

SUMMARY
AMP-activated protein kinase (AMPK) regulates cellular energy homeostasis by inhibiting anabolic and activating catabolic processes. While AMPK activation has been extensively studied, mechanisms that inhibit AMPK remain elusive. Here we report that glycogen synthase kinase 3 (GSK3) inhibits AMPK function. GSK3 forms a stable complex with AMPK through interactions with the AMPK β regulatory subunit and phosphorylates the AMPK α catalytic subunit. This phosphorylation enhances the accessibility of the activation loop of the α subunit to phosphatases, thereby inhibiting AMPK kinase activity. Surprisingly, PI3K-Akt signaling, which is a major anabolic signaling and normally inhibits GSK3 activity, promotes GSK3 phosphorylation and inhibition of AMPK, thus revealing how AMPK senses anabolic environments in addition to cellular energy levels. Consistently, disrupting GSK3 function within the AMPK complex sustains higher AMPK activity and cellular catabolic processes even under anabolic conditions, indicating that GSK3 acts as a critical sensor for anabolic signaling to regulate AMPK.
INTRODUCTION
Differences in nutrient availability trigger cells to activate anabolic programs to promote growth in nutrient-rich conditions, or catabolic programs to sustain survival in nutrient-poor conditions. The insulin/insulin-like growth factor-1 (IGF1) signaling pathway represents a key anabolic pathway that is activated when nutrients are readily available. Upon insulin/IGF1 stimulation, the PI3K-Akt pathway stimulates a variety of anabolic processes that consume cellular ATP. In contrast, the AMPK pathway represents a major catabolic signaling pathway that is activated when cells are metabolically starved. AMPK phosphorylates diverse substrates to stimulate catabolic processes that maintain cellular ATP levels while inhibiting anabolic programs. Although there are a few exceptions, such as glucose transport, gluconeogenesis, and lipolysis in certain tissues, these two pathways generally exert opposite functions in the regulation of metabolic processes. For instance, insulin stimulates biosynthetic pathways to promote protein, glycogen, and lipid synthesis (Samuel and Shulman, 2012), whereas AMPK suppresses these biosynthetic pathways and stimulates autophagy, a bulk protein degradation and recycling pathway triggered under starvation conditions (Hardie et al., 2012).
Under nutrient-rich, anabolic conditions, growth factors stimulate the PI3K-Akt pathway. In turn, activated Akt phosphorylates and inhibits tuberin (TSC2), resulting in the activation of the mammalian mTOR (target of rapamycin) complex 1, which promotes protein and lipid synthesis (Duvel et al., 2010; Peterson et al., 2011). Akt also phosphorylates and inhibits glycogen synthesis kinase 3 (GSK3), thereby stimulating glycogen synthesis (Cross et al., 1995). Conversely, under nutrient-limiting catabolic conditions, AMPK inhibits protein synthesis by phosphorylating TSC2 and Raptor (regulatory-associated protein of mTOR) (Gwinn et al., 2008; Inoki et al., 2003), but stimulates autophagy via ULK1 phosphorylation (Egan et al., 2011; Kim et al., 2011). AMPK also phosphorylates acetyl-CoA carboxylases 1 (ACC1) and 3-hydroxy-3 methylglutaryl CoA reductase (HMGR) to inhibit fatty acid and cholesterol synthesis, respectively (Carling et al., 1989; Clarke and Hardie, 1990). Furthermore, AMPK phosphorylates and inhibits glycogen synthase (GS) to suppress glycogen biosynthesis (Jorgensen et al., 2004).
AMPK is composed of a catalytic α subunit, and β and γ regulatory subunits (Kahn et al., 2005). AMPK activation requires phosphorylation of the activation loop (AL: Thr172) in the kinase domain of the α catalytic subunit and is accomplished by upstream kinases such as LKB1 and CAMKKs (Hawley et al., 2005; Woods et al., 2003). Phosphorylation of the α subunit AL site is essential for AMPK activity. Under catabolic conditions such as starvation or ischemia, ATP is converted to ADP, which leads to subsequent production of AMP through the activation of adenylate kinase. Increased cellular AMP induces allosteric activation of AMPK by binding to the AMPK γ regulatory subunit. Furthermore, ADP also binds to the γ regulatory subunit and induces a conformational change of the α catalytic subunit, which promotes AMPK kinase activity by preventing dephosphorylation of the AL Thr172 site by phosphatases (Oakhill et al., 2011; Xiao et al., 2011).
Although the molecular events underlying AMPK activation under nutrient-limiting catabolic conditions have been well characterized, the mechanisms by which the activity of AMPK is inhibited under nutrient-rich or anabolic conditions have not been well understood. Here we report that glycogen synthase kinase 3 (GSK3) constitutively interacts with the AMPK heterotrimeric kinase complex and inhibits AMPK kinase activity under anabolic conditions. Surprisingly, PI3K-Akt signaling, a canonical inhibitory pathway for GSK3, promotes GSK3-dependent AMPK phosphorylation and inhibition. Thus, we provide an unexpected molecular mechanism whereby the PI3K-Akt pathway and GSK3 collaborate to negatively regulate AMPK activity in vitro and in vivo. We propose that, in addition to the cellular energy levels, GSK3 in the AMPK heterotrimeric complex plays a key role in sensing growth factor signaling to regulate AMPK activity during transitions between catabolic and anabolic states.
RESULTS
GSK3 associates with the AMPK heterotrimeric kinase complex
Recent studies have identified more than thirty AMPK substrates, significantly expanding our understanding of the physiological roles of AMPK (Banko et al., 2011; Hardie et al., 2012). However, studies elucidating the regulatory mechanisms that affect AMPK function remain relatively scant. In this study, we identified proteins that associate with AMPK to discover a stable association between the α subunit of AMPK and glycogen synthase kinase 3β (GSK3β), a kinase that plays diverse roles in catabolic conditions (Cohen and Frame, 2001). Mammalian cells express two isoforms of the AMPK α subunit, α1 and α2. We found that both isoforms of the endogenous AMPK α subunit associate with endogenous GSK3β via cationic interactions and/or hydrogen bonding (Figures 1A and S1A). Subsequent analyses showed that GSK3β was co-IPed with individual AMPK subunits (Figure S1B, S1C, and S1D), and, reciprocally, all three AMPK subunits were co-IPed with GSK3β (Figure 1B), indicating that GSK3β associates with the AMPK heterotrimeric kinase complex. GSK3β was co-IPed with the β subunit in α1/3α 2 null mouse embryonic fibroblast (MEF) cells (Figure 1C), and neither the α nor the γ subunit was essential for the interaction between GSK3β and the AMPK β subunit (Figure 1D), suggesting that GSK3β primarily interacts with the β regulatory subunit of the AMPK complex. Furthermore, the association between endogenous GSK3β and the endogenous AMPK complex was not dependent on cellular energy status, AMPK activity, or serum stimulation, indicating that GSK3β stably and constitutively associates with the AMPK complex (Figure S1E). Mammalian GSK3 consists of two isoforms, GSK3α and GSK3β (Woodgett, 1990). Exogenous GSK3α was also co-IPed with the β subunit of AMPK (Figure S1F), suggesting that both GSK3 isoforms interact with the AMPK complex.
Figure 1. GSK3 associates with the AMPK heterotrimeric kinase complex through the β regulatory subunit.
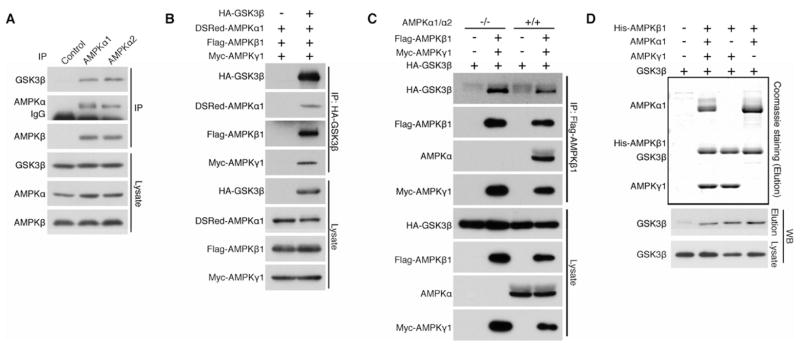
(A) Endogenous AMPK α1 or α2 subunit co-IPs endogenous GSK3β in HEK293T cells.
(B) All three AMPK subunits co-IP with GSK3β in HEK293T cells.
(C) AMPK α1/α2 subunits are dispensable for the interaction between GSK3β and the β/γ heterodimer. HA- GSK3β sufficiently co-IPs with Flag-AMPK β1 subunit in AMPK α1/2 double knockout MEF cells.
(D) AMPK α1 and γ1 subunits are dispensable for the interaction of GSK3β with the β/γ and α/β heterodimers, respectively. His-tagged AMPK β1 subunit and the indicated AMPK subunits were expressed with GSK3β in Sf9 insect cells. GSK3β co-purifies with the His-β1 AMPK subunit (Elution) in the absence of the α1 orγ 1 subunit expression. See also Figures S1.
AMPK α subunit is an atypical substrate of GSK3
The α catalytic subunit of vertebrate AMPK contains candidate sites for GSK3 phosphorylation in a serine/threonine stretch (ST-stretch) near its carboxyl terminus (Figure 2A). The ST-stretch of the mammalian AMPK α subunit contains seven consecutive serine or threonine residues that represent candidate GSK3 phosphorylation sites. Treatment with wild type or constitutively active GSK3β (S9A) that lacks the inhibitory phosphorylation site, but not with kinase inactive GSK3β (GSK3β-KI), retarded the electrophoretic mobility of the HA-AMPK α1 subunit in a λ phosphatase-sensitive manner (Figure S2A), indicating that GSK3 induces the phosphorylation of the AMPK α subunit in cultured cells.
Figure 2. AMPK α subunit is an atypical substrate of GSK3.

(A) Location and sequence conservation of the ST-stretch of the αsubunit.
(B) GSK3β-induced phosphorylation of the α1 subunit is largely abolished in T479A α1 mutant in HEK293T cells. The mobility shift was monitored by Phos-Tag™ acrylamide gel electrophoresis.
(C) GSK3β phosphorylates the α1 subunit in vitro. Myc-tagged wild type or kinase inactive GSK3β was purified from HEK293T cells. A GST-AMPK α1 subunit fragment (aa. 463–520) containing the ST-stretch purified from bacteria was used as α substrate.
(D) T479A mutation of the α subunit abolishes GSK3β-induced AMPK phosphorylation in vitro. *p<0.05, **p<0.01 vs WT or 483A with GSK3, mean±SEM (n=3).
(E) Stoichiometry analysis of GSK3β-induced ST stretch phosphorylation. The indicated polypeptides (10 μM) were subjected to in vitro kinase assay using GSK3β (0.23 μM) purified from Sf21 insect cells. Note that unprimed TSC2 peptide containing GSK3 phosphorylation sites was used in this assay.
(F) GSK3 inhibitor-sensitive Thr479 phosphorylation of the α subunit. Levels of Thr479 phosphorylation of the IPed endogenous AMPK α1 subunit were detected with a phospho-specific Thr479 antibody. CHIR99021 (10 nM for 1 hr) treatment was performed.
(G) Ablation of GSK3 expression abolishes Thr479 phosphorylation of the endogenous AMPK α subunit. GSK3α was knocked down in wild or GSK3β−/− MEF cells.
(H) GSK3β mutant (R96A) phosphorylates Thr479 of the α1 subunit in HEK293T cells. See also Figures S2.
Many GSK3 substrates are first phosphorylated by another protein kinase at a serine or threonine residue (priming site) located four residues C-terminal to the site of GSK3 phosphorylation (Cohen and Frame, 2001). Once primed, GSK3 substrates can be subject to multiple rounds of GSK3 phosphorylation, thereby creating a cluster of phosphorylated residues as seen in some of its substrates such as glycogen synthase (Fiol et al., 1987). To determine if the ST-stretch could be phosphorylated by GSK3, we mutated these serines and threonines individually to alanine and monitored the mobility shift of the α1 mutants. Substitution of Thr479 or Ser475 in the ST-stretch with alanine largely attenuated the GSK3β-induced mobility shift of the α1 subunit, while mutation of the other putative GSK3β sites in the ST-stretch (e.g. T471A) show varying degrees of increased electrophoretic mobility relative to wild type (Figure 2B). These observations suggest that Thr479 might be a priming phosphorylation site for GSK3β, leading to the successive phosphorylation of additional residues, including S475 and T471, in the ST-stretch of the α1 subunit in vivo.
To examine whether GSK3β itself can function to directly phosphorylate Thr479 in the ST-stretch, we performed in vitro kinase assays using a polypeptide containing the ST-stretch (aa. 463–520) purified from bacteria. Generally, bacteria-derived polypeptides are poor substrates for GSK3 in vitro because they lack any post-translational modifications of serine/threonine residues, which are typically required for functional priming sites of GSK3 substrates. However, we found that wild type GSK3β, but not kinase-inactive GSK3β-KI, effectively phosphorylates bacteria-derived α1 polypeptide in vitro, suggesting that the ST-stretch can be phosphorylated by GSK3β alone without first being phosphorylated at a priming site by another kinase (Figure 2C). Consistent with the in vivo observations (Figure 2B), mutation of Thr479 to alanine (T479A) in the α1 polypeptide completely abrogated phosphorylation by GSK3β, while GSK3β-induced phosphorylation was partially inhibited in the S475A or T471A mutant in vitro (Figure 2D). These data suggest that GSK3 alone primes the phosphorylation of the AMPK α1 subunit at Thr479 and then subsequently phosphorylates Ser475 and Thr471 using phosphorylated Thr479 as the priming site. This hypothesis is also supported by stoichiometry analyses in which GSK3β incorporates more than twice the amount of phosphates in the wild type ST-stretch polypeptide (Figure 2E) with a distinct Km (Figure S2B and S2C) compared to the S475A mutant where only Thr479 can be phosphorylated. In addition, GSK3β is able to phosphorylate Tau polypeptide, a representative non-primed substrate (Stoothoff et al., 2005), with similar kinetic constants as those of the S475A mutant polypeptides, but failed to phosphorylate unprimed TSC2 polypeptide, a representative substrate for GSK3 that requires priming phosphorylation, in vitro (Inoki et al., 2006). Collectively, these observations suggest that Thr479 of the α subunit can be phosphorylated by GSK3 in a non-primed manner that does not require priming phosphorylation by another kinase. To examine further GSK3-induced Thr479 phosphorylation of the AMPK α subunit in mammalian cells, we generated a phospho-specific Thr479 antibody (pT479-AMPKa). The specificity of the pT479-AMPK α antibody was confirmed by phosphorylation of the IPed endogenous α1 subunit with α1 or pT479-AMPK α antibody, which was abolished by treatment of lysates with λ phosphatase, or with a GSK3-specific inhibitor CHIR99021 in cultured cells (Figure 2F). Consistently, GSK3β overexpression enhanced Thr479 phosphorylation of both wild type α1 and α2 subunits, but failed to phosphorylate the T479A α1 mutant or the T485A α2 mutant in which Thr485, an equivalent site to Thr479 in the α1 subunit, was substituted with alanine (Figure S2D). In contrast, while GSK3α knockdown or GSK3β knockout partially suppressed endogenous Thr479 phosphorylation of the α subunit, the ablation of both GSK3α and GSK3β completely abolished phosphorylation of the same residue, indicating that both GSK3α and GSK3β contribute to Thr479 phosphorylation (Figure 2G). Finally, the priming-independent Thr479 phosphorylation of the α subunit was tested in mammalian cells using the GSK3β R96A mutant, which bears a mutation in the priming phosphorylation recognition site. The GSK3β R96A mutant is unable to phosphorylate primed substrates but is still capable of phosphorylating non-primed substrates (Frame et al., 2001). Consistent with our in vitro observations, the GSK3β R96A mutant effectively enhanced α1 Thr479 phosphorylation in vivo (Figure 2H), but failed to enhance the phosphorylation of β-catenin (Figure S2E), a representative substrate for GSK3 that requires priming phosphorylation (Liu et al., 2002). Taken together, these data indicate that the α subunit of AMPK is an atypical non-primed substrate for GSK3.
GSK3 inhibits AMPK activity through Thr479 phosphorylation of the α subunit
To explore the functional role of GSK3 phosphorylation in the regulation of AMPK, we examined the effect of GSK3 inhibitors on the kinase activity of α subunit and the phosphorylation status of its activation loop (AL site: Thr172), which is well correlated with in vivo AMPK activity. Interestingly, inhibition of endogenous GSK3 activity by treatment of cells with CHIR99021, BIO (6-bromoindirubin-3′-oxime), or LiCl enhanced phosphorylation of Thr172 of the endogenous AMPK α subunit and increased AMPK kinase activity with a concomitant decrease in Thr479 phosphorylation (Figure 3A). Conversely, GSK3β or GSK3α overexpression decreased AL phosphorylation and AMPK kinase activity with a concomitant enhancement of Thr479 phosphorylation of the α1 subunit (Figure 3B, S3A, 3C, and S3B). However, when Thr479 of the α1 subunit is mutated to alanine (T479A), even overexpression of GSK3β failed to inhibit AL phosphorylation and AMPK kinase activity (Figure 3C and S3B). These data indicate that GSK3 inhibits AMPK by phosphorylating the Thr479 of the α subunit. Since GSK3 also phosphorylates Ser475 and possibly Thr471 of the α1 subunit in a manner dependent on Thr479 phosphorylation in vivo (Figure 2B and S2B), we tested the functional role of these primed phosphorylation sites for AL phosphorylation. Intriguingly, neither T471A nor S475A mutation significantly prevents GSK3-induced reduction of AL phosphorylation (Figure 3D), suggesting that only Thr479 phosphorylation by GSK3 is sufficient to inhibit AL phosphorylation and AMPK activity in vivo.
Figure 3. GSK3-induced Thr479 phosphorylation inhibits AL phosphorylation and kinase activity of the α subunit.
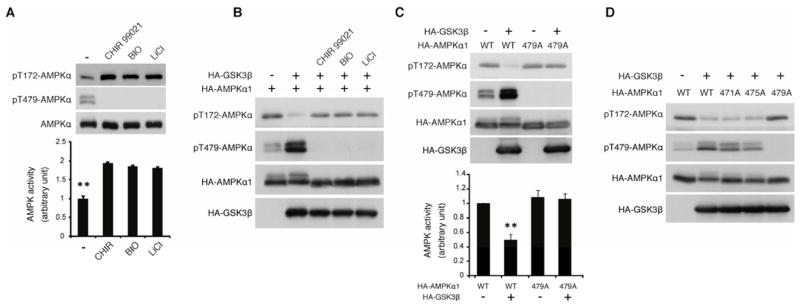
(A) Reciprocal correlation between Thr479 and Thr172 (AL) phosphorylation of the α subunit. Levels of AL and Thr479 phosphorylation and the kinase activity of endogenous AMPK α1 subunit were monitored. Pretreatment with DMSO or GSK3 inhibitors, CHIR99021 (10 nM), BIO (5 μM), or LiCl (20 mM) treatment for 1 hr was performed before harvesting the HEK293T cells. **p<0.01 vs other groups; mean±SEM (n=3).
(B) GSK3β inhibits AL phosphorylation through its kinase activity. Levels of AL and Thr479 phosphorylation were monitored in the presence or absence of GSK3 inhibitors in serum-starved HEK293T cells.
(C) GSK3β inhibits AL phosphorylation via Thr479 phosphorylation. Levels of AL and Thr479 phosphorylation and the kinase activity were monitored in serum-starved HEK293T cells. **p<0.01 vs other groups; mean±SEM (n=3).
(D) GSK3β inhibits AL phosphorylation via Thr479 but not Ser475 or Thr471 phosphorylation. GSK3β-induced reduction of AL phosphorylation in the indicated α1 mutant was monitored in serum-starved HEK293T cells. See also Figures S3.
GSK3-induced Thr479 phosphorylation enhances the phosphatase sensitivity towards the AL site of the α subunit
To explore the molecular mechanisms by which GSK3-induced phosphorylation of the α subunit inhibits AMPK activity, we examined the possible molecular events including 1) a direct influence of GSK3 on the activity of LKB1, a major AL kinase of the α subunit, 2) an interference of LKB1-dependent AL phosphorylation by GSK3-induced Thr479 phosphorylation, and 3) the effect of Thr479 phosphorylation on phosphatase sensitivity towards AL phosphorylation. Our data indicated that GSK3β did not suppress the kinase activity of LKB1 (Figure S4A), and Thr479 phosphorylation of the α1 subunit did not interfere with the phosphorylation of the AL site by overexpressed LKB1 (Figure S4B). These data suggest that Thr479 phosphorylation in the α subunit may enhance the phosphatase sensitivity of the AL phosphorylation site. Previous studies indicated that the replacement of AMP/ADP with ATP on the AMPK γ regulatory subunit increases phosphatase sensitivity of the AL site, possibly through conformational changes of the α-hook, linker, and kinase domains of the α subunit (Oakhill et al., 2011; Xiao et al., 2011). However, GSK3β was still able to inhibit AL phosphorylation of the α subunit when co-IPed with a mutant form of the γ subunit (R152Q/R171Q) that is defective in adenylate nucleotide exchange (Figure 4A). This finding suggests that GSK3-mediated inhibition of AL phosphorylation is independent of the regulatory role of the γ subunit. Additionally, GSK3β also inhibits the AL phosphorylation in an α-hook mutant (Xiao et al., 2011) that mimics the ATP-bound conformations of the linker and kinase domains of the α subunit (Figure S4C). Furthermore, an additional T479A mutation in the α-hook mutant restored decreased AL phosphorylation induced by not only GSK3β but also the α-hook mutation (Figure S4C). These results indicate that GSK3-induced reduction of AL phosphorylation of the α subunit is not mediated by adenylate nucleotide exchange on the γ subunit and the related conformational changes of the α subunit; rather, these data suggest that Thr479 phosphorylation may have a parallel but unique role in enhancing phosphatase sensitivity towards the AL site. Consistent with this model, cells expressing the Thr479 phospho-mimetic α1 mutant (T479E) were partially resistant to the induction of AL phosphorylation by 2-deoxyglucose or glucose depletion, which stimulates AMP/ADP binding to the γ subunit and protects dephosphorylation of the AL site from phosphatases such as PP2Cα (Figure 4B and S4D). To test directly the role of Thr479 phosphorylation in regulating the sensitivity of AMPK to phosphatase, AMPK complexes containing either wild type, T479A, or T479E α subunits were purified from cells overexpressing GSK3β and LKB1, followed by in vitro measurement of their sensitivities to PP2Cα, a known phosphatase for the AL site of the α subunit. Consistent with our hypothesis, the T479A mutation inhibited dephosphorylation of the AL site by PP2Cα (Figure 4C and 4D). The phospho-mimetic mutant (T479E) was as sensitive to PP2Cα as wild type α subunits phosphorylated on Thr479, and more sensitive than T479A (Figure S4E). Furthermore, T479A, but not T479E ST-stretch polypeptide, associated with the kinase domain of the α subunit in vitro (Figure 4E). These data suggest that the ST-stretch may provide a unique phosphorylation-sensitive shield for the AL site: GSK3-dependent Thr479 phosphorylation may inhibit the association of the ST-stretch with the kinase domain, thereby inducing dephosphorylation of the AL site by phosphatase.
Figure 4. GSK3-induced Thr479 phosphorylation enhances the sensitivity of phosphatase towards the AL site of theα subunit.

(A) GSK3β inhibits AL phosphorylation in a manner independent of adenine nucleotide exchange on the γ regulatory subunit in serum-starved HEK293T cells.
(B) The T479E α1 mutant is resistant to the induction of its AL phosphorylation upon glucose starvation. Transfected cells cultured in growth media containing 10% FBS were treated with glucose-free DMEM without serum for 2 hrs. The ratio of phospho-AMPK(T172): total AMPK was determined. Data were expressed as mean±SEM (n=3).
(C and D) The T479A α1 mutant shows resistance to PP2Cα-induced AL de-phosphorylation in vitro. The indicated AMPK complex containing wild type or the T489A α1 subunit was purified from HEK293T cells and incubated with PP2Cα in vitro. 250 nM PP2Cα was used in the time course experiments (C). The PP2Cα titration experiments were incubated for 1 hr (D). Levels of AL phosphorylation were quantified. *p<0.05, **p<0.01 vs WT, mean±SEM (n=3).
(E) T479A, but not T479E, α1 polypeptide interacts with the kinase domain of the α1 subunit. GST pull-down assays were performed using the indicated GST-ST-stretch polypeptides purified from E. coli and cell lysates expressing HA-AMPK α1 kinase domain (aa. 1–312, T172D). Intensity of the pulled down kinase domain was quantified. **p<0.01 vs. other groups, mean±SEM (n=3). See also Figures S4 and Supplemental Methods for additional details.
The PI3K-Akt pathway inhibits AMPK via GSK3-dependent Thr479 phosphorylation of the α subunit
How is GSK3-mediated AMPK inhibition regulated? Interestingly, glucose starvation-induced phosphorylation of the AL site of the AMPK α subunit and of Acetyl-CoA Carboxylase (ACC1/2), a major AMPK substrate, were suppressed by serum stimulation in a phosphoinositide 3-kinase- (PI3K) and GSK3-dependent manner (Figure 5A), suggesting that PI3K and GSK3 function in the same pathway to inhibit AMPK activity. Moreover, the level of Thr479 phosphorylation was dramatically enhanced by both serum and insulin treatment in a GSK3-dependent manner (Figure 5B and S5A). Consistently, insulin-induced AL dephosphorylation of the α subunit and the inhibition of AMPK activity were restored by GSK3 inhibitors (Figure 5C and S5A). In further support of these observations, genetic ablation of both GSK3α and GSK3β abolished insulin-induced AL de-phosphorylation (Figure 5D), and insulin stimulation failed to suppress AMPK kinase activity in GSK3-deficient cells (Figure S5B). Although ablation of GSK3α or GSK3β partially attenuated insulin-induced Thr479 phosphorylation of the AMPK α subunit, ablation of either isoform alone failed to block insulin-induced AL dephosphorylation (Figure S5C), suggesting that both GSK3α and GSK3β contribute to insulin-induced AL dephosphorylation of the AMPK α subunit. Importantly, insulin failed to induce AL dephosphorylation in the T479A mutant of the AMPK α subunit (Figure 5E), indicating that GSK3-dependent Thr479 phosphorylation is essential for insulin-induced AL dephosphorylation. Together, these data indicate insulin/PI3K is required for GSK3 to phosphorylate Thr479 and inhibit AMPK activity.
Figure 5. The PI3K-Akt pathway inhibits AMPK via GSK3-dependent Thr479 phosphorylation of the AMPK α subunit.

(A) Serum stimulation inhibits AL phosphorylation of the endogenous α subunit in a PI3K- and GSK3-dependent manner. Serum- and glucose-starved MEF cells were stimulated with 10% dialyzed serum for the indicated times in the presence or absence of PI3K inhibitor (LY294002: 20 μM; preincubation for 2 hrs) or GSK3 inhibitors (LiCl: 20 mM, BIO: 5 μM; preincubation for 2 hrs). Levels of AL phosphorylation of endogenous AMPK α subunit were quantified. *p<0.05, **p<0.01 vs time 0, mean±SEM (n=3).
(B) Serum stimulation induces GSK3-dependent Thr479 phosphorylation of the α subunit. MEF cells were treated with the indicated concentrations of serum for 30 min in the presence or absence of GSK3 inhibitors.
(C) Insulin enhances GSK3-dependent Thr479 AMPK phosphorylation. The effect of insulin (100 nM for 60 min) on Thr479 or AL phosphorylation, or the kinase activity of the α1 subunit was monitored in the presence or absence of the GSK3 inhibitors in serum-starved HEK293E cells. **p<0.01 vs other groups, mean±SEM (n=3).
(D) Ablation of GSK3 attenuates insulin-induced Thr479 phosphorylation and AL de-phosphorylation. GSK3α was knocked down in GSK3β−/− MEF cells. The effects of insulin (100 nM) on AL and Thr479 phosphorylation of the endogenous α subunit were monitored.
(E) Insulin-dependent Ser485 phosphorylation is required for GSK3-induced Thr479 phosphorylation. HEK293E cells were transfected with the indicated AMPK α1 subunits. The cells were treated with insulin (100 nM) for 60 min.
(F) Active Akt induces AL dephosphorylation through GSK3-dependent Thr479 phosphorylation in HEK293E cells.
(G) Insulin induces AL dephosphorylation in a GSK3-dependent manner in vivo. C57BL/6J male mice starved for 16 hours were treated with insulin (1 mU/g, ip) for 60 min. LiCl (200 μg/g, ip) was injected 2 hrs before obtaining heart tissues. **p<0.01 vs other groups, mean±SEM (n=5).
(H) PI3K activity is required for GSK3-dependent AMPK inhibition in vivo. Wild type and muscle-specific TSC1 knockout male mice were treated as Figure 5G. **p<0.01 vs other groups, NS indicates “not significant”, mean±SEM (n=3). See also Figures S5.
The insulin/PI3K-Akt pathway phosphorylates Ser9 in the N-terminus of GSK3β to block GSK3β from phosphorylating primed substrates (Cross et al., 1995). The phosphorylated N-terminus of GSK3β acts as a pseudo substrate and occupies its phosphate binding pocket through the intra-molecular interaction with the Arg96 (R96) residue of GSK3β. Thus, it has been proposed that GSK3-dependent phosphorylation of non-primed substrates such as Axin is not blocked by Akt-dependent GSK3 phosphorylation (Frame et al., 2001). Indeed, Ser9 of GSK3β co-IPed with the AMPK complex was largely phosphorylated upon insulin treatment (Figure S5D). To examine whether insulin-induced Ser9 phosphorylation of GSK3β modulates its kinase activity towards the AMPK α subunit, we compared the ability of Ser9-phosphorylated GSK3β and non-phosphorylated GSK3β to phosphorylate Thr479 of the ST-stretch in vitro. Although the activity of Ser9-phosphorylated GSK3β towards a primed substrate (e.g. pTSC2) was significantly decreased as expected (Figure S5E), the kinase activity of Ser9-phosphorylated GSK3β for the ST-stretch of the α subunit or another non-primed substrate (Tau) was comparable to the kinase activity of non-phosphorylated GSK3β (Figure S5F and S5G). These data suggest that growth factor-induced phosphorylation of Ser9 does not affect GSK3β to phosphorylate Thr479 of the AMPK α subunit. However, it remains unclear why insulin promotes GSK3-dependent Thr479 phosphorylation of the AMPK α subunit.
Previous studies have demonstrated that Akt enhances Ser485 phosphorylation of the α subunit and inhibits AMPK activity in heart tissue and vascular smooth muscle cells via unknown mechanisms (Horman et al., 2006; Kovacic et al., 2003). We speculated that Akt phosphorylation of the AMPK α subunit might positively influence GSK3-dependent Thr479 phosphorylation in a hierarchical manner, but not in a priming-dependent fashion since R96A GSK3, which is not able to recognize a primed phosphate, still sufficiently phosphorylates Thr479 of the α subunit in vivo. To test this hypothesis, we examined the effect of insulin or active Akt overexpression on the phosphorylation of Thr479 in the S485A α subunit mutant, which is unable to be phosphorylated by Akt. As expected, insulin and active Akt effectively enhanced both Thr479 (GSK3 site) and Ser485 (Akt site) phosphorylation and inhibited AL phosphorylation in the wild type α1 subunit, and the reduction of AL phosphorylation were completely blocked in the T479A mutant (Figure 5E and 5F). Interestingly, they failed to stimulate Thr479 phosphorylation and to dephosphorylate AL phosphorylation in the S485A mutant (Figure 5E and 5F), suggesting that endogenous GSK3-depependent Thr479 phosphorylation requires Akt-dependent Ser485 phosphorylation in vivo. Importantly, the phosphorylation of Ser485 (Akt site) alone is not sufficient to produce AL dephosphorylation because Ser485 is highly phosphorylated by insulin or active Akt in the T479A mutant (Figure 5E and 5F). Finally, Ser485 phosphorylation does not affect the interaction of GSK3β with the AMPK complex (Figure S5H). Collectively, these data demonstrate a novel regulatory mechanism wherein anabolic stimuli such as serum and insulin suppress AMPK activity through an unexpected collaboration between Akt and GSK3, which regulate the phosphatase accessibility towards AL phosphorylation of the α subunit via successive phosphorylations of the ST-stretch. To validate these observations obtained from cell lines, GSK3-mediated AL dephosphorylation of the α subunit was investigated under more physiological conditions. In murine heart tissue and primary hepatocytes, insulin treatment significantly enhanced Thr479 phosphorylation and induced AL dephosphorylation, while LiCl pretreatment abolished the effects of insulin on these phosphorylation sites (Figure 5G and S5I). Furthermore, GSK3-dependent Thr479 phosphorylation in heart tissue requires PI3K-Akt activity, as insulin failed to stimulate Thr479 phosphorylation when the PI3K-Akt pathway was downregulated by the constitutive activation of mTORC1 signaling that occurs in the muscle-specific Tsc1 knockout mice (Harrington et al., 2004; Hsu et al., 2011; Yu et al., 2011) (Figure 5H). The reciprocal relationship between Thr479 and AL site phosphorylation was also observed under other physiological settings such as satiety, where food intake stimulated Thr479 phosphorylation but inhibited AL phosphorylation in a GSK3-dependent manner (Figure S5J). These observations indicate that GSK3 also functions as a critical negative regulator for AMPK activity in vivo.
Loss of GSK3-dependent AMPK inhibition causes metabolic inflexibility
Finally, the functional importance of GSK3 in the regulation of AMPK signaling was interrogated in α1/α2 double knockout MEF cells stably expressing only wild type or T479A mutant α1 subunit (Figure S6). AMPK phosphorylates and inhibits both ACC and Raptor (Gwinn et al., 2008), stimulating fatty acid beta-oxidation (Merrill et al., 1997) and inhibiting protein synthesis, respectively. These processes are important for maintaining cellular energy under stress conditions. In contrast, under anabolic conditions, growth factors such as insulin stimulate fatty acid and protein synthesis. Both ACC and Raptor phosphorylation were diminished in AMPKα1/α2 double knockout MEF cells, confirming that AMPK, but not other kinases, is responsible for these phosphorylation events (Figure 6A). In two reconstituted cell lines, the levels of ACC and Raptor phosphorylation under serum starvation conditions were similar. However, insulin treatment inhibited ACC as well as Raptor phosphorylation in cells expressing the wild type α1 subunit, but not the T479A α1 subunit (Figure 6A), suggesting that GSK3-dependent Thr479 phosphorylation may play an important role in the catabolic-to-anabolic state transition. Consistent with this hypothesis, the rate of protein synthesis was significantly lower in cells expressing the T479A α1 subunit than in cells expressing wild type α1 subunit after serum stimulation (Figure 6B). The data indicate that disrupting GSK3 function in the AMPK complex failed to drive sufficient anabolic responses to growth factor stimuli. We next examined the effect of GSK3-mediated AMPK function in catabolic processes. Recent studies demonstrated that AMPK also plays a role in the induction of autophagy, a major catabolic pathway for protein degradation and recycling (Egan et al., 2011; Kim et al., 2011). Interestingly, cells expressing the T479A mutant α1 subunit showed significantly higher autophagosome formation (Figure 6C) as well as autophagic flux (Figure 6D) compared to wild type cells after serum stimulation. Together, these data demonstrate that GSK3-dependent Thr479 phosphorylation of the α subunit is critical for cells to enter an anabolic state in response to environmental cues.
Figure 6. Loss of GSK3-dependent AMPK inhibition causes metabolic inflexibility.

(A) Insulin fails to suppress ACC1/2 and Raptor phosphorylation in T479A MEF cells. The indicated MEF cells were serum-starved for 16 hours and then stimulated with insulin (100 nM) for 60 min. See also Figures S6.
(B) Lower levels of protein synthesis are observed in T479A MEF cells. The indicated MEF cells were serum-starved for 16 hours and then stimulated with serum (10%) for 4 hours. Nascent protein synthesis was determined (upper panel). The rate of nascent protein synthesis was quantified by the expression of β-actin. *p<0.05, mean±SEM (n=3).
(C) Higher autophagic activity is observed in T479A MEF cells. Autophagosome formation was monitored in the indicated MEF cells in the presence of serum stimulation (10% for 60 min). The number of LC3-puncta per cell was measured and quantified in the indicated MEF cells. **p<0.01, mean±SEM (n=15).
(D) Higher autophagic flux is observed in T479A MEF cells. MEF cells were stimulated with serum (10% for 60 min) in the presence of NH4Cl. The ratio of LC3-II/LC3-I was quantified. *p<0.05, vs WT 30 min, mean±SEM (n=3).
(E) Schematic model of AMPK inhibition by GSK3. Under catabolic conditions (low energy and low PI3K-Akt activity), the binding of AMP or ADP to the γ subunit promotes the association of the α-hook with the γ subunit and the subsequent interaction of the kinase domain (KD) with the β subunit. Simultaneously, the non-phosphorylated ST-stretch and its adjacent region of the α subunit (blue bar) may sterically hinder phosphatase accessibility for the activation loop (left panel). In response to anabolic stimuli (high PI3K-Akt activity), successive phosphorylations on the ST-stretch of the α subunit induced by Akt and GSK3 may promote dissociation of the ST-stretch from the KD, thereby allowing the phosphatase to dephosophorylate AL loop (middle panel). Under anabolic conditions (high energy), the KD of the α subunit may be further exposed to the phosphatase through additional conformational changes in the α-hook and linker region (right panel). A previous structure study proposed that the dissociation of the α-hook from the γ subunit precedes nucleotide exchange (AMP/ADP to ATP) on the γ subunit (Xiao et al., 2011). Whether this steric change of the ST-stretch triggers dissociation of the α hook from the γ subunit remains to be resolved.
DISCUSSION
The data presented in this study reveal an unexpected role for GSK3 in sensing anabolic stimuli to inhibit the AMPK pathway. The ability of endogenous GSK3 to phosphorylate and inhibit the α catalytic subunit of AMPK requires PI3K-Akt signaling, which paradoxically is a major canonical inhibitory pathway for regulation of GSK3 activity in other systems. However, our studies demonstrate a novel mechanism where GSK3 is able to continue to phosphorylate the ST-stretch of the AMPK α subunit and inhibit AMPK activity in a timely manner when cells are ready to transition from catabolic to anabolic conditions.
Recent crystal structures of the mammalian AMPK heterotrimeric complex omit the region that contains the ST-stretch of the α subunit due to its extensive flexibility (Xiao et al., 2011). Interestingly, our study suggests that this flexible region functions as a phosphorylation-sensitive switch that regulates phosphatase accessibility to the kinase domain of the α subunit (Figure 6E). Through an increase in negative charge of the ST-stretch via successive phosphorylations achieved by GSK3 and Akt, this molecular switch plays an important role for the catabolic-to-anabolic conversion in cultured cells. Our data indicate that upon growth factor-mediated PI3K activation, Akt first phosphorylates Ser485 on the ST-stretch of AMPK α subunit, which induces GSK3-dependent Thr479 phosphorylation in a non-primed manner and subsequent dephosphorylation of the AL site of the α subunit. Therefore, although Akt has been widely recognized as a primary suppressor for both GSK3α and β via its Ser21 and Ser9 phosphorylation, respectively, thus predominantly inhibiting the activity of GSK3 to phosphorylate primed substrates, the activity of GSK3 to phosphorylate Thr479 of the AMPK α subunit is nonetheless still maintained. However, the precise molecular mechanism by which Akt-dependent Ser485 phosphorylation enhances GSK3-dependent Thr479 phosphorylation remains elusive. Given that the interaction of endogenous GSK3 with the AMPK complex is not changed by growth factor stimulation (Figure S1E) and Akt-induced Ser485 phosphorylation of the α subunit (Figure S5H), we speculate that Ser485 phosphorylation of the α subunit may induce an intermolecular conformation change that unmasks or exposes the Thr479 residue on the ST-stretch to GSK3. Our data also suggest that upon Thr479 phosphorylation of the α subunit by GSK3, a subsequent conformational change of the ST-stretch plays an important role in regulating the accessibility of phosphatase to the AL site. We found that the phospho-defective, but not the phospho-mimetic, ST-stretch of the α subunit interacts with its kinase domain. Since phosphorylation of the ST-stretch by Akt and GSK3 induces AL dephosphorylation and inhibits the activity of AMPK in vivo, the non-phosphorylated ST-stretch may block the accessibility of phosphatase to the AL site. However, in contradiction to our simple assumption, the phospho-defective (T479A) ST-stretch polypeptide, which has a higher affinity for the kinase domain of the AMPK, did not significantly protect against PP2Cα-induced AL dephosphorylation of the α subunit in vitro (data not shown). These results indicate that the ST stretch polypeptide, which encompasses 57 amino acids near the carboxyl terminus of the AMPK α subunit, is not a region directly responsible for repelling phosphatase accessibility to the AL site. Instead, we posit that regions adjacent to the ST stretch may function as a shield for the AL site from the phosphatases.
Our observations suggest that the PI3K-Akt-GSK3 mediated AMPK inhibition plays important roles in rapid catabolic-to-anabolic conversion such as growth factor-induced protein synthesis and possibly the storage of energy as glycogen. Interestingly, previous studies demonstrated that GSK3 collaborates with or supports AMPK function to inhibit glycogen and protein synthesis under catabolic conditions. Glycogen synthase (GS) is negatively regulated by AMPK- and GSK3-dependent phosphorylation under catabolic conditions where AMPK phosphorylates Ser8, while GSK3 phosphorylates Ser641, Ser645, Ser649, and Ser653 in a manner dependent on CK2-induced Ser657 phosphorylation (Fiol et al., 1987; Jorgensen et al., 2004). Interestingly, maximal activation of GS was observed when both AMPK and GSK3-mediated phosphorylation were blocked in vitro (Skurat et al., 1994). It is likely that upon anabolic stimuli, active Akt inhibits GSK3 to phosphorylate primed phosphorylation sites on GS, and may simultaneously suppress AMPK phosphorylation of GS by inducing GSK3-mediated Thr479 phosphorylation of the AMPK α subunit. Consistent with our hypothesis, a recent study showed that insulin stimulation reverses both GSK3- and AMPK-mediated phosphorylation of GS (Hunter et al., 2011). TSC2, a Tuberous Sclerosis Complex (TSC) gene product, is a critical suppressor of mTORC1 that stimulates translation initiation and protein synthesis (Sengupta et al., 2010). TSC2 is negatively regulated by Akt-dependent phosphorylation under anabolic conditions, but is positively regulated by AMPK and GSK3-dependent phosphorylation under catabolic conditions (Inoki et al., 2006; Manning et al., 2002). Our findings suggest that active Akt may inhibit TSC2 through not only its direct phosphorylation but also attenuation of both AMPK and GSK3-mediated TSC2 phosphorylation under anabolic conditions.
From a pathophysiological point of view, disruption of GSK3 function in the AMPK complex may have beneficial effects in preventing the progression of metabolic disorders such as insulin resistance and obesity because increasing AMPK activity supports glucose oxidation and inhibits lipogenesis (Steinberg and Kemp, 2009). Intriguingly, GSK3 inhibitors have recently been shown to stimulate glucose uptake and suppress certain gluconeogenic gene expression in vitro and in vivo by unknown mechanisms (Dokken et al., 2005; Lochhead et al., 2001; Ring et al., 2003). We speculate that these anti-diabetic properties may be mediated at least in part through the activation of AMPK (Koo et al., 2005; Lochhead et al., 2000). While clarification of the physiological relevance of GSK3-AMPK regulation may require generation of an α1/α2 T479A knock-in mouse model, our biochemical data raise the possibility that facilitating GSK3 dissociation from the AMPK complex may represent a novel therapeutic approach for stimulating AMPK activity without suppressing cellular energy levels.
EXPERIMENTAL PROCEDURE
Kinase assays
For the AMPK kinase assays, HEK293T or MEF cells were lysed with RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA and EGTA, 0.5% deoxycholic acid, 0.1% SDS, 1% Triton X-100, 1 mM DTT, 1mM PMSF, 10 mM pyrophosphate, 50 mM NaF, and protease inhibitors) and AMPK was IPed by HA (for exogenous) or AMPK (for endogenous) antibody with protein G-Sepharose beads. IPed AMPK was subjected to a kinase reaction mixture (18 mM HEPES [pH 7.5], 10 mM MgCl2, 50 μM cold ATP, 1 mM DTT, 1 μCi [γ-32P] ATP) with GST-fused TSC2 fragments (1193–1247 amino acid) purified from bacteria as substrates. Reaction mixtures were then incubated for 30 min at 30°C, terminated with SDS sample buffer, and subjected to SDS-PAGE and autoradiography. For the GSK3β or LKB1 kinase assay, GST-fused AMPK α1 fragments purified from bacteria or full length AMPK α1 purified from Sf9 cells were used as substrates, respectively. Conditions for these kinase reactions were the same as those used for the AMPK kinase assay.
In vitro de-phosphorylation assays
HA-AMPKα1 or indicated HA-AMPKα1 mutants were purified from HEK293T cells in the presence of LKB1 and GSK3β expression. The immunoprecipitates were incubated at 25 °C in a buffer containing 50 mM Tris, [pH 7.5], 100 mM NaCl, 0.1 mM EDTA and 2.5 mM MgCl2, in the presence or absence of recombinant PP2Cα (Calbiochem).
Supplementary Material
HIGHLIGHTS.
GSK3 constitutively interacts with the AMPK complex through the β subunit.
GSK3 phosphorylates the α subunit of AMPK and inhibits its kinase activity.
The PI3K-Akt pathway enhances GSK3-dependent phosphorylation of the α subunit.
GSK3-dependent AMPK inhibition is critical for cells to enter an anabolic state.
Acknowledgments
We thank D. Kwiatkowski for TSC1 flox mice and K. Stankunas for GSK3β−/− MEF. We thank JK. Kim for critical discussions and reading of this paper. We also thank D. Fingar, JL. Smith, Tianqing Zhu, and members of Inoki Lab for helpful discussions and technical assistance. This work was supported by the NIH (DK083491 to K.I, DK061618 to A.R.S, DK077086 to J.L).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villen J, Wang B, Kim SR, Sakamoto K, et al. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D, Clarke PR, Zammit VA, Hardie DG. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem. 1989;186:129–136. doi: 10.1111/j.1432-1033.1989.tb15186.x. [DOI] [PubMed] [Google Scholar]
- Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. Embo J. 1990;9:2439–2446. doi: 10.1002/j.1460-2075.1990.tb07420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Dokken BB, Sloniger JA, Henriksen EJ. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E1188–1194. doi: 10.1152/ajpendo.00547.2004. [DOI] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, Roach PJ. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem. 1987;262:14042–14048. [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMP-Activated Protein Kinase: A Target for Drugs both Ancient and Modern. Chem Biol. 2012;19:1222–1236. doi: 10.1016/j.chembiol.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281:5335–5340. doi: 10.1074/jbc.M506850200. [DOI] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RW, Treebak JT, Wojtaszewski JF, Sakamoto K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes. 2011;60:766–774. doi: 10.2337/db10-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, Andreelli F, Schjerling P, Vaulont S, Hardie DG, Hansen BF, et al. The alpha2–5′AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes. 2004;53:3074–3081. doi: 10.2337/diabetes.53.12.3074. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Coghlan M, Rice SQ, Sutherland C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes. 2001;50:937–946. doi: 10.2337/diabetes.50.5.937. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes. 2000;49:896–903. doi: 10.2337/diabetes.49.6.896. [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, Ma ST, Reeder JW, Samuels I, Slabiak T, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurat AV, Wang Y, Roach PJ. Rabbit skeletal muscle glycogen synthase expressed in COS cells. Identification of regulatory phosphorylation sites. J Biol Chem. 1994;269:25534–25542. [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Stoothoff WH, Cho JH, McDonald RP, Johnson GV. FRAT-2 preferentially increases glycogen synthase kinase 3 beta-mediated phosphorylation of primed sites, which results in enhanced tau phosphorylation. J Biol Chem. 2005;280:270–276. doi: 10.1074/jbc.M410061200. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. Embo J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.