chenyang Zhan and Michael Brenowitz and for their help with informatics, crystallization, crystallography and analytical ultracentrifugation, respectively. We gratefully acknowledge the staff of the X29 beamline at the National Synchrotron Light Source for their assistance in data collection. This work was supported by grants GM094665, GM094662, AI007289, GM096041 and CA13330.
Footnotes
The structure of the Ig-V domain of CRTAM has been deposited to the PDB as entry 3RBG. A computer program implementing the Brotherhood algorithm is available from the Authors on request.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One. 2009;4:e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. The Universal Protein Resource (UniProt) Nucleic acids research. 2005;33:D154–159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G. The JAM family of junctional adhesion molecules. Current opinion in cell biology. 2003;15:525–530. doi: 10.1016/s0955-0674(03)00104-2. [DOI] [PubMed] [Google Scholar]
- Boles KS, Barchet W, Diacovo T, Cella M, Colonna M. The tumor suppressor TSLC1/NECL-2 triggers NK-cell and CD8+ T-cell responses through the cell-surface receptor CRTAM. Blood. 2005;106:779–786. doi: 10.1182/blood-2005-02-0817. [DOI] [PubMed] [Google Scholar]
- Brown D, Trowsdale J, Allen R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue antigens. 2004;64:215–225. doi: 10.1111/j.0001-2815.2004.00290.x. [DOI] [PubMed] [Google Scholar]
- Brown DP, Krishnamurthy N, Sjolander K. Automated protein subfamily identification and classification. PLoS Comput Biol. 2007;3:e160. doi: 10.1371/journal.pcbi.0030160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annual review of immunology. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay K, Lazar-Molnar E, Yan Q, Rubinstein R, Zhan C, Vigdorovich V, Ramagopal UA, Bonanno J, Nathenson SG, Almo SC. Sequence, structure, function, immunity: structural genomics of costimulation. Immunol Rev. 2009;229:356–386. doi: 10.1111/j.1600-065X.2009.00778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL. Analysis of heterogeneous interactions. Methods Enzymol. 2004;384:212–232. doi: 10.1016/S0076-6879(04)84013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SJ, Ikemizu S, Evans EJ, Fugger L, Bakker TR, van der Merwe PA. The nature of molecular recognition by T cells. Nature immunology. 2003;4:217–224. doi: 10.1038/ni0303-217. [DOI] [PubMed] [Google Scholar]
- Dondoshansky I. Blastclust (NCBI Software Development Toolkit) NCBI; Bethesda, MD: 2002. [Google Scholar]
- Dong X, Xu F, Gong Y, Gao J, Lin P, Chen T, Peng Y, Qiang B, Yuan J, Peng X, Rao Z. Crystal structure of the V domain of human Nectin-like molecule-1/Syncam3/Tsll1/Igsf4b, a neural tissue-specific immunoglobulin-like cell-cell adhesion molecule. The Journal of biological chemistry. 2006;281:10610–10617. doi: 10.1074/jbc.M513459200. [DOI] [PubMed] [Google Scholar]
- Eguchi J, Wada J, Hida K, Zhang H, Matsuoka T, Baba M, Hashimoto I, Shikata K, Ogawa N, Makino H. Identification of adipocyte adhesion molecule (ACAM), a novel CTX gene family, implicated in adipocyte maturation and development of obesity. The Biochemical journal. 2005;387:343–353. doi: 10.1042/BJ20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Engel P, Eck MJ, Terhorst C. The SAP and SLAM families in immune responses and X-linked lymphoproliferative disease. Nat Rev Immunol. 2003;3:813–821. doi: 10.1038/nri1202. [DOI] [PubMed] [Google Scholar]
- Fogel AI, Li Y, Giza J, Wang Q, Lam TT, Modis Y, Biederer T. N-glycosylation at the SynCAM (synaptic cell adhesion molecule) immunoglobulin interface modulates synaptic adhesion. The Journal of biological chemistry. 2010;285:34864–34874. doi: 10.1074/jbc.M110.120865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger EHC, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. Humana Press; 2005. [DOI] [PubMed] [Google Scholar]
- Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol. 2000;1:REVIEWS0005. doi: 10.1186/gb-2000-1-5-reviews0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M. Measurement of the effectiveness of transitive sequence comparison, through a third ‘intermediate’ sequence. Bioinformatics. 1998;14:707–714. doi: 10.1093/bioinformatics/14.8.707. [DOI] [PubMed] [Google Scholar]
- Harrison OJ, Vendome J, Brasch J, Jin X, Hong S, Katsamba PS, Ahlsen G, Troyanovsky RB, Troyanovsky SM, Honig B, Shapiro L. Nectin ectodomain structures reveal a canonical adhesive interface. Nature structural & molecular biology. 2012;19:906–915. doi: 10.1038/nsmb.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SS, Chen R. Functional misassignment of genes. Nat Biotechnol. 2001;19:95. doi: 10.1038/84480. [DOI] [PubMed] [Google Scholar]
- John B, Sali A. Detection of homologous proteins by an intermediate sequence search. Protein Sci. 2004;13:54–62. doi: 10.1110/ps.03335004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. Journal of molecular biology. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Analytical ultracentrifugation in biochemistry and polymer science. Cambridge; England: 1992. [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Marsh SG, Parham P, Dupont B, Geraghty DE, Trowsdale J, Middleton D, Vilches C, Carrington M, Witt C, Guethlein LA, et al. Killer-cell immunoglobulin-like receptor (KIR) nomenclature report, 2002. Immunogenetics. 2003;55:220–226. doi: 10.1007/s00251-003-0571-z. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Park J, Teichmann SA, Hubbard T, Chothia C. Intermediate sequences increase the detection of homology between sequences. Journal of molecular biology. 1997;273:349–354. doi: 10.1006/jmbi.1997.1288. [DOI] [PubMed] [Google Scholar]
- Patino-Lopez G, Hevezi P, Lee J, Willhite D, Verge GM, Lechner SM, Ortiz-Navarrete V, Zlotnik A. Human class-I restricted T cell associated molecule is highly expressed in the cerebellum and is a marker for activated NKT and CD8+ T lymphocytes. J Neuroimmunol. 2006;171:145–155. doi: 10.1016/j.jneuroim.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Pegg SC, Babbitt PC. Shotgun: getting more from sequence similarity searches. Bioinformatics. 1999;15:729–740. doi: 10.1093/bioinformatics/15.9.729. [DOI] [PubMed] [Google Scholar]
- Pillai S, Netravali IA, Cariappa A, Mattoo H. Siglecs and immune regulation. Annual review of immunology. 2012;30:357–392. doi: 10.1146/annurev-immunol-020711-075018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. Protein structures sustain evolutionary drift. Fold Des. 1997;2:S19–24. doi: 10.1016/s1359-0278(97)00059-x. [DOI] [PubMed] [Google Scholar]
- Salamov AA, Suwa M, Orengo CA, Swindells MB. Combining sensitive database searches with multiple intermediates to detect distant homologues. Protein Eng. 1999;12:95–100. doi: 10.1093/protein/12.2.95. [DOI] [PubMed] [Google Scholar]
- Scanlan MJ, Ritter G, Yin BW, Williams C, Jr, Cohen LS, Coplan KA, Fortunato SR, Frosina D, Lee SY, Murray AE, et al. Glycoprotein A34, a novel target for antibody-based cancer immunotherapy. Cancer immunity. 2006;6:2. [PubMed] [Google Scholar]
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J Appl Crystallogr. 2008;41:641–643. [Google Scholar]
- Streydio C, Lacka K, Swillens S, Vassart G. The human pregnancy-specific beta 1-glycoprotein (PS beta G) and the carcinoembryonic antigen (CEA)-related proteins are members of the same multigene family. Biochem Biophys Res Commun. 1988;154:130–137. doi: 10.1016/0006-291x(88)90660-2. [DOI] [PubMed] [Google Scholar]
- Takai Y, Miyoshi J, Ikeda W, Ogita H. Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol. 2008;9:603–615. doi: 10.1038/nrm2457. [DOI] [PubMed] [Google Scholar]
- van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annual review of immunology. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, Lu LF, Gondek D, Wang Y, Fava RA, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic acids research. 2008;36:D13–21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Vendome J, Shapiro L, Ben-Shaul A, Honig B. Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature. 2011;475:510–513. doi: 10.1038/nature10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani U, Terman JR. The semaphorins. Genome biology. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JH, Sidhu SS, Chan AC. Regulation of a late phase of T cell polarity and effector functions by Crtam. Cell. 2008;132:846–859. doi: 10.1016/j.cell.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- Zhang X, Schwartz JC, Almo SC, Nathenson SG. Expression, refolding, purification, molecular characterization, crystallization, and preliminary X-ray analysis of the receptor binding domain of human B7-2. Protein Expr Purif. 2002;25:105–113. doi: 10.1006/prep.2002.1616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.