

Abstract
The histone acetyltransferase Rtt109 is the sole enzyme responsible for acetylation of histone H3 lysine 56 (H3K56) in fungal organisms. Loss of Rtt109 renders fungal cells extremely sensitive to genotoxic agents, and prevents pathogenesis in several clinically important species. Here, via a high throughput chemical screen of >300,000 compounds, we discovered a chemical inhibitor of Rtt109 that does not inhibit other acetyltransferase enzymes. This compound inhibits Rtt109 regardless of which histone chaperone cofactor protein (Asf1 or Vps75) is present, and appears to inhibit Rtt109 via a tight-binding, uncompetitive mechanism.
Keywords: Saccharomyces cerevisiae, Candida albicans, Histone acetyltransferase, Molecular Libraries Probe Production Centers Network (MLPCN)
In fungi, H3K56 acetylation (H3K56ac) quantitatively marks newly synthesized, soluble histones, stimulating their association with assembly proteins that deposit them onto DNA 1–5. Although H3K56ac has been detected in metazoan organisms, it is far less abundant 6, 7, and therefore does not appear to serve as a quantitative marking system for nascent histones.
Not only is the function of H3K56ac specific to the fungal Kingdom, but so is the enzyme that creates it. In all fungi studied to date, acetylation of H3K56 is catalyzed solely by the histone acetyltransferase (HAT) Rtt109 8–11. Rtt109 has very limited primary sequence similarity to other HAT enzymes 12. However, the tertiary fold of Rtt109 is similar to p300 13–16, a HAT important for N-terminal histone tail acetylations related to transcriptional activation in metazoans 17. Nevertheless, the kinetic mechanisms of Rtt109 and p300 differ in several important ways. Rtt109 uses a sequential catalytic mechanism, forming a ternary intermediate complex with both histone and acetyl-CoA substrates before catalysis 18. In contrast, p300 operates with a Theorell-Chance (“hit-and-run”) mechanism in which the enzyme binds acetyl-coA first, followed by transient association with the protein substrate 19. Furthermore, the active sites of these two enzymes display dramatically different electrostatic characteristics 13. Finally, p300 inhibitors, including the bisubstrate mimic Lys-CoA, do not affect Rtt109 catalysis 14, 20. Therefore, Rtt109 displays significant differences from its closest mammalian homolog, p300, in terms of both its structure and its biological function.
Rtt109 by itself is a poor enzyme, but can be activated by either of two different histone chaperone proteins, Asf1 and Vps75 9. These cofactors stimulate modification of distinct substrate lysines. For example, Asf1 is required for acetylation of H3K56 by Rtt109 in vivo 8, 21. In contrast, Vps75 stimulates Rtt109 to acetylate H3K9, H3K23 and H3K27 but is not required for H3K56 acetylation in vivo 22–24, despite its ability to stimulate H3K56 acetylation in vitro 9.
Cells of any fungal species that are incapable of acetylating H3K56 are extremely sensitive to DNA damage 10, 11, 25–27. We previously demonstrated that deletion of RTT109 in the pathogen Candida albicans dramatically reduces mortality of mice subjected to systemic candidiasis 11. Subsequent studies confirmed that H3K56 acetylation is the crucial function of Rtt109 in C. albicans, because the HST3 gene encoding the deacetylase that removes this modification is essential for C. albicans viability, but only if Rtt109 is present 28. Notably, the poor pathogenicity of rtt109−/− cells correlates with an inability to withstand phagocyte-generated reactive oxygen species 11.
In humans, systemic candidiasis results in approximately 40% mortality, despite currently available anti-fungal medications 29. Unfortunately, anti-fungal drug resistance is common in this organism 30, 31. Therefore, discovery of antifungal therapeutics with novel targets is a high medical priority. Because Rtt109 is required for C. albicans pathogenesis and is conserved structurally and functionally only within the fungal kingdom, we reasoned that a specific inhibitor of Rtt109 could provide a novel path to a new class of antifungal therapeutics that would not impair HATs found in mammalian hosts 32.
The Rtt109-Vps75 complex displays a catalytic efficiency (kcat/Km) 20-fold greater than Rtt109/Asf1-mediated catalysis, and Rtt109 tightly binds to Vps75 (Kd = ~10–23 nM; 9, 18, 22), allowing easy co-expression and purification from bacteria. Therefore, we developed a high throughput screen (HTS) for small molecule inhibitors of the HAT activity of recombinant Rtt109-Vps75 complexes, and tested for inhibition of Rtt109/Asf1 as a secondary criterion.
In the primary HTS, coenzyme A (CoA) molecules produced during the acetyltransferase reaction were detected using a fluorescent malemide (ThioGlo1) that reacts with the sulfhydryl group on CoA (33, see Methods). We screened 363,843 small molecules at a compound concentration of 25 µM in single-point HAT assays in a 1536-well plate format. Each compound was assayed twice, and 224 out of 333,734 compounds that generated two data points resulted in >50% inhibition (0.07% hit rate). 30,109 compounds generated a single data point, of which 313 yielded >50% inhibition (1% hit rate). The HTS campaign and retests are stored under Pubchem Summary AID: 540342.
Of these initial 537 hits, 449 compounds were readily available for re-testing in an 8-point, 2-fold dose titration. During these retests, the free sulfhydryl groups of released CoA were detected using Ellman’s reagent (5,5'-dithiobis-(2-nitrobenzoic acid); DTNB) via optical absorbance rather than via fluorescence using ThioGlo1. This alternate detection strategy was designed to eliminate non-specific fluorescence quenchers obtained in the initial screen. In the retests, we observed that 82 compounds produced a dose-dependent response with 50% inhibition values ≤ 20 µM. Twenty-eight were chosen to re-test from powders based on their medicinal chemistry potential. The identity (correct mass) and purity (>80%) of powder samples were confirmed with HPLC/MS. Again, these compounds were tested in 8-point, 2-fold dose response curves. Nine compounds exhibited 50% inhibition values below 10 µM and were further characterized for their specificity.
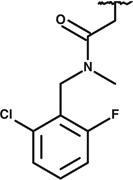
A crucial criterion for our Rtt109 inhibitors was that they should not affect the HAT activity of the structurally related HAT, mammalian p300. We therefore performed single point enzyme assays comparing Rtt109-Vps75 and p300 in the presence of 0.5 – 2 µM of each of the nine remaining candidate compounds. Only a single compound, termed compound 1 in this paper, inhibited Rtt109 without inhibiting p300 (Figure 1 and data not shown). Compound 1 has the chemical formula C22H23ClFN3O3 (IUPAC name N-[(2-chloro-6-fluorophenyl)methyl]-2-(2,5-dioxo-4-phenyl-4-propylimidazolidin-1-yl)-N-methylacetamide), and is listed as PubChem Compound CID 4785700, Substance SID 49676148 (Figure 1a). Notably, current listings for PubChem Bioactivity assays show it has been tested in 417 assays, but was active in only two. One was our screen (PubChem BioAssay AID 588764), and the other assay was for inhibition of the interaction between the tudor domain of histone methyltransferase JMJD2A and a histone H4 peptide trimethylated on lysine 20 (PubChem BioAssay AID 504339). However, in the latter case, the IC50 value was reported to be 25 µM. Therefore, the present results are the most potent effects reported for this compound to date.
Figure 1. Compound 1 is a specific inhibitor of Rtt109 histone acetyltransferase activity.

(a) Molecular structure of compound 1. (b) Dose response for compound 1 inhibition of acetylation by Rtt109-Vps75, p300, and Gcn5. 50 nM Rtt109-Vps75 and 15 µM H3n21 peptide were pre-incubated with either DMSO or compound 1 at the indicated concentrations for 5 minutes at 30°C. Reactions were initiated by addition of 30 µM acetyl-Coenzyme A. Released Coenzyme A was quantified every 2 minutes during the initial 10 minutes to determine reaction rates. A nonlinear curve fit of these data indicates 50% inhibition at 56 nM, R2 = 0.94. Alternatively, HAT reactions were performed as described above with either 15 µg/ml p300 or 3.1 µg/ml (61 nM) Gcn5 as indicated. (c) Acetylation of H3K56 is inhibited by compound 1. Histone acetyltransferase reactions were performed with 50 nM Rtt109, 300 µM (H3-H4)2 tetramers, 50 nM Vps75 or 400 nM Asf1N. Reactions were pre-incubated with DMSO or 500 nM compound 1 for 5 minutes prior to initiation with 15 µM acetyl-Coenzyme A (or water). After incubation at 30°C for 30 minutes, acetylated H3K56 was detected by ELISA using H3K56ac-specific rabbit anti-serum. Signal was normalized to that observed in the presence of DMSO + acetyl-CoA. (d) HAT activity of p300 is not inhibited by compound 1. Reactions were performed as in panel (c) with 15 µg/ml p300, and acetylated lysine was detected by ELISA using a polyclonal anti-acetyllysine antibody.
To assess the potency of compound 1 for Rtt109-Vps75 inhibition, we performed HAT reactions with varying concentrations of compound 1 and determined rates during the initial linear phase of the reactions. Each reaction rate was calculated via linear regression of product formed versus time (mean R2 = 0.91, ranging from 0.77–0.99). Reaction velocities were plotted against compound 1 concentration on a semi-log scale (Figure 1b), revealing a dose response over approximately two orders of magnitude, from 10 nM – 1 µM. A non-linear fit of the data (R2 = 0.94; GraphPad Prism) suggests that 50% inhibition of Rtt109-Vps75 occurred at 56 +/− 1.3 nM. In contrast to Compound 1’s potent effects on Rtt109-Vps75, it did not significantly inhibit the HAT activity of either p300 or Gcn5. As above, each reaction rate was determined via linear regression of product formed versus time (for p300, mean R2 = 0.98, ranging from 0.97–0.99; for Gcn5 mean R2 = 0.97, ranging from 0.94–0.99). High concentrations of Compound 1 (up to 100 µM) did not reduce enzyme reaction rates below 80% activity, with no apparent trend as Compound 1 concentration was increased (Figure 1c). Due to solubility limitations, Compound 1 was not tested above 100 µM. However, the higher concentrations of the inhibitor are still far above the enzyme concentrations in these experiments (50 nM for Rtt109-Vps75; 61 nM for Gcn5). We conclude that Compound 1 has a greater than two order of magnitude preference for inhibition of Rtt109.
An Rtt109 inhibitor could affect catalysis stimulated by either Vps75 or Asf1, but not necessarily both. To compare inhibition of acetylation by Rtt109-Vps75 and Rtt109/Asf1, we performed single-point HAT assays using (H3-H4)2 tetramers as the substrates. Acetylated H3K56 was detected by ELISA using a modification-specific anti-H3K56ac antibody. Histone tetramer substrates rather than H3 N-terminal peptides were required for these experiments because Asf1 stimulates H3K56 acetylation but does not stimulate acetylation on the N-terminal tail of histone H3 22, 23. In these assays, we observed that Compound 1 inhibited acetylation by both Rtt109-Vps75 and Rtt109/Asf1 comparably (Figure 1d). Importantly, these data also confirm that acetylation of H3K56 is inhibited by Compound 1, which had not been examined in the ThioGlo1-based assays that used the N-terminal histone H3n21 peptide substrate. Additionally, we also tested whether Compound 1 would affect p300 HAT activity on (H3-H4)2 tetramers, using the same HAT-ELISA methodology with antibodies that detect acetylation of lysine residues without regard to site specificity. Consistent with our previous experiments with the peptide substrate, we observed that Compound 1 had no inhibitory effect on p300 (Figure 1e). We conclude that Compound 1 is a specific inhibitor of Rtt109 but not p300, and can inhibit Rtt109 activity regardless of cofactor protein or substrate lysine.
The one other activity listed in PubChem for compound 1 is inhibition of a tudor domain-histone peptide interaction (PubChem BioAssay AID 504339). This suggested that compound 1 might function as a competitive inhibitor of interactions between Rtt109 and one or both of its substrates. To test this idea, we titrated Ac-CoA or the H3n21 peptide in the presence of varying concentrations of the inhibitor. We first performed experiments in which H3n21 concentrations were held constant, and Ac-CoA was titrated. 50 nM enzyme and peptide were pre-mixed, and reactions were initiated with acetyl-coenzyme A, and reaction rates were plotted and fit to the Michaelis-Menten equation (Figure 2a). Our value for Km in the absence of inhibitor (1.47 +/− 0.33 µM), was similar to that observed previously (1.0 +/− 0.2 µM, 22). Compound 1 clearly reduced Vmax, and its effects on Km were most clearly displayed by analysis on a double reciprocal (Lineweaver-Burk) plot of 1/v versus 1/[Ac-CoA] (Figure 2b). In this type of plot, x-intercept values are −1/Km for each inhibitor concentration, and the y-intercept values are 1/Vmax. The slopes of the lines are very similar, indicating that increasing concentrations of compound 1 reduced both Vmax and Km values.
Figure 2. Titration of acetyl-coenzyme A and peptide substrates.

(a–b) acetyl-CoA titration. 50 nM Rtt109-Vps75 was pre-incubated with 15 µM H3n21 peptide substrate. Reactions were initiated with the indicated amounts of acetyl-CoA and reaction rates were measured at 30°C over 10 minutes (for individual reactions, the linear regression R2 ranged from 0.93 – 0.99). (a) Michaelis-Menton curve. Compound 1 reduced both the Vmax (2.52 +/− 0.23 nM/s; 1.46 +/− 0.08 nM/s; 0.68 +/− 0.09 nM/s for 0, 30, and 60 µM compound 1, respectively) and Km (1.47 +/− 0.33 µM; 0.86 +/− 0.15 µM; 0.68 +/− 0.32 µM for 0, 30, and 60 µM compound 1, respectively). (b) Lineweaver-Burk reciprocal plot of data in panel a. (c–d) Peptide titration. 100 nM Rtt109-Vps75 was incubated with varying amounts of H3n21 peptide substrate (5–50 µM). The reaction was initiated with 30 µM acetyl-CoA and reaction rates were measured at 30°C for 10 minutes (linear regression R2 ranged from 0.96 to 0.99). (c) Michealis-Menton curve. Vmax = 150 +/− 10, 95 +/− 7, 37 +/− 3 nM/s and Km= 22.1 +/− 3.0, 15.3 +/− 2.6, 7.9 +/− 1.8 µ M for reactions with 0, 30 nM and 200 nM compound 1, respectively. (d) Lineweaver-Burk reciprocal plot of data in panel c.
Conversely, we also performed experiments in which initial Ac-CoA concentrations were high and constant and the H3n21 peptide concentrations were varied (Figure 2c). We observed a kcat for Rtt109-Vps75 with our peptide substrate in absence of inhibitor equal to 0.15 +/− 0.01 s−1, similar to the previously observed value of 0.13 +/− 0.04 s−1 22. We note that our observed Km in the absence of inhibitor was 22 +/− 3 µM, about three-fold less than the reported 75 +/− 15 µM 22. However, we note that in these experiments, the peptide substrate was 21 residues long, one residue longer than that used in the previous study, suggesting the added peptide length strengthened binding to Rtt109-Vps75. Consistent with this interpretation, with a 20-mer peptide we observed a Km of 102 µM (data not shown). As in the Ac-CoA titrations, in these peptide titrations reciprocal plot analysis again demonstrated that both the Vmax and Km were reduced in the presence of increasing concentrations of compound 1 (Figure 2d). Therefore, titration of both substrates, Ac-CoA and the H3n21 histone peptide, yielded similar inhibition profiles.
Reduction of Vmax is expected for an inhibitor compound. However, a competitive inhibitor is expected to increase the apparent Km. In contrast, the observed reduction in Km for both the AcCoA and peptide substrates would be consistent with an uncompetitive mechanism of inhibition. This occurs when the inhibitor binds to enzyme-substrate complexes, effectively removing bound enzyme molecules from the reaction. Such an inhibitor reduces the number of active enzyme molecules remaining, resulting in an apparent low concentration of substrates required for catalysis. We therefore tested for tight binding of Rtt109 by compound 1 by dialyzing mixtures after binding for four hours prior to enzymatic assay. Enzyme activity in the presence of dimethyl sulfoxide (DMSO) vehicle was diminished after four hours, perhaps due to denaturation and/or loss of binding to the Vps75 cofactor under these dilute conditions (Figure 3a). However, normalizing these data to the activity observed in the presence of each DMSO control, we observed that dialysis did not diminish the percent of activity inhibited by compound 1 (Figure 3b). These data suggest that compound 1 is a tight or perhaps irreversible inhibitor.
Figure 3. Compound 1 is a tight-binding, time-dependent inhibitor of Rtt109.

(a) Inhibition by compound 1 is stable during dialysis. Concentrated binding mixtures (1 µM Rtt109-Vps75, added to 1.5 µM compound 1 or DMSO) were assembled, and histone acetylation kinetics were analyzed prior to or after 4 hrs of dialysis of these mixtures as described in the Materials and Methods. The pmol of product CoA formed by each reaction mixture is shown. Despite the decrease in enzyme activity during dialysis, compound 1 still inhibits product formation. (b) Normalization of data from panel A to activities observed in absence of inhibitor. Reactions containing compound 1 displayed 47% activity without dialysis, and 33% after dialysis. Because 4 hrs of dialysis did not diminish the percent inhibition by compound 1, these data suggest tight binding of compound 1 to Rtt109-Vps75. (c) Titration of compound 1 in enzyme reactions containing 150 nM Rtt109-Vps75 (triangle points with dotted line non-linear curve fit; rate R2 values > 0.94 for all reactions). Curve of inhibition by compound 1 in reactions containing 50 nM Rtt109-Vps75 from Figure 1b (solid line) is shown for comparison. (d) Time course of inhibition. Ten-fold-concentrated binding mixtures (1 µM Rtt109-Vps75 added to 1.0 µM compound 1 or DMSO) were assembled, and incubated for 0, 12, 24, 40 or 60 minutes on ice. Histone acetylation reactions were initiated with 12.5 µM acetyl CoA and 15 µM H3n21 peptide in nine volumes of reaction buffer. The degree of inhibition continued to increase during the entire 60 minute period analyzed. (e) Titration of compound SAR9 (see Table 1) at indicated concentrations in enzyme reactions containing 50 nM Rtt109-Vps75. Curve of inhibition by compound 1 from Figure 1b is shown for comparison.
Consistent with these data suggesting a tight inhibition mechanism, we found that the concentration of compound 1 required for 50% inhibition depended on the enzyme concentration. Our previous experiments in the presence of 50 nM Rtt109-Vps75 demonstrated 50% inhibition at a concentration of ~56 nM (Figure 1B). However, increasing the enzyme concentration increased the amount of compound 1 required for similar levels of inhibition (Figure 3c). Another characteristic of tight-binding inhibitors is that they often display time-dependent inhibition. To test for this, we assayed aliquots from a large reaction mixture at various times after addition of compound 1 to Rtt109-Vps75 (Figure 3d). Indeed, the degree of inhibition increased during the entire 60 minutes of this experiment, indicating time-dependent inhibition. Together, our data demonstrate that compound 1 is not a competitive inhibitor of either Ac-CoA or peptide substrates (Figure 2). Instead, compound 1 appears to behave like a tight-binding inhibitor.
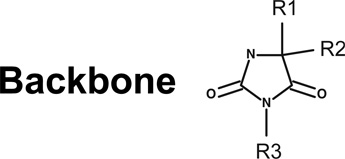
To determine whether particular moieties within compound 1 are associated with its inhibitory activity, we ordered nine commercially available related compounds, termed SAR2-SAR10 (Table 1). Each commercially obtained analog was confirmed have the expected mass and at least 80% purity by HPLC/MS. Some of these compounds were extremely similar to compound 1, notably SAR9, which differs by a single methylene group. To make a rapid initial assessment of these compounds, we first tested their effects on acetylation by Rtt109-Vps75 complexes in single-point HAT reactions, in which we quantified the amount of CoA formed by the end of a 30-minute reaction. Strikingly, only a single compound, SAR9, displayed any inhibition, and it was not as potent as compound 1 (data not shown). To more thoroughly characterize SAR9, we then measured its effects on reaction rates in the range of 100 nM – 1 µM, in the presence of 50 nM Rtt109-Vps75. We observed that SAR9 displayed a poor dose response over this concentration range, with minimal inhibition even at the highest concentration tested (Figure 3e); at these concentrations, compound 1 is a much more effective inhibitor (Figure 1b). Therefore, despite the minimal chemical differences between SAR compounds tested and compound 1, only compound 1 displayed strong inhibitory activity. We conclude that the pharmacophore of compound 1 appears to be limited in its ability to withstand modification.
Table 1.
SAR analysis of compound 1
 |
||||
|---|---|---|---|---|
| PubChem CID | R1= | R2= | R3= | |
| Compound 1 | 4785700 | CH3CH2CH2- |  |
 |
| SAR2 | 45837676 | Same as compound 1 |
Same as compound 1 |
 |
| SAR3 | 42897940 |  |
Same as compound 1 |
|
| SAR4 | 24249195 | 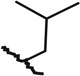 |
Same as compound 1 |
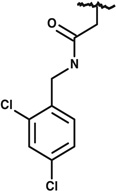 |
| SAR5 | 24234374 | CH3- |  |
Same as compound 1 |
| SAR6 | 4084701 | CH3CH2- | 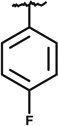 |
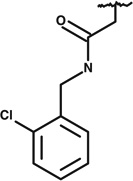 |
| SAR7 | 17492362 | CH3CH2- | 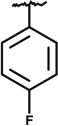 |
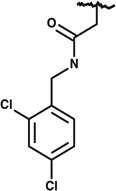 |
| SAR8 | 3997132 | CH3CH2CH2CH2- | Same as compound 1 |
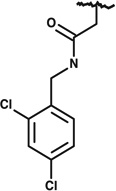 |
| SAR9 | 4787324 | CH3CH2- | Same as compound 1 |
Same as compound 1 |
| SAR10 | 16250795 | Same as compound 1 |
Same as compound 1 |
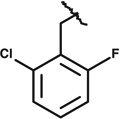 |
This is the first report of a specific small molecule inhibitor for Rtt109, an enzyme required for genome stability and fungal pathogenesis 10, 11, 25. Compound 1 inhibits Rtt109 when stimulated by either of its protein cofactors, Vps75 or Asf1 (Figure 1). Because Rtt109 is thought to bind to these proteins via distinct surfaces, it appears unlikely that compound 1 dislodges these cofactor proteins from the enzyme. We also observe that compound 1 is not a competitive inhibitor of Rtt109’s association with either of its substrates (Figure 2). Instead, it reduces Vmax and Km with respect to both substrates, and appears to be a tight-binding inhibitor that displays time-dependent inhibition of the enzyme (Figure 3). Additionally, compound 1 does not appear to be a promiscuous protein denaturant, because of its specificity for Rtt109 but not the p300 or Gcn5 HAT enzymes (Figure 1).
The goal in finding Rtt109 inhibitors is to provide a lead compound for medicinal chemistry alterations leading to a novel anti-fungal drug. However, we have not observed that compound 1 is able to affect cellular levels of H3K56ac or sensitivity to the DNA damaging agent camptothecin in either C. albicans or Saccharoyces cerevisiae (data not shown). Our inability to detect effects of compound 1 in vivo could result from the efficient multidrug efflux pumps in fungi 34, or rapid metabolism of the compound in cells into non-inhibitory byproducts. Indeed, our observation that small modifications of the structure of compound 1 lead to pronounced loss of inhibitory activity (Figure 3e) would provide many routes for metabolic inactivation. Therefore, we have discovered a novel, enzyme-specific inhibitor of Rtt109 which will be a valuable tool for probing enzyme mechanism, but which also faces challenges in an in vivo setting.
Methods are found in the Supplementary Material
Supplementary Material
Acknowledgements
This work was supported by NIH grants R21 NS66432 and R01 GM055712 to PDK, and F31 AI078726 to JLS. We thank Amie C. Dehner for assistance with protein preparation, Dr. William Youngsaye for consultation regarding the cherry picks and Drs. Craig Peterson and Sharon Cantor for Gcn5 and p300 expression constructs.
Abbreviations
- H3K56
histone H3 lysine 56
- HAT
histone acetyltransferase
- HTS
high throughput screen
- CoA
coenzyme A
- Ac-CoA
acetyl-coenzyme A
- DTNB
5,5'-dithiobis-(2-nitrobenzoic acid)
- HPLC/MS
High pressure liquid chromatography/ mass spectrometry
- PBS
phosphate-buffered saline
- TBS
Tris-buffered saline
- PMSF
phenylmethanesulfonylfluoride
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- ELISA
enzyme-linked immunosorbant assay
- NEM
N-ethylmaleimide
- DMSO
dimethyl sulfoxide
- BSA
bovine serum albumin
- HRP
horseradish peroxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, Verreault A. Curr Biol. 2006;16:1280. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 2.Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. Cell. 2008;134:244. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Cell. 2008;134:231. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan T, Liu CL, Erkmann JA, Holik J, Grunstein M, Kaufman PD, Friedman N, Rando OJ. PLoS Genetics. 2008;4:e1000270. doi: 10.1371/journal.pgen.1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erkmann JA, Kaufman PD. DNA Repair (Amst) 2009;8:1371. doi: 10.1016/j.dnarep.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu Y, Song C, Zhang Q, DiMaggio PA, Garcia BA, York A, Carey MF, Grunstein M. Mol Cell. 2012;46:7. doi: 10.1016/j.molcel.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drogaris P, Villeneuve V, Pomies C, Lee EH, Bourdeau V, Bonneil E, Ferbeyre G, Verreault A, Thibault P. Sci Rep. 2012;2:220. doi: 10.1038/srep00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider J, Bajwa P, Johnson FC, Bhaumik SR, Shilatifard A. J Biol Chem. 2006;281:37270. doi: 10.1074/jbc.C600265200. [DOI] [PubMed] [Google Scholar]
- 9.Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, Kaufman PD. Mol Cell. 2007;25:703. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xhemalce B, Miller KM, Driscoll R, Masumoto H, Jackson SP, Kouzarides T, Verreault A, Arcangioli B. J Biol Chem. 2007;282:15040. doi: 10.1074/jbc.M701197200. [DOI] [PubMed] [Google Scholar]
- 11.Lopes da Rosa J, Boyartchuk VL, Zhu LJ, Kaufman PD. Proc Natl Acad Sci U S A. 2010;107:1594. doi: 10.1073/pnas.0912427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bazan JF. Cell Cycle. 2008;7:1884. doi: 10.4161/cc.7.12.6074. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Tang Y, Cole PA, Marmorstein R. Curr Opin Struct Biol. 2008;8:741. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Y, Holbert MA, Wurtele H, Meeth K, Rocha W, Gharib M, Jiang E, Thibault P, Verrault A, Cole PA, Marmorstein R. Nat Struct Mol Biol. 2008;15:738. doi: 10.1038/nsmb.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stavropoulos P, Nagy V, Blobel G, Hoelz A. Proc Natl Acad Sci U S A. 2008;105:12236. doi: 10.1073/pnas.0805813105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin C, Yuan YA. Structure. 2008;16:1503. doi: 10.1016/j.str.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Li Q. Epigenetics. 2011;6:957. doi: 10.4161/epi.6.8.16065. [DOI] [PubMed] [Google Scholar]
- 18.Albaugh BN, Kolonko EM, Denu JM. Biochemistry. 2010;49:6375. doi: 10.1021/bi100381y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA. Nature. 2008;451:846. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- 20.Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, Sun Y, Meyers DJ, Marmorstein R, Mahadevan LC, Alani RM, Cole PA. Chem Biol. 2010;17:471. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Recht J, Tsubota T, Tanny JC, Diaz RL, Berger JM, Zhang X, Garcia BA, Shabanowitz J, Burlingame AL, Hunt DF, Kaufman PD, Allis CD. Proc Natl Acad Sci U S A. 2006;103:6988. doi: 10.1073/pnas.0601676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berndsen CE, Tsubota T, Lindner SE, S L, Holton JM, Kaufman PD, Keck JL, Denu JM. Nature Structural & Molecular Biology. 2008;15:948. doi: 10.1038/nsmb.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fillingham J, Recht J, Silva AC, Suter B, Emili A, Stagljar I, Krogan NJ, Allis CD, Keogh MC, Greenblatt JF. Mol Cell Biol. 2008;28:4342. doi: 10.1128/MCB.00182-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burgess RJ, Zhou H, Han J, Zhang Z. Mol Cell. 2010;37:469. doi: 10.1016/j.molcel.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masumoto H, Hawke D, Kobayashi R, Verreault A. Nature. 2005;436:294. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 26.Hyland EM, Cosgrove MS, Molina H, Wang D, Pandey A, Cottee RJ, Boeke JD. Mol Cell Biol. 2005;25:10060. doi: 10.1128/MCB.25.22.10060-10070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Driscoll R, Hudson A, Jackson SP. Science. 2007;315:649. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wurtele H, Tsao S, L�pine G, Mullick A, Tremblay J, Drogaris P, Lee EH, Thibault P, Verreault A, Raymond M. Nature Medicine. 2010;16:774. doi: 10.1038/nm.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gudlaugsson O, Gillespie S, Lee K, Vande Berg J, Hu J, Messer S, Herwaldt L, Pfaller M, Diekema D. Clin Infect Dis. 2003;37:1172. doi: 10.1086/378745. [DOI] [PubMed] [Google Scholar]
- 30.Cowen LE, Anderson JB, Kohn LM. Annu Rev Microbiol. 2002;56:139. doi: 10.1146/annurev.micro.56.012302.160907. [DOI] [PubMed] [Google Scholar]
- 31.Cannon RD, Lamping E, Holmes AR, Niimi K, Tanabe K, Niimi M, Monk BC. Microbiology. 2007;153:3211. doi: 10.1099/mic.0.2007/010405-0. [DOI] [PubMed] [Google Scholar]
- 32.Lopes da Rosa J, Kaufman PD. Biochim Biophys Acta. 2012;1819:349. doi: 10.1016/j.bbagrm.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trievel RC, Li FY, Marmorstein R. Anal Biochem. 2000;287:319. doi: 10.1006/abio.2000.4855. [DOI] [PubMed] [Google Scholar]
- 34.Ramage G, Bachmann S, Patterson TF, Wickes BL, L�pez-Ribot JL. Journal of Antimicrobial Chemotherapy. 2002;49:973. doi: 10.1093/jac/dkf049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.