

Abstract
Chemoresistance, due to oxidative stress resistance or upregulation of Bcl-2, contributes to poor outcome in the treatment of hematological malignancies. In this study, we utilize the copper chelator drug, ATN-224 (choline tetrathiomolybdate), to induce cell death in oxidative stress resistant cells and cells overexpressing Bcl-2 by modulating the cellular redox environment and causing mitochondrial dysfunction. ATN-224 treatment decreases superoxide dismutase 1 (SOD1) activity, increases intracellular oxidants and induces peroxynitrite-dependent cell death. ATN-224 also targets the mitochondria, decreasing both cytochrome c oxidase (CcOX) activity and mitochondrial membrane potential (ΔΨm). The concentration of ATN-224 required to induce cell death is proportional to SOD1 levels, but independent of Bcl-2 status. In combination with doxorubicin, ATN-224 enhances cell death. In primary B cell acute lymphoblastic leukemia (B-ALL) patient samples, ATN-224 decreases the viable cell number. Our findings suggest that ATN-224’s dual targeting of SOD1 and CcOX is a promising approach for treatment of hematological malignancies either as an adjuvant or as a single agent.
Introduction
Homeostasis of the redox environment is maintained by balancing the generation of reactive oxygen species (ROS) and their removal by antioxidant defense enzymes. Disruption of this homeostasis, either through increased ROS production and/or decreased removal, results in oxidative stress. In comparison to their normal counterparts, cancer cells have increased oxidative stress [1], which can promote proliferation or survival [2]. The ability of cancer cells to adapt to increased ROS generation and maintain redox homeostasis is, in part, through the induction of ROS-scavenging enzymes [3]. Increases in oxidative stress have been associated with lymphoid malignancies [4]. In primary human lymphocytic leukemia cells, Hileman et al. measured an increase in oxidative stress as well as upregulated superoxide dismutase (SOD) and catalase, the antioxidant defense enzymes responsible for detoxification of superoxide and hydrogen peroxide, respectively [5]. Upregulation of antioxidant defenses can impact drug resistance. In a lymphoma model, our laboratory previously demonstrated that upregulation of SOD and catalase results in oxidative stress resistance and multi-drug chemoresistance [6].
Recent evidence suggests that the anti-apoptotic function of Bcl-2 (B cell lymphoma/leukemia 2) is partially dependent on the ability of Bcl-2 to regulate the redox environment [7]. Bcl-2 is commonly overexpressed in lymphoid malignancies and is associated with chemoresistance [8, 9]. The Bcl-2 family consists of pro-apoptotics, such as BH3 only proteins, and anti-apoptotics, such as Bcl-2, Bcl-xL and Mcl-1, that interact with one another to regulate mitochondrial outer membrane permeabilization (MOMP), a key event in the mitochondrial intrinsic death pathway [9]. The canonical function of Bcl-2 is to prevent MOMP through direct interactions with pro-apoptotic proteins, while the noncanonical function of Bcl-2 is to regulate mitochondrial respiration [10], which may account for its ability to alter the redox environment. Chen et al. showed that Bcl-2 regulates the activity of cytochrome c oxidase (CcOX), a redox driven proton pump, through direct and indirect interactions with the CcOX subunits Va and Vb, respectively. In response to oxidative stress, mitochondrial redox homeostasis is maintained in cells with Bcl-2 upregulation; mitochondrial redox homeostasis is not maintained in cells without Bcl-2 upregulation [11].
Targeting the ability of cancer cells to adapt to and survive oxidative stress is an appealing therapeutic strategy. In theory, tumor cells with intrinsically higher oxidant levels than their normal counterparts would be more vulnerable to the toxic effects of agents that increase oxidants [3]. Based on this idea, we hypothesized that ATN-224 (choline tetrathiomolybdate), a copper chelator drug that targets the copper-dependent enzymes SOD1 and CcOX [12], would induce cell death in lymphoma, a tumor type associated with increased oxidative stress [4]. In this study, we found that ATN-224 induced oxidant-dependent cell death in lymphoma cell culture models at low nanomolar concentrations. ATN-224-induced cell death was independent of oxidative stress resistance or increased Bcl-2. Primary B-ALL cells, a clinically relevant model of a tumor type that has elevated Bcl-2, were similarly sensitive to ATN-224. These data suggest that: 1) ATN-224, an agent that has been tested in clinical trials for solid tumors and multiple myeloma [13], has potential for the treatment of resistant lymphoid malignancies; and 2) lymphoid malignancies in general are sensitive to agents that increase oxidants.
Materials and Methods
Drug Treatments and Reagents
ATN-224 was provided by Dr. Andrew Mazar (Northwestern University, Evanston, IL). The EC50 was determined for each cell type and all subsequent experiments were carried out using the EC50 unless otherwise stated. MnTE-2-PyP5+ and MnTBAP3− were provided by Dr. Ines Batinić-Haberle (Duke University, Durham, NC). MnTE-2-PyP5+ was synthesized as described [14]. MnTBAP3− was synthesized, purified and analyzed by several methods, including LCMS as described [15, 16]. The purity and stability of both compounds were addressed by the studies of Reboucas et al. [16, 17]. FeTM-4-PyP5+, which exists as a monohydroxo species (OH)FeTM-4-PyP4+ at pH 7.8 [14], was purchased from Cayman (Ann Arbor, MI). Concentrations of all metalloporphyrins used in this study were chosen based on preliminary studies. All other drugs and chemicals were purchased from Sigma Aldrich Co. (St. Louis, MO) unless otherwise stated.
Cell Culture
Murine thymic lymphoma WEHI7.2 cells, WEHI7.2 cells overexpressing Bcl-2 [18] (Hb12 cells) and WEHI7.2 cells selected for resistance to 200 μM H2O2 [19] (200R cells) were maintained as previously described [19, 20]. For one week prior to each experiment, 200R cells were cultured in medium without H2O2.
Human histiocytic lymphoma U937 cells were obtained from Dr. Terry Landowski (University of Arizona). Human acute T cell lymphoblastic leukemia Molt-4 cells were obtained from the Arizona Lymphoid Tissue and Blood Repository. Cells were maintained in suspension in RPMI 1640 (Cell Gro, Manassas, VA) supplemented with 10% fetal bovine serum (Gemini, Sacremento, CA), 2 mM L-glutamine (Invitrogen) and 50 U/mL each of penicillin and streptomycin (Invitrogen) at 37°C in a 5% CO2 humidified environment. Molt-4 ρ0 cells were obtained from Dr. Lionel Lewis (Dartmouth, Hanover, NH) and were maintained in the same medium supplemented with 50 μg/mL uridine and 100 μg/mL sodium pyruvate.
Primary Acute Leukemia Cells
Primary human samples with the diagnosis of precursor B cell acute lymphoblastic leukemia (B-ALL) were obtained from the Arizona Lymphoid Tissue and Blood Repository in accordance with the University of Arizona regulations for the use of primary human tissue under an IRB approved protocol. The cells were thawed and resuspended in Iscoves’s modified Dulbecco’s medium (IMDM; Invitrogen) supplemented with 20% fetal bovine serum (Gemini) and 1% glutamine (Invitrogen). Viable B-cell content of the sample was analyzed at thaw. Cells were incubated at 37°C in a 5% CO2 humidified environment in the presence of vehicle control or ATN-224 for 24 hours. Cells were labeled for 20 min with both phycoerythrin-labeled anti-CD20 (AbD Serotec, Raleigh, NC), to identify B-cells, and 7-Aminoactinomycin D (7-AAD) (R & D Systems Inc., Minneapolis, MN), a membrane impermeant dye that intercalates with DNA in dying cells. CD20-positive/7-AAD-negative cells were considered viable B cells and measured using FACScan flow cytometer with Cell Quest software (Becton Dickinson, Franklin Lakes, NJ). A minimum of 5,000 events were analyzed per sample. The percentage of viable B cells in the sample incubated for 24 hours in the absence of drug was set to 100%.
Cell Viability Measurements
The number of viable treated cells, relative to control treated cells, were measured after 48 or 72 hours of treatment in murine and human cell lines, respectively, using the Non-radioactive Cell Proliferation Assay (MTS) according to the manufacturer’s instructions (Promega Corp., Madison, WI). Absorbance was read at 490 nm using a Synergy HT plate reader (BioTek Instruments, Winooski, VT). The MTS assay was used to determine the estimated ATN-224 concentration needed to decrease the number of viable cells by 50 percent (EC50) and the effect of drugs alone or in combination with ATN-224. Viable cell number was also determined by propidium iodide (Molecular Probes, Eugene, OR) a membrane impermeant dye that intercalates with DNA in dying cells, which was measured using an EPICS XL-MCL flow cytometer (Coulter, Corp., Miami, FL). Ten thousand cells were analyzed per sample. Numbers less than 5% different were considered within the error of the machine after calibration for this assay.
Trypan Blue Staining
The number of viable treated cells, relative to control treated cells, was measured after 24 and 48 hours of treatment in human Molt-4 ρ0 cells, using trypan blue. An equal volume of cells was mixed with an equal volume of trypan blue and transferred to a hemocytometer. Cells that excluded dye were counted as viable.
Protein Levels
Cellular and mitochondrial protein levels were measured using the BCA Protein Assay kit (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer’s instructions. Absorbance was read at 562 nm using a Synergy HT plate reader (BioTek Instruments).
Superoxide Dismutase 1 (SOD1) and Cytochrome c Oxidase (CcOX) Activity
SOD1 was measured using the SOD Determination kit (Sigma), according to the manufacturer’s instructions. Diethyldithiocarbamate (DDC) treatment of a control sample [21] was used to confirm that the measured SOD activity was specific to SOD1. CcOX was measured by monitoring oxidation of reduced cytochrome c, using a protocol adapted from Zhang et al. [22]. Briefly, mitochondria were collected using the Mitochondrial Isolation kit for Cultured Cells (Thermo Fisher Scientific) and resuspended in respiration medium. Potassium cyanide (KCN) was used to confirm the measured activity was specific to CcOX. SOD1 and CcOX activity were normalized to cellular and mitochondrial protein, respectively.
Caspase 3 Activity
Caspase 3 activity was measured using an assay dependent on the enzymatic cleavage of a synthetic caspase 3 specific substrate, Ac-DEVD-p-nitroanilide (pNA) (Enzo Life Science, Plymouth Meeting, PA), as described previously [23]. Caspase 3 activity was normalized to cellular protein.
Measurements of Reactive Species
Reactive oxygen/nitrogen species were measured using the fluorescent probe TEMPO-9-AC (Molecular Probes). Cells were incubated with 10 μM TEMPO-9-AC for 1 hour at 37°C in a 5% CO2 humidified environment. Cells were then washed with PBS, resuspended in PBS and transferred to a black well plate. TEMPO-9-AC (Ex: 360/Em: 460) fluorescence was measured using a Synergy HT plate reader (Bio Tek Instruments). Fluorescence was normalized to cellular protein.
Measurement of Nitrotyrosine
Nitrotyrosine levels were assessed by flow cytometry. Cells were fixed in 4% formaldehyde for 10 minutes, permeabilized in 90% methanol for 30 minutes, and blocked in incubation buffer (5% BSA in PBS) for 10 minutes. Cells were then incubated with anti-nitrotyrosine (EMD Millipore, Billerica, MA) for 60 minutes, washed, incubated with Alexa Fluor® 647-linked anti-mouse Ig (Cell Signaling, Danvers, MA) for 30 minutes in the dark, washed and then resuspended in PBS. Fluorescence was measured using an EPICS XL-MCL flow cytometer (Coulter, Corp.). A minimum of 10,000 events were analyzed per sample. Fluorescence was normalized to control-treated cells.
ΔΨm
The fluorescent probe JC-1 (Molecular Probes) was used to measure ΔΨm. Cells were incubated with 2 μg/mL JC-1 for 30 minutes at 37°C in a 5% CO2 humidified environment. Cells were then washed with PBS, resuspended in PBS and transferred to a black well plate. JC-1 J-aggregates (Ex: 560/Em: 595) were measured using a Synergy HT plate reader (Bio Tek Instruments). Fluorescence was normalized to cellular protein.
Immunoblots
Proteins from total cell lysates were separated by SDS-PAGE and transferred to PVDF membrane using standard protocols. Blots were probed with antibodies for SOD1 (AbCam, Cambridge, MA), Bcl-2 (BD Pharmingen, San Diego, CA), CcOX Va, CcOX Vb (Invitrogen). Proteins were detected by incubating with either horseradish peroxidase-linked anti-rabbit Ig or horseradish peroxidase-linked anti-mouse Ig (Cell Signaling), where appropriate, and visualized by chemiluminescence (Perkin Elmer, Waltham, MA). Blots were also probed with anti-β actin (AbCam) as a loading control. To visualize multiple bands on the same blot, blots were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific) before being probed with a new antibody.
Drug Synergy Analysis
To determine whether the interactions between ATN-224 and doxorubicin were antagonistic, additive or synergistic the following mathematical drug synergy model was used:
where Fa is the fractional response to drug A alone, Fb is the fractional response of drug B alone, and ER is the expected response when the two drugs interact in an additive manner [24]. If the observed response (OR) is less than the ER, the interaction between the two drugs is antagonistic. If the OR is equal to the ER, the interaction of the two drugs is additive. If the OR is greater than the ER, the interaction between the two drugs is synergistic.
Statistics
Means were compared using student’s t-tests with the algorithm in Excel (Microsoft Corp., Redmond, WA). Means were considered significantly different when p ≤ 0.05. When a comparison required multiple t-tests, the Dunn-Bonferroni method was used to control for type I error [25].
Results
Oxidant/drug resistant profile of the cell culture model system
To determine the ability of ATN-224 to overcome oxidative stress resistance and elevated Bcl-2 we utilized the WEHI7.2 and WEHI7.2 variant cell culture model. The WEHI7.2 variants include WEHI7.2 cells selected for resistance to hydrogen peroxide (200R) and WEHI7.2 cells overexpressing Bcl-2 (Hb12 cells) (Figure 1A). Our laboratory has previously reported that the 200R cells are oxidative stress resistant and have increased resistance to chemotherapeutics [6, 19]. These data are shown in Table 1 and 2 for reference purposes.
Figure 1. ATN-224 induced cell death in WEHI7.2 and WEHI7.2 variant cells.
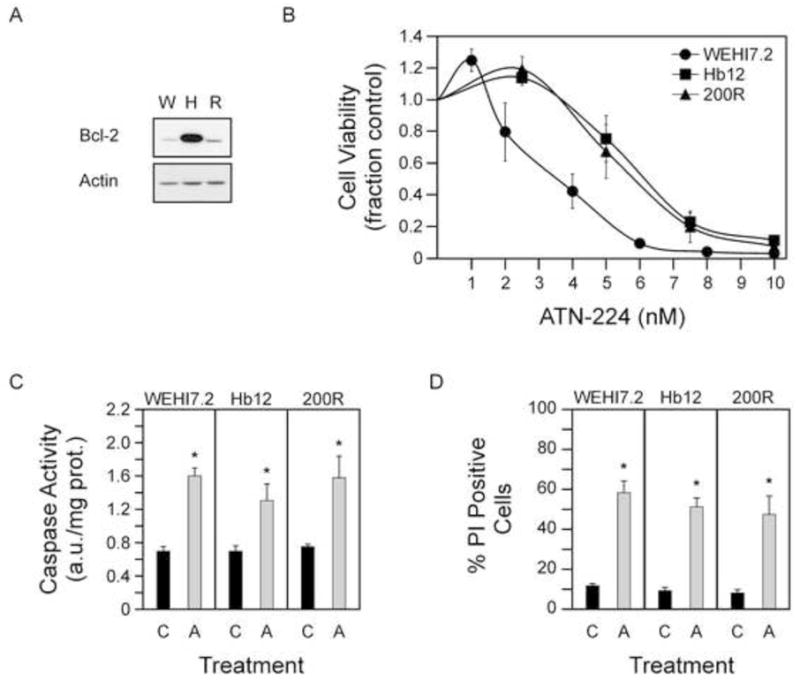
A. Immunoblot showing Bcl-2 (26 kDa) protein levels in WEHI7.2 (W), Hb12 (H) and 200R (R) cells. Immunoblot showing actin (42 kDa) protein levels to demonstrate similar loading. B. Cell viability in WEHI7.2 and WEHI7.2 variant cells treated with nanomolar concentrations of ATN-224. C. Caspase 3 activity measured in WEHI7.2 (24 h), Hb12 (18 h) and 200R (30 h) cells treated with vehicle (C) or ATN-224 (A). D. Propidium iodide uptake in WEHI7.2 (30 h), Hb12 (30 h) and 200R (36 h) cells treated with vehicle (C) or ATN-224 (A). All values are mean + SEM (n ≥). *, significantly different from vehicle treated control cells (p ≤ 0.05).
Table 1.
Oxidant sensitivity in the WEHI7.2 cells and the WEHI7.2 variants as measured by EC50 values
| Cell Variant | H2O2 (μM) | tert-butyl hydroperoxide (μM) | 4-hydroxynonenol (μM) | paraquat (μM) |
|---|---|---|---|---|
| WEHI7.2# | 105.37 ± 2.94 | 0.939 ± 0.045 | 12.67 ± 1.23 | 32.73 ± 6.68 |
| 200R# | 1049.26 ± 9.83* | 1.098 ± 0.068 | 20.33 ± 0.83* | 77.00 ± 8.96* |
| Hb12 | 531.70 ± 21.02* | 1.215 ± 0.029* | 30.52 ± 1.05* | 316.03 ± 25.42* |
Values represent the mean ± SEM (n ≥ 3).
These data have been previously published (6).
denotes significantly greater than WEHI7.2 values (p ≤ 0.05).
Table 2.
Hematological chemotherapeutic sensitivity in the WEHI7.2 cells and the WEHI7.2 variants.
| Cell Variant | Cyclophosphamide EC50 (mM)a | Doxorubicin EC50 (nM)a | Doxorubicin EC90 (nM)a | Vincristine EC50 (nM)a | Vincristine EC90 (nM)a | Dexamethasone (% apoptosis)b |
|---|---|---|---|---|---|---|
| WEHI7.2# | 1.82 ± 0.03 | 8.60 ± 0.58 | 23.74 ± 0.65 | 2.37 ± 0.17 | 4.26 ± 0.34 | 23.79 ± 1.30 |
| 200R# | 5.71 ± 0.18* | 11.40 ± 1.11 | 28.66 ± 0.72* | 9.01 ± 3.90 | 48.43 ± 3.53* | 10.16 ± 0.28* |
| Hb12 | 6.34 ± 0.39* | 23.36 ± 0.84* | 63.43 ± 2.80* | 2.51 ± 0.30* |
Values represent the mean ± SEM (n ≥ 3).
Values are the mean percent apoptosis after a 24h treatment with 1 μM dexamethasone corrected for % apoptosis in vehicle-treated cells (n = 3).
These data have been previously published (6).
denotes significantly different from WEHI7.2 values (p ≤ 0.05).
To evaluate whether the Hb12 cells have a similar resistance profile we first measured cell viability following treatment with multiple oxidative stress inducing agents: hydrogen peroxide; tert-butyl-hydroperoxide, which is involved in lipid peroxidation and oxidation of thiols; 4-hydroxynonenol, a lipid peroxidation byproduct that damages proteins and lipids; and paraquat, which generates superoxide [6]. To establish resistance we compared the concentrations needed to decrease the number of viable cells by 50 percent (EC50) in the Hb12 cells to that in the parental WEHI7.2 cells. As indicated by the higher EC50 values, the Hb12 cells demonstrated decreased sensitivity to hydrogen peroxide, tert-butyl-hydroperoxide, 4-hydroxynonenol and paraquat (Table 1), indicating that overexpression of Bcl-2 in the WEHI7.2 cells results in oxidative stress resistance.
Next we measured cell viability following treatment with multiple chemotherapeutics commonly used to treat non-Hodgkin lymphoma: cyclophosphamide, an alkylating agent; doxorubicin, a DNA intercalating agent; vincristine, a mitotic inhibitor; and dexamethasone, a synthetic glucocorticoid. The Hb12 cells exhibited decreased sensitivity to cyclophosphamide, doxorubicin, vincristine and dexamethasone (Table 2) indicating that overexpression of Bcl-2 in the WEHI7.2 cells also results in chemoresistance.
ATN-224 induces cell death in oxidative stress resistant lymphoma cells
To determine the effect of ATN-224 on the WEHI7.2 and WEHI7.2 variant cells, we measured cell viability following ATN-224 treatment. In the WEHI7.2 and WEHI7.2 variant cells, nanomolar concentrations of ATN-224 decreased the number of viable cells (Figure 1B). The EC50 values for the WEHI7.2, Hb12 and 200R cells were 3.17 ± 0.27 nM, 5.84 ± 0.34 nM and 5.25 ± 0.32 nM, respectively. Although the EC50 values for the WEHI7.2 variant cells were significantly higher in comparison to the WEHI7.2 parental cells (p ≤ 0.05), the Hb12 and 200R cells were sensitive to low nanomolar concentrations. These results indicate that oxidative stress resistant cells are sensitive to ATN-224.
To determine whether the decrease in viable cell number was attributed to cell death, we measured caspase 3 activity and propidium iodide (PI) uptake, two markers of cell death. ATN-224 treatment in the WEHI7.2 and WEHI7.2 variant cells resulted in significant increases in caspase 3 activities (Figure 1C) and PI uptake (Figure 1D). These results indicate that ATN-224 is capable of inducing cell death in oxidative stress resistant cells at nanomolar concentrations.
ATN-224 targets SOD1 and increases superoxide levels
The primary target of ATN-224 is SOD1 in other cell types [26]. Our laboratory has previously shown that SOD activity is significantly increased in the 200R cells [6, 19]. To determine whether SOD activity was increased in the Hb12 cells, we measured the activity and found that it was significantly increased (Figure 2A). Since the WEHI7.2 variant cells have significantly increased SOD activity and significantly higher ATN-224 EC50 values, we tested whether ATN-224 was targeting SOD1. In the WEHI7.2 and WEHI7.2 variant cells, ATN-224 treatment resulted in a time dependent decrease in SOD1 activity; the decrease was significant by 3 h after ATN-224 addition and was nearly abolished by 12 h (Figure 2B). To rule out whether these decreases in activity were due to protein degradation, we looked at the protein levels of SOD1 following ATN-224 treatment. The levels of SOD1 did not change; indicating the decrease in SOD1 activity was not due to protein degradation, but most likely the loss of the redox-active metal center (Figure 2C). These results suggest that the response to ATN-224 is roughly proportional to SOD activity.
Figure 2. ATN-224 targets SOD1 and increases ROS.
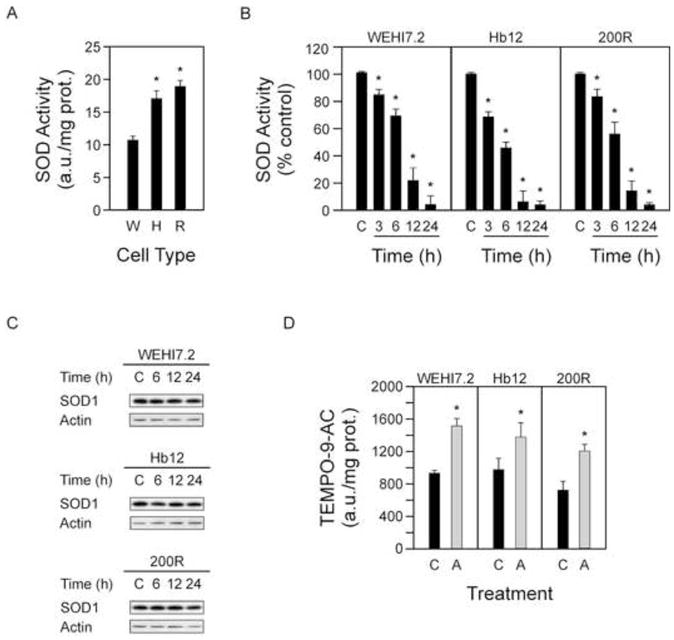
A. SOD activity in WEHI7.2 (W), Hb12 (H) and 200R (R) cells. B. SOD1 activity in WEHI7.2 and WEHI7.2 variant cells treated with ATN-224 at 3, 6, 12 and 24 h time points, relative to vehicle (C). C. Immunoblots showing SOD1 (18 kDa) protein levels in the WEHI7.2 and WEHI7.2 variant cells treated with vehicle (C) or ATN-224 at 6, 12 and 24 h time points. Immunoblots showing actin (42 kDa) protein levels to demonstrate similar loading. D. TEMPO-9-AC fluorescence in WEHI7.2 (18 h), Hb12 (12 h) and 200R (24 h) cells treated with vehicle (C) or ATN-224 (A). All values are mean + SEM (n ≥). *, significantly different from vehicle-treated control cells (p ≤ 0.05).
SOD1 is responsible for the dismuation of superoxide, thus decreases in SOD1 activity should lead to increased levels of superoxide and peroxynitrite, the reaction product of superoxide and nitric oxide that has been implicated in cell death signaling [27]. To determine whether ATN-224 treatment causes an increase in these species prior to cell death, we assessed their levels at time points preceeding caspase 3 activation and the appearance of PI positive cells, employing the fluorescent spin trap TEMPO-9-AC. We measured a significant increase in TEMPO-9-AC fluorescence in the WEHI7.2 and WEHI7.2 variant cells following ATN-224 treatment (Figure 2D). The increased levels of superoxide/peroxynitrite preceed caspase 3 activation and the appearance of PI positive cells, consistent with superoxide/peroxynitrite being involved in ATN-224 induced cell death.
ATN-224 induces peroxynitrite-dependent cell death
To determine which reactive species is responsible for ATN-224 induced cell death we used a series of metalloporphyrins. First we tested whether treatment with either MnTE-2-PyP5+ or (OH)FeTM-4-PyP4+, two porphyrins that can act as SOD mimetics to scavenge superoxide, but are also capable of scavenging peroxynitrite [14, 28], were protective. When added in combination with ATN-224, both porphyrins blocked the effect of ATN-224 in the WEHI7.2 and WEHI7.2 variant cells (Figure 3A). These results indicate that superoxide and/or peroxynitrite are involved in ATN-224 induced cell death.
Figure 3. ATN-224 induced cell death is peroxynitrite-dependent.
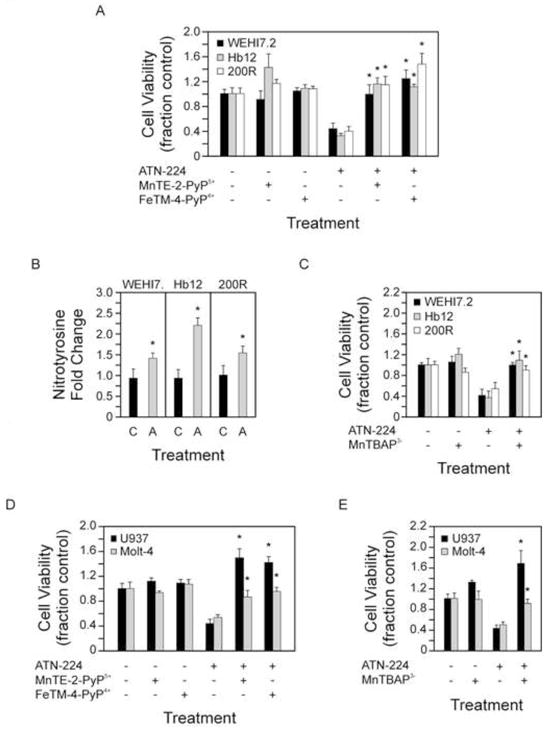
A. Cell viability in WEHI7.2 and WEHI7.2 variant cells treated with vehicle, ATN-224, 50 nM MnTE-2-PyP5+, 1 μM (OH)FeTM-4-PyP4+, or a combination of ATN-224 and either 50 nM MnTE-2-PyP5+ or 1 μM (OH)FeTM-4-PyP4+ for 48 h. B. Nitrotyrosine levels in WEHI7.2 and WEHI7.2 variant cells treated with vehicle (C) or ATN-224 (A) for 24 h. C. Cell viability in WEHI7.2 and WEHI7.2 variant cells treated with vehicle, ATN-224, 100 μM MnTBAP3− or a combination of ATN-224 and 100 μM MnTBAP3− for 48 h. D. Cell viability in U937 and Molt-4 cells treated with vehicle, ATN-224, 50 nM MnTE-2-PyP5+, 1 μM (OH)FeTM-4-PyP4+, or a combination of ATN-224 and either 50 nM MnTE-2-PyP5+ or 1 μM (OH)FeTM-4-PyP4+ for 72 h. E. Cell viability in U937 and Molt-4 cells treated with vehicle, ATN-224, 50 μM MnTBAP3− or a combination of ATN-224 and 50 μM MnTBAP3− for 72 h. All values are mean + SEM (n ≥). *, significantly different from ATN-224 treated control cells (p ≤ 0.05).
Superoxide is a short lived radical that is rarely linked directly to the induction of cell death [10]. Peroxynitrite, a downstream product of superoxide, is a highly reactive oxidant that can nitrate biomolecules, specifically tyrosine residues, and can readily induce cell death [27]. To assess the involvement of peroxynitrite, the levels of nitrotyrosine were measured following ATN-224 treatment. We detected a significant increase in nitrotyrosine levels in the WEHI7.2 and WEHI7.2 variant cells treated with ATN-224 (Figure 3B). These results indicate protein nitration in ATN-224 treated cells. We tested the ability of MnTBAP3−, which is not an SOD mimetic and is specific for peroxynitrite [29], to protect cells from ATN-224. In the WEHI7.2 and WEHI7.2 variants, MnTBAP3− abrogated the effect of ATN-224 (Figure 3C). MnTBAP3− has a 1000–2000 fold lower efficiency for peroxynitrite binding than MnTE-2-PyP5+, based on the rate constants, kred, for peroxynitrite reduction [30]. This fits with the higher concentration needed to achieve the same result as MnTE-2-PyP5+. Taken together these results indicate that ATN-224 induced cell death is peroxynitrite-dependent.
To determine whether these findings extend to human models and other types of hematological malignancies we tested ATN-224 in combination with either MnTE-2-PyP5+, (OH)FeTM-4-PyP4+ or MnTBAP3− in the histiocytic lymphoma U937 and acute T cell lymphoblastic leukemia Molt-4 cells (ATN-224 EC50 values 16 nM and 30 nM, respectively). All porphyrins blocked the effect of ATN-224 in the U937 and Molt-4 cells (Figure 3D-3E). These results indicate that ATN-224 is capable of inducing peroxynitrite-dependent cell death in human hematological malignancies.
ATN-224 induces cell death independent of Bcl-2 degradation
In other cell types, increased oxidants cause Bcl-2 degradation through the ubiquitin-proteasomal pathway [31]. The ability of ATN-224 to induce death in cells overexpressing the anti-apoptotic Bcl-2 suggests that Bcl-2 may be an indirect target of ATN-224. To determine whether ATN-224 treatment degrades Bcl-2, thus contributing to ATN-224 induced cell death, we compared the Bcl-2 protein levels in the WEHI7.2 and WEHI7.2 variant cells following ATN-224 treatment. Bcl-2 levels remained unaffected before and after increases in caspase 3 activities were detected (Figure 4A), therefore Bcl-2 degradation is not involved in ATN-224 induced cell death. These results indicate that ATN-224 induces cell death through a mechanism other than Bcl-2 degradation.
Figure 4. ATN-224 effects on the mitochondria.
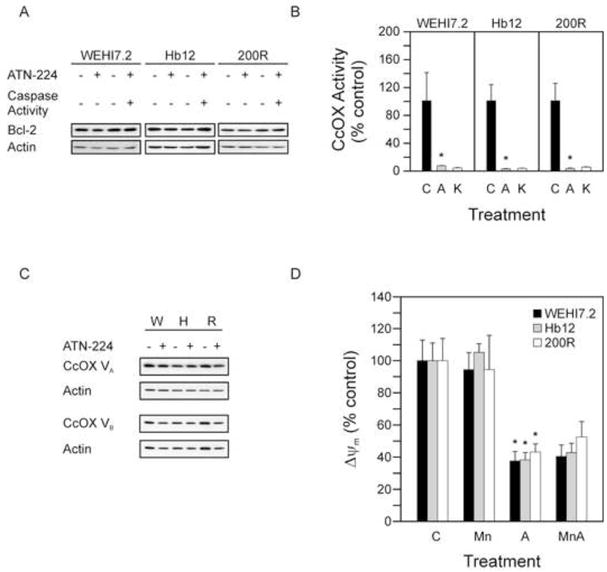
A. Immunoblot showing Bcl-2 (26 kDa) protein levels in WEHI7.2 and WEHI7.2 variant cells treated with vehicle or ATN-224 before and after caspase 3 activation. B. CcOX activity in WEHI7.2 and WEHI7.2 variant cells treated with vehicle (C) or ATN-224 (A) for 12 h. KCN (K) included as a positive control. C. Immunoblots showing CcOX subunits Va (16.8 kDa) and Vb (13.7 kDa) protein levels in WEHI7.2 and WEHI7.2 variant cells treated with vehicle control or ATN-224. Immunoblots showing actin (42 kDa) protein levels to demonstrate similar loading. D. JC-1 aggregates in WEHI7.2 and WEHI7.2 variant cells treated with vehicle (C), 50 nM MnTE-2-PyP5+ (Mn), ATN-224 (A) or a combination of 50 nM MnTE-2-PyP5+ and ATN-224 (MnA) for 6 h. All values are mean + SEM (n ≥). *, significantly different from vehicle treated control cells (p ≤ 0.05).
ATN-224 inhibits CcOX and decreases ΔΨm
Bcl-2 regulates the redox environment and prevents cell death [10]. This may, to some extent, be attributed to Bcl-2’s regulation of mitochondrial respiration through CcOX [11]. Using isolated mitochondria Juarez et al. showed that ATN-224 partially inhibited CcOX activity [12]. Since we measured minimal SOD1 activity after 12 h of ATN-224 treatment, we assessed CcOX activity at this time point to determine whether ATN-224 is targeting CcOX. ATN-224 treatment in the WEHI7.2 and WEHI7.2 variant cells abolished CcOX activity (Figure 4B). To rule out whether these decreases in activities were due to protein degradation we looked at the protein levels of the two copper subunits of CcOX, Va and Vb (Figure 4C). Immunoblots of CcOX Va and CcOX Vb confirm that the reduction in CcOX activity is not due to protein degradation. These results indicate that CcOX is a target of ATN-224.
CcOX tightly controls ΔΨm [32]. To determine whether loss of CcOX activity affects ΔΨm, we measured ΔΨm in WEHI7.2 and WEHI7.2 variant cells following treatment with either MnTE-2-PyP5+, ATN-224 or a combination of MnTE-2-PyP5+ and ATN-224. An approximate 60% decrease in ΔΨm was observed as early as 6 h (Figure 4D) in both the combination of MnTE-2-PyP5+ and ATN-224, and ATN-224 alone. These results suggest that ability of ATN-224 to overcome Bcl-2 may, in part, be due to its ability to efficiently target CcOX thus disrupting the ability of Bcl-2 to modulate mitochondrial respiration.
ATN-224 induces death through dual targeting of SOD1 and CcOX
We have shown that both SOD1 and CcOX are targets of ATN-224. To elucidate the relative importance of either SOD1 and/or CcOX we attempted to target each individually. We first tested whether inhibition of CcOX induced cell death by treating cells with 2 mM KCN, the concentration used for inhibition in the CcOX activity assay. After a 48 h treatment, there was no significant increase in propidium iodide uptake in the WEHI7.2 or WEHI7.2 variant cells (Figure 5A). These data suggest targeting CcOX alone is not enough to induce cell death.
Figure 5. Inhibition of SOD1 is important for ATN-224 induced cell death.
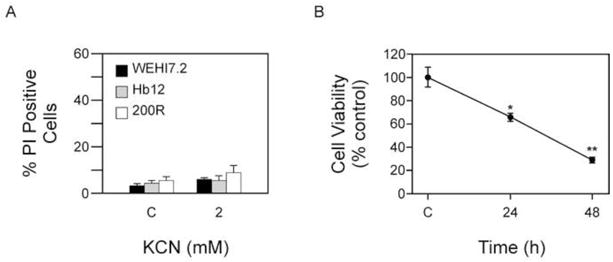
A. Propidium iodide uptake in WEHI7.2 and WEHI7.2 variant cells treated with vehicle (C) or 2 mM KCN for 48 h. B. Tyrpan blue staining in Molt-4 ρ0 cells treated with ATN-224 for 24 and 48 h. All values are mean + SEM (n ≥). *, significantly different from vehicle treated control cells and ** significantly different from 24 and 48 h ATN-224 treated cells (p ≤ 0.05).
To measure the importance of inhibiting SOD1 we used Molt-4 ρ0 cells, which have previously been shown to have minimal CcOX activity and decreased ΔΨm [33]. In culture these cells do not have a greater baseline amount of cell death than the parental cells (data not shown). We predicted that if inhibiting SOD1 contributed to the observed cell death, the Molt-4 ρ0 cells should be sensitive to ATN-224. In the Molt-4 ρ0 cells treated with 30 nM ATN-224 we measured a ~35% decrease in cell viability at 24 h, which further decreased to ~70% at 48 h (Figure 5B), indicating the Molt-4 ρ0 cells were sensitive to ATN-224. Taken together these results suggest that inhibiting SOD1 is important for the observed effect; however, CcOX inhibition may also contribute to ATN-224 induced cell death
Superoxide enhances ATN-224 induced cell death
The ability of ATN-224 to decrease SOD1 activity may result in increased sensitivity to ROS, superoxide specifically. To determine whether ATN-224 sensitizes cells to superoxide we tested ATN-224 in combination with paraquat, a compound that produces superoxide [34]. Treatment of the WEHI7.2 cells and WEHI7.2 variants with ATN-224 in combination with paraquat resulted in a significant increase in caspase 3 activities, in comparison to ATN-224 alone (Figure 6A).
Figure 6. ATN-224 enhances paraquat and chemotherapeutic drug-induced cell death.
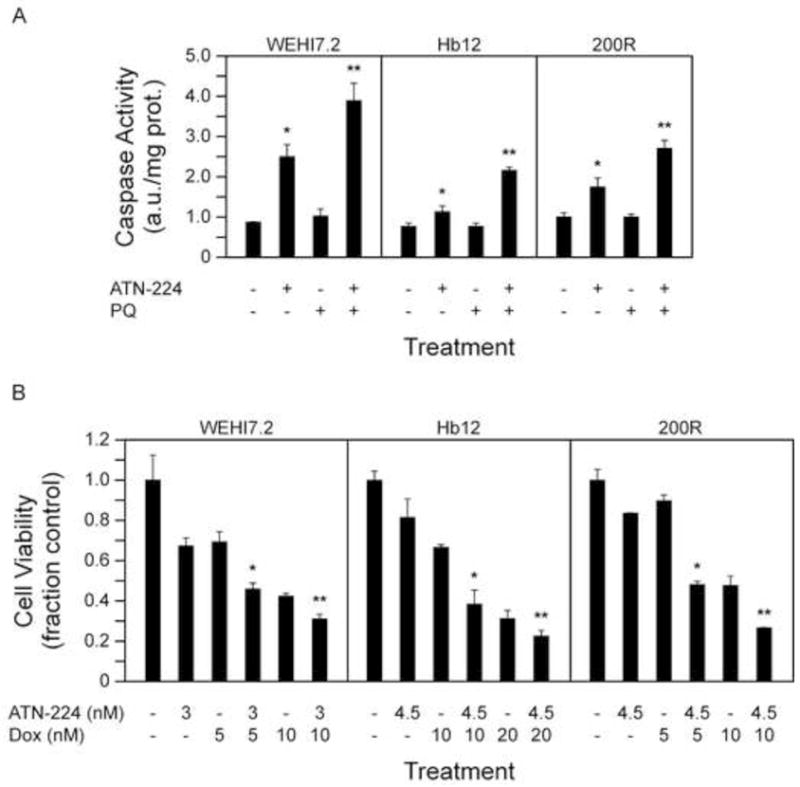
A. Caspase 3 activity measured in WEHI7.2 (24 h), Hb12 (18 h) and 200R (30 h) cells treated with vehicle, ATN-224, 50 μM paraquat (PQ) or a combination of ATN-224 and paraquat. B. Cell viability in WEHI7.2 and WEHI7.2 variant cells treated with vehicle, ATN-224 (3 nM WEHI7.2; 4.5 nM Hb12 and 200R), doxorubicin (5 nM and 10 nM WEHI7.2 and 200R; 10 nM and 20 nM Hb12) or a combination of ATN-224 and doxorubicin. All values are mean + SEM (n ≥). *, significantly different from vehicle treated control cells (p ≤ 0.05). **, significantly different from ATN-224 treated cells (p ≤ 0.05).
These data suggest that ATN-224 has the potential to enhance the effect of current chemotherapeutics, such as doxorubicin, which produce superoxide [35]. To determine whether ATN-224 sensitized the cells to doxorubicin we combined slightly lower concentrations of ATN-224 (3 nM for WEHI7.2 cells; 4.5 nM for Hb12 and 200R cells) with two concentrations of doxorubicin (5 nM and 10 nM for WEHI7.2 and 200R cells; 10 nM and 20 nM for Hb12). The combination of ATN-224 with doxorubicin resulted in an enhanced effect, in comparison to either drug alone (Figure 6B). To determine whether the enhanced effect was additive or synergistic we calculated the estimated response (ER) and compared it to the observed response (OR) using the Chou and Talalay model [24]. The combination of ATN-224 with doxorubicin was at least additive in the WEHI7.2 and WEHI7.2 variant cells (OR = ER), and at some concentrations synergistic, in the WEHI7.2 variants (OR > ER) (data not shown). These results suggest ATN-224 has potential as an adjuvant to ROS-implicated therapeutics.
ATN-224 induces cell death in primary B-ALL cells
ATN-224 induced peroxynitrite-dependent cell death, independent of Bcl-2 status, at nanomolar concentrations in cell culture models of T cell lymphoid malignancies. To determine the ability of ATN-224 to induce cell death in primary patient samples we tested ATN-224 in pre-treatment, precursor B cell acute lymphoblastic leukemia (B-ALL), which commonly overexpresses Bcl-2 [36]. We treated five B-ALL patient samples with 12.5 nM and 25 nM concentrations of ATN-224 for 24 h. We measured a decrease in the number of viable cells in all five B-ALL patient samples (Figure 7). There appears to be a concentration dependent decrease occurring in most of the patient samples, with responses ranging from 11–51% at 12.5 nM and 14–46% at 25 nM. These results imply that ATN-224 has therapeutic potential in the treatment of B cell lymphoid malignancies, and may be particularly useful in those patients whose malignancies overexpress Bcl-2.
Figure 7. ATN-224 induces cell death in primary B-ALL cells.
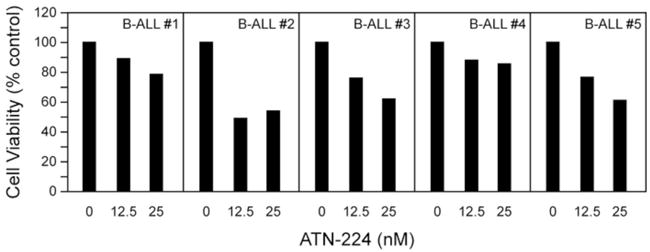
7-AAD uptake in primary B-ALL cells with vehicle control or ATN-224 (12.5 nM and 25 nM). Each bar represents a single measurement.
Discussion
These data suggest that use of a copper chelator drug to inhibit copper-dependent enzymes (SOD1 and CcOX) involved in the regulation of the redox environment, has the potential to overcome oxidative stress resistance and induce death in hematological malignancies with known drug resistant phenotypes. ATN-224 modulates the cellular redox environment by targeting SOD1, resulting in increased superoxide and inducing peroxynitrite dependent cell death. ATN-224 modulates the mitochondrial redox environment by inhibiting CcOX, which has the potential to increase oxidant production from the mitochondria. The combination of ATN-224 with doxorubicin enhances the effect of doxorubicin, suggesting ATN-224 has potential as an adjuvant to ROS-implicated chemotherapeutics. In addition to inducing cell death in cell culture models, nanomolar concentrations of ATN-224 induced cell death in primary B-ALL patient samples. Our data suggest that ATN-224 has the clinical potential for the treatment of B and T cell hematological malignancies.
Inhibition of SOD1 is a successful strategy for inducing cell death in oxidative stress resistant lymphoma cells. SOD1 is a therapeutic target being explored in many types of cancer: lung [37], ovarian [38], breast [39], prostate [40] and chronic lymphocytic leukemia [5]. SOD1 is the primary antioxidant defense enzyme responsible for dismutation of superoxide to hydrogen peroxide and oxygen in the cytosol. Unlike targeting catalase or glutathione peroxidase, which both convert hydrogen peroxide to water and oxygen, there is no other antioxidant defense enzyme in the cytosol to compensate for a sudden decrease in SOD1 activity. In WEHI7.2 cells overexpressing catalase, CAT38 and CAT2 [41], nanomolar concentrations of ATN-224 decreased cell viability (data not shown), indicating that targeting SOD1 may circumvent catalase overexpression and potentially other antioxidant defense enzymes, which are downstream of SOD1. Hileman et al. showed targeting SOD1 selectively induced cell death in primary lymphocytic leukemia cells and ovarian cancer cells, but not their normal counterparts [5]. In human umbilical vein endothelial cells (HUVEC) ATN-224 decreased SOD1 activity, increased superoxide and inhibited proliferation but did not induce cell death [12]. This suggests that ATN-224 selectively induces cell death in tumor cells.
Peroxynitrite is the major oxidant responsible for ATN-224 induced cell death. Scavenging superoxide, a peroxynitrite precursor, or peroxynitrite directly was shown to be completely protective. Our finding that peroxynitrite is the effector oxidant, rather than the previously assumed superoxide [12], helps to suggest critical targets of ATN-224 treatment and tumor types that could benefit from ATN-224 treatment. Peroxynitrite can target MnSOD (SOD2) and the mitochondrial permeability transition (MPT) pore, resulting in mitochondrial dysfunction and induction of cell death [27]. The MPT pore is susceptible to oxidation by peroxynitrite, which may allow for the release of cytochrome c and the activation of caspases, resulting in programmed cell death [27]. The opening of this pore could circumvent the overexpression of anti-apoptotic proteins such as Bcl-2, Bcl-xL and Mcl-1. Peroxynitrite also inactivates SOD2 [27]. In certain malignant tumors high levels of SOD2 are associated with poor prognosis and in cell models overexpression of SOD2 causes increased resistance to oxidants and ROS-implicated therapeutics [42]. Inactivation of SOD2 by peroxynitrite could overcome SOD2 overexpression and sensitize cells to ROS-generating therapeutics. Our data suggest that ATN-224 could prove effective in tumors with increased SOD or Bcl-2.
Although our data are consistent with inhibition of SOD1 as the most important effect of ATN-224 for inducing cell death, inhibition of CcOX may also play a role. One possibility is that inhibition of CcOX and resulting mitochondrial dysfunction “primes” the cells for apoptosis [43]. In primary leukemia and multiple myeloma cells Chonghaile et al. demonstrated that mitochondrial “priming”, which describes the proximity to the apoptotic threshold, correlates to better response and clinical outcome [43]. Although ATN-224 is a moderate to poor inhibitor of CcOX at millimolar concentrations in normal hepatic mitochondria [12], in our tumor model, ATN-224 abolished CcOX activity in the WEHI7.2 and WEHI7.2 variants. Treatment with ATN-224 decreased ΔΨm to nearly the same extent in the WEHI7.2 and WEHI7.2 variants. Use of ATN-224 to target CcOX and decrease ΔΨm, instead of using promiscuously interacting BH3 peptides [43], may be another, more specific way to “prime” mitochondria and thus enhance the efficacy of cytotoxic agents like doxorubicin, vincristine and etoposide. Although we have focused on inhibiton of SOD1 and CcOX as targets of ATN-224 there are other copper-dependent enzymes that may also contribute to the observed effect [44]. This possibility remains to be tested.
Overexpression of Bcl-2 is a major mechanism of chemoresistance in lymphoid malignancies. The development of BH3 mimetics, that directly target the canonical function of Bcl-2, are in clinical trials [45]. While these mimetics have shown promise, upregulation of other anti-apoptotic proteins, like Mcl-1, result in chemoresistance [46]. Here we report another approach to circumvent Bcl-2 overexpression, through the modulation of the redox environment. The inability of Bcl-2 to maintain redox homeostasis, in response to increased oxidative stress, may be attributed to ATN-224’s ability to target CcOX [11]. Disrupting the ability of Bcl-2 to maintain mitochondrial respiration, through the regulation of CcOX activity, appears to have potential as a novel therapuetic stragety for circumventing Bcl-2 overexpression.
Our data suggest that ATN-224 has pontential as an adjuvant in combination with ROS-implicated chemotherapeutics. There are several ROS-implicated chemotherapeutics used to treat lymphoid malignancies, such as bortezomib, gluocorticoids and aresenic trioxide [20, 47, 48]. Enhancing the efficacy of these drugs would be of great benefit to the patient, especially when lower doses could be used. In the case of doxorubicin, where cardiotoxicity is a dose limiting factor [49], decreasing the dose while achieving similar if not better efficacy could improve patient outcome. In ovarian cancer Kim et al. showed that another analogue of tetrathiomolybdate enhanced the effect of mitomycin C, fenretinide and 5-fluorouracil [50], suggesting ATN-224 also has potential as an drug adjuvant in solid tumors.
Redox modulating drugs mainly achieve efficacy in combination with other chemotherapeutics. In our model, ATN-224 was able to induce cell death at nanomolar concentrations in hematological malignancies, both in cell culture and in primary patient samples as a single agent. Hematological malignancies arise at sites of chronic inflammation, thus exposing them to higher levels of oxidative stress [2]. In response to increased oxidative stress, cells upregulate antioxidant defense enzymes [3], suggesting these malignanices could exhibit chemoresistance. Many hematological malignancies overexpress Bcl-2, which is associated with drug resistance. The ability of ATN-224 to induce cell death directly or act as a chemosensitizer in tumor cells with known chemoresistance mechanisms indicates that ATN-224 has clinical potential for the treatment of hematological malignancies.
Highlights.
ATN-224 decreases SOD1 and CcOX activity in lymphoma cell culture models.
ATN-224 induces death independent of antioxidant defense enzyme and Bcl-2 levels.
ATN-224 increases oxidants and induces peroxynitrite-dependent cell death.
Primary B cell acute lymphoblastic leukemia cells were sensitive to ATN-224.
Acknowledgments
We thank John Fitch and Paula Campbell at the AZCC Cytometry Core Facility for their assistance and the Arizona Lymphoid Tissue and Blood Repository which is supported by 1P50CA130805-04. This work was supported by National Cancer Institute Grants CA09213 (KL) and CA71768 (MMB). APM was supported by U01 CA151461-02, P50 HL107186-01 and H Foundation Funds. IBH acknowledges her general research funds. LMR and MET were supported by Arizona Cancer Center Support Grant CA023074 and Lymphoma SPORE CA 130805.
Footnotes
Conflicts of interest:
APM is a consultant to Wilson Therapeutics AB, who is developing ATN-224 for Wilson’s disease, and has a small amount of equity in that company. All other authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gibellini L, Pinti M, Nasi M, De Biasi S, Roat E, Bertoncelli L, Cossarizza A. Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin. Cancers. 2010;2:1288–1311. doi: 10.3390/cancers2021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 4.Chandra J. Oxidative stress by targeted agents promotes cytotoxicity in hematologic malignancies. Antioxid Redox Signal. 2009;11:1123–1137. doi: 10.1089/ars.2008.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 6.Tome ME, Frye JB, Coyle DL, Jacobson EL, Samulitis BK, Dvorak K, Dorr RT, Briehl MM. Lymphoma cells with increased anti-oxidant defenses acquire chemoresistance. Exp Ther Med. 2012;3:845–852. doi: 10.3892/etm.2012.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voehringer DW, Meyn RE. Redox aspects of Bcl-2 function. Antioxid Redox Signal. 2000;2:537–550. doi: 10.1089/15230860050192314. [DOI] [PubMed] [Google Scholar]
- 8.Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Low IC, Kang J, Pervaiz S. Bcl-2: a prime regulator of mitochondrial redox metabolism in cancer cells. Antioxid Redox Signal. 2011;15:2975–2987. doi: 10.1089/ars.2010.3851. [DOI] [PubMed] [Google Scholar]
- 10.Krishna S, Low IC, Pervaiz S. Regulation of mitochondrial metabolism: yet another facet in the biology of the oncoprotein Bcl-2. Biochem J. 2011;435:545–551. doi: 10.1042/BJ20101996. [DOI] [PubMed] [Google Scholar]
- 11.Chen ZX, Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010;17:408–420. doi: 10.1038/cdd.2009.132. [DOI] [PubMed] [Google Scholar]
- 12.Juarez JC, Betancourt O, Jr, Pirie-Shepherd SR, Guan X, Price ML, Shaw DE, Mazar AP, Donate F. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin Cancer Res. 2006;12:4974–4982. doi: 10.1158/1078-0432.CCR-06-0171. [DOI] [PubMed] [Google Scholar]
- 13.Donate F, Juarez JC, Burnett ME, Manuia MM, Guan X, Shaw DE, Smith EL, Timucin C, Braunstein MJ, Batuman OA, Mazar AP. Identification of biomarkers for the antiangiogenic and antitumour activity of the superoxide dismutase 1 (SOD1) inhibitor tetrathiomolybdate (ATN-224) Br J Cancer. 2008;98:776–783. doi: 10.1038/sj.bjc.6604226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batinic-Haberle I, Benov L, Spasojevic I, Hambright P, Crumbliss AL, Fridovich I. The Relationship Between Redox Potentials, Proton Dissociation Constants of Pyrrolic Nitrogens, and in Vitro and in Vivo Superoxide Dismutase Activites of Maganese(III) and Iron(III) Cationic and Anionic Porphyrins. Inorg Chem. 1999;38:4011–4022. [Google Scholar]
- 15.Reboucas JS, Spasojevic I, Batinic-Haberle I. Pure manganese(III) 5,10,15,20-tetrakis(4-benzoic acid)porphyrin (MnTBAP) is not a superoxide dismutase mimic in aqueous systems: a case of structure-activity relationship as a watchdog mechanism in experimental therapeutics and biology. J Biol Inorg Chem. 2008;13:289–302. doi: 10.1007/s00775-007-0324-9. [DOI] [PubMed] [Google Scholar]
- 16.Reboucas JS, Spasojevic I, Batinic-Haberle I. Quality of potent Mn porphyrin-based SOD mimics and peroxynitrite scavengers for pre-clinical mechanistic/therapeutic purposes. J Pharm Biomed Anal. 2008;48:1046–1049. doi: 10.1016/j.jpba.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinto VH, Carvalhoda-Silva D, Santos JL, Weitner T, Fonseca MG, Yoshida MI, Idemori YM, Batinic-Haberle I, Reboucas JS. Thermal stability of the prototypical Mn porphyrin-based superoxide dismutase mimic and potent oxidative-stress redox modulator Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin chloride, MnTE-2-PyP(5+) J Pharm Biomed Anal. 2012 doi: 10.1016/j.jpba.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci U S A. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tome ME, Briehl MM. Thymocytes selected for resistance to hydrogen peroxide show altered antioxidant enzyme profiles and resistance to dexamethasone-induced apoptosis. Cell Death Differ. 2001;8:953–961. doi: 10.1038/sj.cdd.4400904. [DOI] [PubMed] [Google Scholar]
- 20.Tome ME, Jaramillo MC, Briehl MM. Hydrogen peroxide signaling is required for glucocorticoid-induced apoptosis in lymphoma cells. Free Radic Biol Med. 2011;51:2048–2059. doi: 10.1016/j.freeradbiomed.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iqbal J, Whitney P. Use of cyanide and diethyldithiocarbamate in the assay of superoxide dismutases. Free Radic Biol Med. 1991;10:69–77. doi: 10.1016/0891-5849(91)90023-v. [DOI] [PubMed] [Google Scholar]
- 22.Zhang S, Ding JH, Zhou F, Wang ZY, Zhou XQ, Hu G. Iptakalim ameliorates MPP+-induced astrocyte mitochondrial dysfunction by increasing mitochondrial complex activity besides opening mitoK(ATP) channels. J Neurosci Res. 2009;87:1230–1239. doi: 10.1002/jnr.21931. [DOI] [PubMed] [Google Scholar]
- 23.Jaramillo MC, Frye JB, Crapo JD, Briehl MM, Tome ME. Increased manganese superoxide dismutase expression or treatment with manganese porphyrin potentiates dexamethasone-induced apoptosis in lymphoma cells. Cancer Res. 2009;69:5450–5457. doi: 10.1158/0008-5472.CAN-08-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 25.Myers JaWA. Research Design and Statistical Analysis. Routledge; 2002. [Google Scholar]
- 26.Lowndes SA, Sheldon HV, Cai S, Taylor JM, Harris AL. Copper chelator ATN-224 inhibits endothelial function by multiple mechanisms. Microvasc Res. 2009;77:314–326. doi: 10.1016/j.mvr.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 28.Spasojevic I, Chen Y, Noel TJ, Yu Y, Cole MP, Zhang L, Zhao Y, St Clair DK, Batinic-Haberle I. Mn porphyrin-based superoxide dismutase (SOD) mimic, MnIIITE-2-PyP5+, targets mouse heart mitochondria. Free Radic Biol Med. 2007;42:1193–1200. doi: 10.1016/j.freeradbiomed.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batinic-Haberle I, Cuzzocrea S, Reboucas JS, Ferrer-Sueta G, Mazzon E, Di Paola R, Radi R, Spasojevic I, Benov L, Salvemini D. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic Biol Med. 2009;46:192–201. doi: 10.1016/j.freeradbiomed.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batinic-Haberle I, Reboucas JS, Spasojevic I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal. 2010;13:877–918. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azad N, Iyer AK, Manosroi A, Wang L, Rojanasakul Y. Superoxide-mediated proteasomal degradation of Bcl-2 determines cell susceptibility to Cr(VI)-induced apoptosis. Carcinogenesis. 2008;29:1538–1545. doi: 10.1093/carcin/bgn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pacelli C, Latorre D, Cocco T, Capuano F, Kukat C, Seibel P, Villani G. Tight control of mitochondrial membrane potential by cytochrome c oxidase. Mitochondrion. 2011;11:334–341. doi: 10.1016/j.mito.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Armand R, Channon JY, Kintner J, White KA, Miselis KA, Perez RP, Lewis LD. The effects of ethidium bromide induced loss of mitochondrial DNA on mitochondrial phenotype and ultrastructure in a human leukemia T-cell line (MOLT-4 cells) Toxicol Appl Pharmacol. 2004;196:68–79. doi: 10.1016/j.taap.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Krall J, Bagley AC, Mullenbach GT, Hallewell RA, Lynch RE. Superoxide mediates the toxicity of paraquat for cultured mammalian cells. J Biol Chem. 1988;263:1910–1914. [PubMed] [Google Scholar]
- 35.Luanpitpong S, Chanvorachote P, Nimmannit U, Leonard SS, Stehlik C, Wang L, Rojanasakul Y. Mitochondrial superoxide mediates doxorubicin-induced keratinocyte apoptosis through oxidative modification of ERK and Bcl-2 ubiquitination. Biochem Pharmacol. 2012;83:1643–1654. doi: 10.1016/j.bcp.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campana D, Coustan-Smith E, Manabe A, Buschle M, Raimondi SC, Behm FG, Ashmun R, Arico M, Biondi A, Pui CH. Prolonged survival of B-lineage acute lymphoblastic leukemia cells is accompanied by overexpression of bcl-2 protein. Blood. 1993;81:1025–1031. [PubMed] [Google Scholar]
- 37.Somwar R, Erdjument-Bromage H, Larsson E, Shum D, Lockwood WW, Yang G, Sander C, Ouerfelli O, Tempst PJ, Djaballah H, Varmus HE. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc Natl Acad Sci U S A. 2011;108:16375–16380. doi: 10.1073/pnas.1113554108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown DP, Chin-Sinex H, Nie B, Mendonca MS, Wang M. Targeting superoxide dismutase 1 to overcome cisplatin resistance in human ovarian cancer. Cancer Chemother Pharmacol. 2009;63:723–730. doi: 10.1007/s00280-008-0791-x. [DOI] [PubMed] [Google Scholar]
- 39.Cicek M, Iwaniec UT, Goblirsch MJ, Vrabel A, Ruan M, Clohisy DR, Turner RR, Oursler MJ. 2-Methoxyestradiol suppresses osteolytic breast cancer tumor progression in vivo. Cancer Res. 2007;67:10106–10111. doi: 10.1158/0008-5472.CAN-07-1362. [DOI] [PubMed] [Google Scholar]
- 40.Davoodpour P, Landstrom M. 2-Methoxyestradiol-induced apoptosis in prostate cancer cells requires Smad7. J Biol Chem. 2005;280:14773–14779. doi: 10.1074/jbc.M414470200. [DOI] [PubMed] [Google Scholar]
- 41.Tome ME, Baker AF, Powis G, Payne CM, Briehl MM. Catalase-overexpressing thymocytes are resistant to glucocorticoid-induced apoptosis and exhibit increased net tumor growth. Cancer Res. 2001;61:2766–2773. [PubMed] [Google Scholar]
- 42.Kinnula VL, Crapo JD. Superoxide dismutases in malignant cells and human tumors. Free Radic Biol Med. 2004;36:718–744. doi: 10.1016/j.freeradbiomed.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 43.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P, Tai YT, Mitsiades CS, Matulonis UA, Drapkin R, Stone R, Deangelo DJ, McConkey DJ, Sallan SE, Silverman L, Hirsch MS, Carrasco DR, Letai A. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turski ML, Thiele DJ. New roles for copper metabolism in cell proliferation, signaling, and disease. J Biol Chem. 2009;284:717–721. doi: 10.1074/jbc.R800055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- 48.Woo SH, Park IC, Park MJ, Lee HC, Lee SJ, Chun YJ, Lee SH, Hong SI, Rhee CH. Arsenic trioxide induces apoptosis through a reactive oxygen species-dependent pathway and loss of mitochondrial membrane potential in HeLa cells. Int J Oncol. 2002;21:57–63. [PubMed] [Google Scholar]
- 49.Hequet O, Le QH, Moullet I, Pauli E, Salles G, Espinouse D, Dumontet C, Thieblemont C, Arnaud P, Antal D, Bouafia F, Coiffier B. Subclinical late cardiomyopathy after doxorubicin therapy for lymphoma in adults. J Clin Oncol. 2004;22:1864–1871. doi: 10.1200/JCO.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 50.Kim KK, Lange TS, Singh RK, Brard L, Moore RG. Tetrathiomolybdate sensitizes ovarian cancer cells to anticancer drugs doxorubicin, fenretinide, 5-fluorouracil and mitomycin C. BMC Cancer. 2012;12:147. doi: 10.1186/1471-2407-12-147. [DOI] [PMC free article] [PubMed] [Google Scholar]