

Abstract
Brain development is a complex and dynamic process, and many environmental factors have been found to influence the normal development of neural pathways. Cumulative evidence suggests that metabolic hormones that regulate the hypothalamic circuits that control energy homeostasis function in much the same way that sex steroids act on sexually dimorphic circuits. For example, although the effects of the adipocyte-derived hormone leptin were originally thought to be limited to the neural control of energy homeostasis in adult animals, it is now becoming increasingly clear that leptin can also determine patterns of neurogenesis, axon growth, and synaptic plasticity in the developing hypothalamus. More recent studies have also extended the role of the metabolic hormones ghrelin and insulin in various aspects of brain development. Examining how metabolic hormones control hypothalamic development will help our understanding of the developmental origin of adult metabolic diseases and, hopefully, improve our ability to predict adverse outcomes.
Keywords: leptin, ghrelin, insulin, hypothalamus, arcuate nucleus, obesity, development, neurogenesis, plasticity, axon guidance
1. Introduction
One of the brain's core functions is to help the body maintain homeostasis. The hypothalamus plays an essential role in this function by ensuring that physiological responses remain in tune with environmental demands. Accordingly, the regulation of hypothalamic circuits is influenced by signals that carry information about nutritional and metabolic states. The hormones leptin, ghrelin, and insulin are particularly well positioned to communicate environmental nutrient availability to the hypothalamus. These hormones, secreted in proportion to body energy stores and/or nutritional state, regulate energy balance by influencing feeding and/or energy expenditure. The arcuate nucleus of the hypothalamus (ARH) appears to be a predominant site for the integration of these peripheral blood-borne signals. In considering ARH neurons involved in the regulation of energy balance, primary importance has been given to the neurons that produce both neuropeptide Y (NPY) and agouti-related peptide (AgRP) and the neurons that contain proopiomelanocortin (POMC)-derived peptides, in part because they can directly respond to leptin, ghrelin, and insulin (Williams and Elmquist, 2012). Each of these neuronal populations provides overlapping projections to other key parts of the hypothalamus, including the paraventricular nucleus of the hypothalamus (PVH), the dorsomedial nucleus of the hypothalamus (DMH), and the lateral hypothalamic area (LHA), exerting opposing effects on feeding and energy expenditure. These adult functional properties are progressively elaborated in response to evolving developmental events that include distinct sets of critical periods designed to ensure that the environmental responsiveness is preserved throughout life. Hormones are particularly important in this regard, and they appear to exert major structural effects during hypothalamic development. Sex hormones have been the traditional focus because of their well-established capacity to promote sexual differentiation during the critical period of development. For example, hormones such as estrogen, testosterone, and progesterone can cause permanent changes in the architecture of limbic-hypothalamic circuits by specifying cell number, the density of axonal connections, dendritic architecture and neurotransmitter phenotype (de Vries and Simerly, 2002; McCarthy, 2008; McEwen, 1992; Simerly, 2002). This review discusses recent findings on how metabolic hormones also exert profound organizational effects by directing key developmental processes in the developing hypothalamus.
2. Development of metabolically relevant hypothalamic circuits
2.1. Determination of cell numbers
Before considering the structural actions of metabolic hormones in the developing hypothalamus, it seems important that we have a good understanding of the timelines and cellular processes of normal hypothalamic development. In mice and rats, the hypothalamus begins to form during mid-gestation and continues to develop during the postnatal period (see (Markakis, 2002) and (Bouret, 2010) for review). During this extended developmental period, a variety of processes shape the hypothalamic nuclei involved in the control of feeding and energy balance. The formation of hypothalamic pathways is characterized by various developmental processes that fall into three major categories: 1) neurogenesis, 2) neuronal migration, and 3) the formation of functional circuits that include axon growth and synaptogenesis. Neurons that form hypothalamic nuclei originate from the proliferative zone of the third ventricle (Altman and Bayer, 1986; Shimada and Nakamura, 1973). Classic experimental tools for the study of neurogenesis have included the administration of the thymidine analog BrdU during critical windows of development and have revealed that the majority of neurons that compose the hypothalamus are born between embryonic day (E) 12 and E14 in mice (Ishii and Bouret, 2012; Padilla et al., 2010; Shimada and Nakamura, 1973) and between E12 and E17 in rats (Altman and Bayer, 1986). In mice, neurons found in the DMH, PVH, and LHA are born between E12-E14. The ARH and VMH nuclei show a relatively longer neurogenic period. Many neurons in these nuclei are born on E12, but some neurons are generated as late as E16. Although the birthdate of specific neuronal populations has not been fully established, we know that the majority of leptin-responsive neurons in the adult hypothalamus are born during a developmental period that is largely restricted to E12 (Ishii and Bouret, 2012). Additionally, Padilla et al. recently reported that POMC neurons in the ARH are born primarily on E12 and that a subpopulation of embryonic Pomc-expressing precursors subsequently adopts an NPY phenotype in adult mice (Padilla et al., 2010). These observations are consistent with the early determination of cell fate and the fact that specific sets of hypothalamic neurons may share a common ontological lineage. Part of the complex process of defining hypothalamic cell numbers also includes the proper migration of neurons from their sites of origin (i.e., the proliferative zone of the third ventricle) to their final positions in the mature hypothalamus. This developmental process occurs primarily during late gestation in rodents (McClellana et al., 2006).
2.2. Establishment of axonal projections
In both rats and mice, the hypothalamus is still relatively immature at birth and continues to develop during the first two weeks of postnatal life when neurons send axonal projections to their target sites and form functional synapses. In part because of its importance for the regulation of feeding, the first detailed study that utilized axonal labeling defined the ontogeny of projection pathways from the ARH. These neuroanatomical studies revealed that ARH projections are immature at birth and develop mainly during the second week of life in mice (Bouret et al., 2004a). ARH projections extend through the periventricular zone of the hypothalamus to provide inputs to the DMH by P6, followed by inputs to the PVH between P8 and P10. Projections from the ARH to the LHA develop significantly later, with the mature pattern of innervation is first apparent on P12 (Bouret et al., 2004a). Terminals containing AgRP/NPY are found in the DMH, PVH, and LHA of rat neonates in a pattern that coincides with the innervation of axons from the ARH (Grove et al., 2003; Nilsson et al., 2005). In contrast to the development of projections from the ARH, efferents from the DMH to the PVH and LHA are fully established by P6 (Bouret et al., 2004a). Projections from the VMH also develop prior to those from the ARH, and by P10, VMH fibers supply strong inputs to the LHA, whereas, at this age, the LHA is almost devoid of fibers from the ARH (Bouret et al., 2004a). Similarly, the neurohypophyseal pathway from the PVH to the median eminence appears to be largely formed by birth (Daikoku et al., 1984).
2.3. Synapse formation
The formation of synapses, which are the neurobiological substrates of almost all cell-to-cell communication, follows the development of axonal projections. However, we still know relatively little about the exact time point (if any) at which the assembly of these circuits is fully established. It appears that hypothalamic synaptogenesis is an active process that, in rodents, continues well past the third week of postnatal life. For example, Matsumoto and collaborators reported a gradual increase in the number of synapses in the ARH from birth to adulthood (Matsumoto and Arai, 1976). Ultrastructural analysis of synapses within the rat ARH revealed very few axodendritic or axosomatic synapses on P5, whereas by P20 (weaning), about one-half of the synapses found in adult animals are already formed. The number of ARH synapses continues to increase after weaning to reach an adult-like pattern by P45 (Matsumoto and Arai, 1976). Supporting these observations, work from Colmers' laboratory reported an increase in GABAergic terminals that contact PVH parvocellular neurons between P7-P9 and P30-P40 (Melnick et al., 2007). These findings indicate that the period of hypothalamic plasticity extends well beyond embryonic and early postnatal life and strongly suggest that exposure to environmental cues during adolescence and early adulthood can also have an influence on hypothalamic organization and subsequent function.
3. Leptin
3.1. Expression pattern of leptin during perinatal life
Leptin is one of the first major metabolic hormones to appear during development. White adipose tissue (the main source of leptin production in adult animals) is minimal at early ages, yet mouse fetuses do contain significant leptin levels in their blood as early as E12 (Udagawa et al., 2006a) (Ishii and Bouret, unpublished data). Various tissues produce leptin during embryonic development. On E13, high levels of leptin gene expression are found in the fetal liver and cartilage-bone structures, followed by cardiac expression between E16 and E18 (Hoggard et al., 1997). In addition to being produced by the embryo itself, dams also contain high levels of leptin during pregnancy, but whether maternal leptin crosses the placenta and reaches the embryo during early-mid gestation (i.e., when brain development is initiated) remains controversial. Circulating leptin levels increase markedly during the postnatal period and exhibit a distinct surge between P8 and P12 in mice (Ahima et al., 1998). These significantly higher levels of postnatal circulating leptin are associated with greater expression of leptin mRNA in both white (abdominal) and brown (interscapular) adipose tissue (Devaskar et al., 1997). There is a coordinated decrease in levels of leptin mRNA and leptin peptide after weaning, i.e., when pups switch from maternal milk to an adult diet (Devaskar et al., 1997). Analysis of leptin transport across the BBB also reveals that the hormone can reach the brain at early ages (Pan et al., 2008). Consistent with these findings, the short form of the leptin receptor, LepRa, which is considered to be one of the main transporters for leptin across the BBB (Hileman et al., 2000; Kastin et al., 1999), is expressed in brain microvessels at birth (Pan et al., 2008).
3.2. Leptin receptor expression in the developing brain
The leptin receptor exists in multiple alternatively spliced isoforms, of which only the long form (LepRb) associates with Janus kinase 2 (JAK2) to mediate intracellular signaling. Upon leptin binding, LepRb initiates multiple intracellular signal transduction pathways that result in the activation of STAT family transcription factors, extracellular signal-regulated kinases (ERK), and phosphoinositol-3 kinase. Leptin receptors, including LepRb, are detected in the mouse brain as early E10 (Hoggard et al., 1997; Udagawa et al., 2000). However, during embryonic and early postnatal life, LepRb mRNA expression is restricted to the ependymal cells of the third ventricle (Carlo AS, 2007; Cottrell et al., 2009). Supporting the idea that these ependymal cells do contain a functional leptin receptor during early life, the injection of leptin into P4 mice leads to strong induction of SOCS3 mRNA in the cells lining the third ventricle (Cottrell et al., 2009). These findings are particularly interesting because, as discussed above, neurons that compose various hypothalamic nuclei in adults are primarily derived from precursors that originate from these ependymal cells lining the third ventricle (Altman and Bayer, 1986; Shimada and Nakamura, 1973). LepRb mRNA becomes more abundant in various parts of the hypothalamus after P10, including in the ARH, VMH, DMH, and LHA (Caron et al., 2010) (Figure 1). LepRb mRNA is also transiently elevated in other regions of the postnatal mouse brain, such as the cortex, hippocampus, and laterodorsal nucleus of the thalamus (Caron et al., 2010). However, in contrast to hypothalamic neurons, neurons located in these brain structures do not exhibit pSTAT3 immunoreactivity after peripheral leptin administration (Caron et al., 2010), implicating that leptin may not reach these brain regions at early ages.
Figure 1. Secretory pattern of metabolic hormones during neonatal life.

Elevated leptin levels are distinctly observed during the first two weeks of postnatal life in rodents. Ghrelin levels are also regulated neonatally but, in contrast to leptin, ghrelin levels are low during the first postnatal week and increase gradually thereafter to reach a plateau during the second week of postnatal life. Insulin levels remain relatively unchanged throughout postnatal life. The long form of the leptin receptor (LepRb), as well as the ghrelin (GHSR) and insulin (InsR) receptors, are expressed and competent in the developing arcuate nucleus (ARH), yet none of these hormones modulate feeding during neonatal life. Instead of regulating food intake and body weight, leptin and ghrelin appear to be important neurodevelopmental cues that influence hypothalamic development.
3.3. Empirical studies supporting a role for leptin in brain development
Long before the discovery of leptin, Bereiter and Jeanrenaud (Bereiter and Jeanrenaud, 1979, 1980) reported that the brains of genetically obese and diabetic mice (Lepob/Lepob and Leprdb/Leprdb mice, respectively) were structurally different from those of control mice. They observed a reduction in cell density in various brain regions, including the hypothalamus. They also reported that Lepob/Lepob mice have alterations in the dendritic orientation of hypothalamic neurons, suggesting widespread structural abnormalities in these mice. Twenty years later, after the discovery that Lepob/Lepob mice actually lack leptin, Ahima and colleagues demonstrated that these mutant mice exhibited reduced brain weight and an immature pattern of expression of synaptic and glial proteins (Ahima et al., 1999). Furthermore, they showed that Lepob/Lepob mice have elevated levels of growth-associated protein in the neocortex and hippocampus and a reduction in syntaxin-1, synaptosomal-associated protein-25, and synaptobrevin expression (Ahima et al., 1999). These landmark studies led the authors to predict a role for leptin in brain development.
3.4. Hypothalamic Actions
The availability of Lepob/Lepob mice, combined with the wide range of methods for visualizing neuronal circuits, made possible the direct assessment of the impact of leptin on hypothalamic development. Axonal labeling of ARH axons with the anterograde tracer DiI revealed that leptin deficiency causes a disruption in formation of projections from the ARH to each major target nucleus (Bouret et al., 2004b). For example, the density of ARH axons innervating the PVH is approximately 10-fold lower in Lepob/Lepob mice compared to wild-type littermates (Bouret et al., 2004b). Another important observation is that the disruption of ARH pathways in Lepob/Lepob mice appears to be permanent, as there are fewer ARH fibers in each terminal field even on P60, a stage considered to be mature regarding the regulation of energy homeostasis in mice. But perhaps the most important discovery was that leptin acts primarily during a restricted neonatal period to exert its developmental effects on ARH neural projections. Peripheral leptin injections from P4 through P12 in Lepob/Lepob mice restore the density of ARH fibers innervating the PVH to a density that was comparable to that of wild-type littermates (Bouret et al., 2004b). In contrast, the treatment of adult Lepob/Lepob mice with leptin for 20 days was relatively ineffective because it did not increase the density of either aMSH or AgRP fibers in the PVH to levels that are characteristic of wild-type mice. Thus, the developmental activity of leptin in ARH projection pathways appears to be restricted to a neonatal window of maximum sensitivity that corresponds to a period of elevated leptin secretion. Based on these observations it is also not surprising that leptin also restores normal brain weight in Lepob/Lepob mice but only when the hormone is injected during early life (Steppan and Swick, 1999). Together, these findings demonstrate that leptin is required for normal postnatal development of ARH projections and suggest that the postnatal leptin surge is indeed a key developmental signal affecting the architecture of hypothalamic circuits that mediate energy balance (Figure 2).
Figure 2. Hormonal mechanisms underlying the development of hypothalamic feeding circuits.

The adipocyte-derived hormone leptin represents a powerful neurotrophic agent that promotes the formation of neural projections from the arcuate nucleus of the hypothalamus (ARH). A key factor in controlling the development of ARH neural projections is the expression of the long form of the leptin receptor (LepRb) by ARH neurons. More specifically, both LepRb->ERK and LepRb->STAT3 signaling pathways appear to play a distinct role in the development of hypothalamic feeding circuits. Autophagy (a cell-intrinsic and metabolic sensing cellular process that degrades cytoplasmic material) is also implicated in the development of ARH neural projections, and it is possible that some of the neurotrophic effects of leptin are mediated through autophagy activation.
3.5. Intracellular pathways that mediate the trophic effects of leptin
One key factor in controlling the development of hypothalamic circuits is the expression of LepRb by ARH neurons (Figure 2). The developing ARH contains a high density of neurons that express LepRb (Caron et al., 2010), and the administration of leptin to mouse neonates results in the activation of major LepRb signaling pathways, including pSTAT3, pERK and pAkt (Bouret et al., 2012; Caron et al., 2010). The exact sites of action for the developmental effects of leptin remain to be investigated, but they appear to include a direct action on ARH neurons. Indeed, ARH projections to each major target region, including projections to extra-hypothalamic regions known to be insensitive to leptin (Caron et al., 2010), appear to be reduced in Lepob/Lepob mice (Bouret et al., 2004b). In addition, leptin induces neurite extension from isolated organotypic explants of the ARH, further supporting the notion that leptin acts directly on LepRb-containing ARH neurons to promote axon growth and illustrating the trophic activity of leptin on ARH neurons (Bouret et al., 2004b). Furthermore, mice or rats that lack functional LepRb signaling (Leprdb/Leprdb mice and fa/fa rats, respectively) display a reduced density of ARH projections to the PVH (Bouret and Simerly, 2007; Bouret et al., 2012).
As noted above, LepRb belongs to the cytokine family and activates various intracellular signaling pathways, such as STAT3, MAP kinase/ERK1/2 and Akt, in neonatal ARH neurons (Bouret et al., 2012; Louis et al., 2011). Although LepRb->pSTAT3 represents a crucial mediator of leptin action on energy balance in mature animals (Bates et al., 2003), this signaling pathway is not solely required to mediate the neurodevelopmental action of leptin. Both mice lacking LepRb->pSTAT3 or LepRb->pERK signaling (s/s and l/l mice, respectively) display the disrupted development of ARH neural projections and are not capable of exhibiting neurite outgrowth in vitro when exposed to leptin (Bouret et al., 2012). However, not all regions that express LepRb respond to the trophic action of leptin. For example, the DMH contains a substantial density of neurons that express leptin receptors during postnatal life (Caron et al., 2010), yet its projections to the PVH are normal in Lepob/Lepob mice (Bouret et al., 2004b). Other cell-intrinsic metabolic-sensing pathways have been implicated in the development of hypothalamic feeding circuits. These include autophagy, a cellular process that degrades cytoplasmic materials, including organelles and misfolded proteins (Coupe et al., 2012). Because leptin directly promotes axon growth from ARH neurons (Bouret et al., 2004b) and because leptin promotes autophagy (Malik et al., 2011), it is possible that some of the neurotrophic effects of leptin might be mediated through autophagy activation (Figure 2).
3.6. Extra-hypothalamic actions
The neurodevelopmental actions of leptin are not restricted to hypothalamic development. Soon after the discovery that leptin influences the establishment of hypothalamic neural projections, various groups reported a role for leptin in the development of the hippocampus and the cortex. For example, via the MAPK/ERK pathway, direct exposure of hippocampal neurons to leptin increases the motility and density of dendritic filopodia, with consequences on synapse morphology (O'Malley et al., 2007). Work from Udagawa and colleagues has also shown a role for leptin during cortical development (Udagawa et al., 2006b). The cortex expressed high levels of LepRb mRNA during development (Caron et al., 2010; Udagawa et al., 2000), and leptin deficiency results in a reduction in the number of cortical neurons born during embryonic life. Of the potential mechanisms underlying leptin's control of cell number during development, a significant alteration in neurogenesis has found the most experimental support. The use of neuronal birthdating methods, such as using 5-bromodeoxyridine (BrdU), provided evidence that leptin influences the cortical cell number by promoting neurogenesis (Udagawa et al., 2006b). Leptin also appears to influence axonal growth in the developing cortex. Leptin causes a marked increase in expansion of the axonal growth cone of primary cultures of embryonic cortical neurons (Valerio et al., 2006). Similar to the axonotrophic action of leptin on hypothalamic neurons, the LepRb->MAPK and LepRb->Akt signaling pathways are the main LepRb signaling pathways involved in the induction of cortical axon outgrowth (Valerio et al., 2006). At the cellular level, the effects of leptin on axon growth appear to be mediated, at least in part, through an action on growth associated protein 43 (GAP-43). LepRb co-localizes with GAP-43 at the level of axonal growth cones (Shanley et al., 2002), and the expression of GAP-43 is regulated by leptin in developing cortical neurons (Valerio et al., 2006).
4. Ghrelin
4.1. Developmental regulation of ghrelin expression
Following its discovery in 2000, it soon became evident that ghrelin may play a role during development. Ghrelin is expressed in the embryo as early as the morula stage and continues to be expressed in the developing embryo and fetus. In rodents, high levels of ghrelin mRNA are detected in fetuses at E12, and E17 fetuses contain significant levels of acylated (the “active” form) and desacylated (the “inactive” form) ghrelin in their blood (Nakahara et al., 2006; Torsello et al., 2003). In the fetus, the pancreas appears to be a major source of ghrelin expression during perinatal life. Ghrelin mRNA and protein are found at high levels in the fetal pancreas, whereas low levels of ghrelin are detected in the fetal stomach (Chanoine and Wong, 2004; Wierup et al., 2002). These observations suggest that, in contrast to the adult, the source of circulating fetal ghrelin may be the pancreas, not the stomach. However, stomach ghrelin expression increases gradually after birth to reach adult-like levels by 3-5 weeks of life (Hayashida et al., 2002; Torsello et al., 2003). Simultaneously, pancreatic ghrelin expression declines progressively from birth to weaning and becomes barely detectable in the adult pancreas (Wierup et al., 2002).
4.2. Expression of ghrelin receptor during development
Ghrelin mediates its effects through binding on growth hormone secretagogue receptors (GHSRs), a G-protein coupled receptor (Sun et al., 2004). Molecular methods showed that the central nervous system expresses high levels of Ghsr mRNA throughout pre-and post-natal life (Inoue et al., 2010; Nakahara et al., 2006), but a detailed distribution of GHSR expression in the neonatal brain is still lacking. In addition, whether central GHSR expression is subjected to developmental regulations is undetermined. Nevertheless, systematic analysis of Ghsr mRNA using in situ hybridization in adult brains shows that the ghrelin receptor is primarily expressed in the hypothalamus, specifically in the nuclei involved in food intake and body weight regulation, such as the arcuate, ventromedial, dorsomedial, and paraventricular nuclei of the hypothalamus (Mitchell and Bouret S, 2001; Zigman et al., 2006). Additionally, GHSRs are expressed in other metabolically relevant brain structures, such as the ventral tegmental area and the nucleus of the tractus solitarius (Mitchell and Bouret S, 2001; Zigman et al., 2006).
4.3. Ghrelin's effects on the developing hypothalamus
Accumulating evidence suggests that there are differences in the biological actions of ghrelin between adults and neonates. For example, in sharp contrast to the potent orexigenic effects of ghrelin on adults, exogenous ghrelin does not significantly promote milk intake nor body weight in the first 2-3 postnatal weeks in rats and mice (Piao et al., 2008; Steculorum and Bouret, 2011a). A possible explanation for this lack of response is that the neonatal brain is relatively insensitive to ghrelin and may present ghrelin resistance. However, GHSRs are found in the nuclei known to regulate feeding, including in the ARH (Figure 1), and acute peripheral ghrelin treatment activates ARH neurons (as evidenced by the induction of cFos immunoreactivity) during early postnatal life (Steculorum and Bouret, 2011a). Further supporting the functional role of ghrelin in the postnatal ARH, the mRNA levels of Pomc and Npy mRNA in the ARH are decreased and increased, respectively, following acute ghrelin injection to mouse pups at P10 (Steculorum and Bouret, 2011a). Collectively, these results support the hypothesis that ghrelin receptors are present and functional in the developing hypothalamus and that the role of ghrelin during neonatal life may differ from the one in adults.
4.4. Effects of ghrelin on brain growth and neurogenesis
Much of what we know about the role of ghrelin in brain development has been inferred from classic studies that used experimental tools such as the thymidine analog bromodeoxyuridine, which monitors cell proliferation and neurogenesis. These studies have demonstrated that the exposure of cells derived from a variety of brain regions to ghrelin promotes cell proliferation. For example, when cells from the traditional ghreli target areas of the adult brain (i.e., the hypothalamus, dorsal motor nucleus of the vagus, and nucleus of the solitary tract) are incubated in vitro with ghrelin, cell proliferation increases in a dose-dependent manner (Inoue et al., 2010; Zhang et al. 2005; Zhang et al., 2004). Importantly, many of the resultant newborn cells acquire a neuronal and/or glial phenotype. Additionally, the neurogenic effects of ghrelin appear to be widespread because ghrelin also induces cell proliferation in the developing spina cord in vitro (Sato et al., 2006). These same in vitro studies further showed that ghrelin exerts its maximal neurogenic effect on fetal neural tissues. The exposure of either cultured spinal cord or hypothalamic cells to ghrelin markedly increases cell proliferation but these effects were greater when the cells were taken from E17 embryos instead of P2 pups (Inoue et al., 2010). Intriguingly, both acyl and des-acyl ghrelin promote cell proliferation and neurogenesis, with the acylated form of ghrelin exerting greate proliferative effects as opposed to the non-acylated form of ghrelin (Nakahara et al. 2006; Sato et al., 2006). These observations suggest that the proliferative effects o ghrelin do not require the octanoic acid modification of ghrelin and that ghrelin act through both a GHSR-dependent and GHSR-independent mechanisms to mediate it neurodevelopmental effects.
5. Insulin
5.1. Developmental regulation of insulin and insulin receptors
Measurable levels of insulin are detected in embryonic tissues as early as at E8. Insulin levels increase thereafter, reaching a peak at E9 and returning to basal levels at E12 (Spaventi et al., 1990). However, the pancreas, which is the main source of insulin production in adults, is still very immature at E8-E9, suggesting that the insulin detected at this early stage may be extra-pancreatic. Supporting this hypothesis, insulin expression was detected in tissues other than the pancreas during early embryonic life. Notably, insulin expression was observed in the central nervous system of mouse embryos at E9, an age that also corresponds to the elevated levels of insulin found in embryonic tissues (Deltour et al., 1993). It is also important to note that maternal insulin cannot cross the placental barrier. However, maternal glucose is actively transported to the fetus, where it can stimulate insulin secretion early during fetal development (Baumann et al., 2002). Pathological conditions, such maternal insulin deficiency and maternal hyperinsulinemia (that results in insulin resistance), lead to maternal hyperglycemia, which in turn induces compensatory fetal and neonatal hyperinsulinemia (Desoye et al., 2011). This developmental hyperinsulinemia is considered to be a main contributor to the perinatal programming for obesity and diabetes (Martin-Gronert and Ozanne, 2005; Paderson, 1971; Paderson and Osler, 1961). During postnatal life, insulin levels remain relatively stable (Bouret, unpublished data) and do not exhibit major developmental regulation, unlike other metabolic hormones such as leptin and ghrelin.
The insulin receptor is a receptor tyrosine kinase, and its function in the regulation of peripheral glucose homeostasis is well studied. The presence of insulin receptor in the central nervous system was first reported decades ago (Havrankova et al., 1979), and its distribution is widespread. In general, insulin receptor expression is higher at early stages and lower in the adult, reaching its maximal level during the first week of postnatal life (Figure 1). Brain regions that display relatively high densities of binding sites throughout embryonic and postnatal development and in adulthood include the hypothalamus, olfactory bulb, cortex, choroid plexus, and cerebellum (Kar et al., 1993). Other brain regions, such as the caudate-putamen neuroepithelium or some brain stem nuclei express high densities of insulin receptors, but only for a restricted developmental period restricted to the late embryonic and early postnatal period (i.e., E18 to P7). Insulin binding to these brain sites declines gradually to low levels over the course of brain maturation (Kar et al., 1993).
5.2. Role of Insulin and insulin receptors in brain development and plasticity
Insulin has long known to be implicated in growth and development. Maternal injections of insulin between gestational day 15 and 20, a critical period for hypothalamic development, induced obesity in the offspring (Jones et al., 1995). The metabolic abnormalities observed in the offspring of insulin-injected dams are accompanied by increased hypothalamic norepinephrine levels (Jones et al., 1995) and increased density of norepinephrine-containing fibers innervating the PVH (Jones et al., 1996). The manipulation of maternal insulin levels can also be performed experimentally by injecting streptozotocin (STZ), a pancreatic beta cell toxin, during gestation. STZ injections result in hypoinsulinemia and are associated with a reduced density of POMC- and NPY-containing fibers that innervate the PVH (Steculorum and Bouret, 2011b). These data indicate that changes in insulin levels during neonatal development can influence axon growth in the offspring (Figure 3). However, studying insulin's influence on axon development in vivo has turned out to be an extremely difficult problem, and progress has been relatively slow, given the obvious importance of the end points. Adding to the complexity is the fact that insulin injection also results in a marked decrease in circulating glucose levels, making it difficult to study the effects of insulin independently of glucose. Nevertheless, early in vitro studies revealed that insulin represents a powerful neurotrophic agent. The exposure of isolated organotypic brain explants (derived from the preoptic area, hypothalamus, or cerebral cortex) to insulin results in a marked induction of axon growth. Further in vitro studies indicate that insulin exerts more widespread structural effects, including the regulation of synaptic plasticity, dendritic outgrowth, and involvement in neuronal survival. At the subcellular level, the insulin receptor is a component of synapses, where it localizes at the postsynaptic density in cultured hippocampal neurons, suggesting a role for insulin in regulating synaptic function and/or morphology (Abbott et al., 1999). Of particular interest, IRSp53, an insulin receptor substrate enriched in the brain, localizes to synapses as a component of the postsynaptic density (PSD) (Abbott et al., 1999). The overexpression of IRSp53 increases spine density in cultured hippocampal neurons and induces filipodium formation and neurite outgrowth in N1E-115 neuroblastoma cells. In contrast, RNA interference knockdown of the IRSp53 protein decreases spine density and alters spine morphogenesis (Choi et al., 2005; Govind et al., 2001; Miki and Takenawa, 2002). Moreover, IGF-1 null mice display a marked reduction in dendritic arbor length and complexity as well as spine density of pyramidal neurons (Cheng et al., 2003), further supporting the idea that insulin receptors play a role in dendritic arbor development.
Figure 3. Developmental programming of hypothalamic feeding pathways.
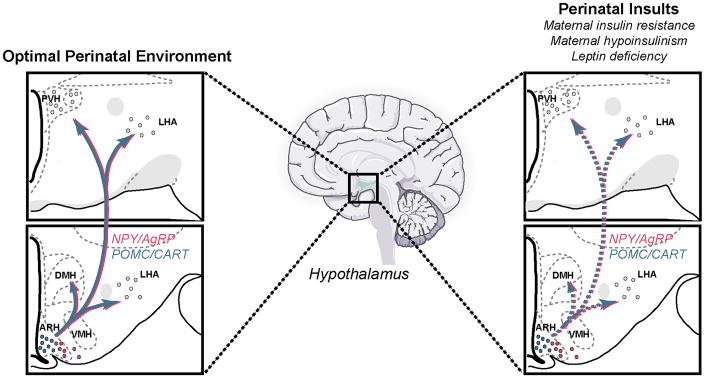
The developmental programming of hypothalamic neural systems by the perinatal hormonal environment represents a possible mechanism by which alterations in maternal and/or postnatal nutrition predispose the offspring to obesity. The development of the arcuate nucleus (ARH), which contains a subset of neurons that are devoted to metabolic regulation, such as neuropeptide Y (NPY) and agouti-related peptide (AgRP)-coexpressing neurons and proopiomelanocortin (POMC)-containing neurons, appears to be highly sensitive to changes in the nutritional environment. Structural defects have been observed in the hypothalamus of animals subjected to an altered nutritional environment. These effects appear to be mediated, to some extent, by abnormal leptin and insulin secretion and/or signaling during critical periods of fetal and/or postnatal development.
In addition to its involvement in neural connectivity, insulin also plays a role in brain development and plasticity by influencing the cell number in the hypothalamus, possibly by regulating neurogenesis. Maternal hypoinsulinemia induced by STZ injection results in an increased number of neuropeptide Y-, POMC- and galanin-containing neurons in the ARH (Franke et al., 2005; Plagemann et al., 1999a; Plagemann et al., 1998; Steculorum and Bouret, 2011b). Moreover, insulin receptor-deficient mice display an increased number of POMC neurons during adulthood, which can partially be rescued by the specific reconstitution of the insulin receptor on POMC neurons (Plum et al., 2012). Importantly, the impaired number of ARH neurons in offspring of diabetic mothers can be prevented by the normalization of gestational hyperglycemia by pancreatic islet transplantation (Franke et al., 2005), suggesting that insulin and/or glucose levels are critical for normal hypothalamic neurons determination. In addition, cross-fostering experiments indicate that pups born from control mothers and raised by diabetic mothers have an altered number of arcuate neurons (Fahrenkrog et al., 2004), showing the importance of the postnatal diabetic environment in influencing hypothalamic cell numbers. Supporting this observation, intra-hypothalamic injections of insulin during early post-natal life are associated with morphological alterations of hypothalamic nuclei (including the ARH and VMH) and life-long metabolic disturbances (Plagemann A et al., 1992v; Plagemann et al., 1999b). In addition to being positively correlated with the number of neurons, insulin levels during early postnatal life are also positively correlated with the number of astrocytes. For example, while hypoinsulinemic pups born to protein-restricted mothers display a decrease in both neurons and astrocytes (Plagemann et al., 2000), hypoinsulinemic offspring of diabetic dams display an increased number of neurons and astrocytes (Plagemann A et al., 1992; Plagemann et al., 1998). Because insulin stimulates GFAP expression (Toran-Allerand et al., 1991), insulin levels may directly and/or indirectly modulate the number of hypothalamic neurons, possibly by stimulating astrocytogenesis.
6. Possible synergetic effects of metabolic hormones in influencing hypothalamic development
Metabolic hormones may also act in concert to direct hypothalamic growth. This idea is supported by the pioneer work of Torran-Allerand and colleagues. More than two decades ago, they found that the incubation of explants from the fetal hypothalamus with insulin induces neuritic extension. Incubation with estradiol alone induces similar neurotrophic effects. However, sequential incubation with insulin and estrogen produces a synergetic enhancement of hypothalamic neurite outgrowth that is stronger than the additive impact of the two hormones acting independently (Toran-Allerand et al., 1988). Because insulin and leptin share common intracellular signaling pathways (including MAPK, Akt, and mTOR) (Carvalheira et al., 2005), it is therefore possible that these two metabolic hormones may also act synergistically to mediate their neurotrophic effects on the hypothalamus.
7. Neuroplastic response of the mature hypothalamus to metabolic cues
7.1. Synaptic response
The mature hypothalamus appears to be sensitive, in terms of neuroplastic response, to a variety of metabolic hormones, including leptin. However, the degree and the nature of leptin's neuroplastic actions differ markedly between adults and neonates. For example, whereas leptin administration to adult Lepob/Lepob mice does not have a significant impact on ARH axonal projections to its target nucleus, it results in changes in synaptic organization within the ARH (Pinto et al., 2004). The excitatory and inhibitory synaptic inputs to the POMC and NPY neurons are markedly altered in adult Lepob/Lepob mice; leptin deficiency increases excitatory inputs on NPY/AgRP neurons while it decreases excitatory synaptic inputs to POMC neurons. Acute leptin injection in adult Lepob/Lepob mice reverses these effects, both at the electrophysiological and ultrastructural levels. This reversal is also very rapid, occurring within hours of leptin administration (Pinto et al., 2004). Together, these data illustrate that the adult hypothalamus remains plastic, keeping the critical “plastic” window open for at least some components of the hypothalamic pathways.
7.2. Neurogenic response
Recent work has also highlighted the importance of leptin-receptor hypothalamic progenitor neurons in influencing hypothalamic cellularity. To test this hypothesis, Fliers and colleagues micro-implanted neural progenitors that expressed leptin receptors into the hypothalamus of newborn Leprdb/Leprdb mice (Czupryn et al., 2011). Remarkably, not only did these hypothalamic grafts differentiate into electrophysiologically and histochemically determined neuronal cell types, but these donor cells also integrated into functional neural circuits and micro-implanted cells received functional synaptic connections. Physiologically, Leprdb/Leprdb mice that received hypothalamic grafts displayed a 20% decrease of body weight, demonstrating the functional relevance of these observations (Czupryn et al., 2011).
In addition, recent evidence has suggested that constitutive hypothalamic neurogenesis may be enhanced by external cues. Low rates of neurogenesis are observed in the mature hypothalamus under basal conditions (Kokoeva et al., 2005; Kokoeva et al., 2007) and median eminence tanycytes appear to be a possible source of these newborn neurons (Lee et al., 2012). Injections of ciliary neurotrophic factor (CNTF) into adult obese mice induced marked neurogenesis in the hypothalamus (Kokoeva et al., 2005). This CNTF-induced neurogenesis has consequences on the regulation of energy balance, and it participates in the weight loss effects of CNTF in obese mice (Kokoeva et al., 2005). Moreover, in mature mice, leptin appears to increase the formation of new neurons in the hippocampus, another well-known neurogenic structure (Zhao et al., 2008). Peripheral administration of leptin to adult mice results in increased cell proliferation in the dentate gyrus, and many of these newborn cells do become neurons that functionally integrate into the hippocampal circuitry (Garza et al., 2008). Of note, hypothalamic neurogenesis can also be induced in neurodegenerative conditions, such as in mutant mice, in which AgRP neurons undergo progressive neurodegeneration due to the deletion of mitochondrial transcription factor A (Pierce and Xu, 2010). These studies indicate that neurogenesis may serve as a compensatory mechanism that contributes to the plastic control of energy balance in response to environmental insults.
8. Conclusion
Data from a variety of studies indicate that we cannot take what we know about the action of metabolic hormones in the brain and extrapolate it to the perinatal brain. It is now clear that metabolic hormones can produce pleiotropic effects on functions that are well outside those that they have traditionally been thought to regulate. Metabolic hormones are particularly important in this regard and appear to be crucial in determining a variety of developmental processes in the hypothalamus, including neurogenesis, axon growth, and synaptic plasticity. The perinatal period represents the period of maximal sensitivity to the structural actions of metabolic hormones. Nevertheless, there is also clear evidence that the mature brain remains sensitive to the neuroplastic actions of metabolic hormones. A better understanding of the mechanisms that mediate the organizational effects of leptin, ghrelin, and insulin is clearly needed. Similarly, it is important that we increase our knowledge about the periods of maximal vulnerability of the brain to changes in metabolic hormone levels in order to open new avenues for understanding perinatally acquired predispositions to adult metabolic diseases.
Highlights.
Metabolic hormones can cause permanent structural changes in the neonatal hypothalamus
Leptin, ghrelin, and insulin can determine patterns of neurogenesis and axon-growth
The mature brain remains sensitive to the neuroplastic action of metabolic hormones
Acknowledgments
I would like to thank members of my laboratory for their active participation in the studies discussed in this review. Research in my laboratory has been funded by grants from the National Institutes of Health (Grant DK84142), the “Fondation pour la Recherche Médicale”, the EU FP7 integrated project (grant agreement n° 266408, “Full4Health”), and the “Agence Nationale de la Recherche” (Grants ANR-08-JCJC-0055-01 and ANR-11-BSV1-021-02).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott MA, Wells DG, Fallon JR. The Insulin Receptor Tyrosine Kinase Substrate p58/53 and the Insulin Receptor Are Components of CNS Synapses. The Journal of Neuroscience. 1999;19:7300–7308. doi: 10.1523/JNEUROSCI.19-17-07300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima R, Bjorbaek C, Osei S, Flier J. Regulation of neuronal and glial proteins by leptin: implications for brain development. Endocrinology. 1999;140:2755–2762. doi: 10.1210/endo.140.6.6774. [DOI] [PubMed] [Google Scholar]
- Ahima R, Prabakaran D, Flier J. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. 1998;101:1020–1027. doi: 10.1172/JCI1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J, Bayer SA. The development of the rat hypothalamus. Adv Anat Embryol Cell Biol. 1986;100 [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MGJ. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Baumann M, Deborde S, Illsley N. Placental glucose transfer and fetal growth. Endocr. 2002;19:13–22. doi: 10.1385/ENDO:19:1:13. [DOI] [PubMed] [Google Scholar]
- Bereiter D, Jeanrenaud B. Altered neuroanatomical organization in the central nervous system of the genetically obese (ob/ob) mouse. Brain Res. 1979;165:249–260. doi: 10.1016/0006-8993(79)90557-2. [DOI] [PubMed] [Google Scholar]
- Bereiter D, Jeanrenaud B. Altered dendritic orientation of hypothalamic neurons from genetically obese (ob/ob) mice. Brain Res. 1980;202:201–206. [PubMed] [Google Scholar]
- Bouret S, Simerly RB. Development of Leptin-Sensitive Circuits. J Neuroendocrinology. 2007;19:575–582. doi: 10.1111/j.1365-2826.2007.01563.x. [DOI] [PubMed] [Google Scholar]
- Bouret SG. Development of Hypothalamic Neural Networks Controlling Appetite. In: Elmadfa, editor. Forum Nutr Karger, Basel. 2010. pp. 84–93. [DOI] [PubMed] [Google Scholar]
- Bouret SG, Bates SH, Chen S, Myers MG, Simerly RB. Distinct Roles for Specific Leptin Receptor Signals in the Development of Hypothalamic Feeding Circuits. The Journal of Neuroscience. 2012;32:1244–1252. doi: 10.1523/JNEUROSCI.2277-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Formation of Projection Pathways from the Arcuate Nucleus of the Hypothalamus to Hypothalamic Regions Implicated in the Neural Control of Feeding Behavior in Mice. J Neurosci. 2004a;24:2797–2805. doi: 10.1523/JNEUROSCI.5369-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Trophic Action of Leptin on Hypothalamic Neurons That Regulate Feeding. Science. 2004b;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- Carlo AS, M W, Williams LM. Early developmental expression of leptin receptor gene and [125I]leptin binding in the rat forebrain. J Chem Neuroanat. 2007;33:155–163. doi: 10.1016/j.jchemneu.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Caron E, Sachot C, Prevot V, Bouret SG. Distribution of Leptin-Sensitive Cells in the Postnatal and Adult Mouse Brain. Journal of Comparative Neurology. 2010;518:459–476. doi: 10.1002/cne.22219. [DOI] [PubMed] [Google Scholar]
- Carvalheira JBC, Torsoni MA, Ueno M, Amaral ME, Araújo EP, Velloso LA, Gontijo JAR, Saad MJA. Cross-Talk between the Insulin and Leptin Signaling Systems in Rat Hypothalamus. Obesity. 2005:48–57. doi: 10.1038/oby.2005.7. [DOI] [PubMed] [Google Scholar]
- Chanoine JP, Wong ACK. Ghrelin Gene Expression Is Markedly Higher in Fetal Pancreas Compared with Fetal Stomach: Effect of Maternal Fasting. Endocrinology. 2004;145:3813–3820. doi: 10.1210/en.2004-0053. [DOI] [PubMed] [Google Scholar]
- Cheng CM, Mervis RF, Niu SL, Salem N, Witters LA, Tseng V, Reinhardt R, Bondy CA. Insulin-like growth factor 1 is essential for normal dendritic growth. Journal of Neuroscience Research. 2003;73:1–9. doi: 10.1002/jnr.10634. [DOI] [PubMed] [Google Scholar]
- Choi J, Ko J, Racz B, Burette A, Lee JR, Kim S, Na M, Lee HW, Kim K, Weinberg RJ, Kim E. Regulation of Dendritic Spine Morphogenesis by Insulin Receptor Substrate 53, a Downstream Effector of Rac1 and Cdc42 Small GTPases. The Journal of Neuroscience. 2005;25:869–879. doi: 10.1523/JNEUROSCI.3212-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell EC, Cripps RL, Duncan JS, Barrett P, Mercer JG, Herwig A, Ozanne SE. Developmental changes in hypothalamic leptin receptor: relationship with the postnatal leptin surge and energy balance neuropeptides in the postnatal rat. Am J Physiol Regul Integr Comp Physiol. 2009;296:R631–639. doi: 10.1152/ajpregu.90690.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupe B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG. Loss of Autophagy in Pro-opiomelanocortin Neurons Perturbs Axon Growth and Causes Metabolic Dysregulation. Cell Metabolism. 2012;15:247–255. doi: 10.1016/j.cmet.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czupryn A, Zhou YD, Chen X, McNay D, Anderson MP, Flier JS, Macklis JD. Transplanted Hypothalamic Neurons Restore Leptin Signaling and Ameliorate Obesity in db/db Mice. Science. 2011;334:1133–1137. doi: 10.1126/science.1209870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku S, Okamura Y, Kawano H, Tsuruo Y, Maegawa M, Shibasaki T. Immunohistochemical study on the development of CRF-containing neurons in the hypothalamus of the rat. Cell Tissue Res. 1984;238:539–544. doi: 10.1007/BF00219870. [DOI] [PubMed] [Google Scholar]
- de Vries GJ, Simerly RB. Anatomy, Development, and Function of Sexually Dimorphic Neural Circuits in the Mammalian Brain. In: Pfaff D, Arnold A, Etgen A, Farhbach S, Rubin R, editors. Hormones, Brain and Behavior. Academic Press; San Diego: 2002. [Google Scholar]
- Deltour L, Leduque P, Blume N, Madsen O, Dubois P, Jami J, Bucchini D. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proceedings of the National Academy of Sciences. 1993;90:527–531. doi: 10.1073/pnas.90.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desoye G, Gauster M, Wadsack C. Placental transport in pregnancy pathologies. The American Journal of Clinical Nutrition. 2011 doi: 10.3945/ajcn.110.000851. [DOI] [PubMed] [Google Scholar]
- Devaskar S, Ollesch C, Rajakumar R, Rajakumar P. Developmental changes in ob gene expression and circulating leptin peptide concentration. Biochemical and Biophysical Research Communications. 1997;238:44–47. doi: 10.1006/bbrc.1997.7237. [DOI] [PubMed] [Google Scholar]
- Fahrenkrog S, Harder T, Stolaczyk E, Melchior K, Franke K, Dudenhausen JW, Plagemann A. Cross-Fostering to Diabetic Rat Dams Affects Early Development of Mediobasal Hypothalamic Nuclei Regulating Food Intake, Body Weight, and Metabolism. The Journal of Nutrition. 2004;134:648–654. doi: 10.1093/jn/134.3.648. [DOI] [PubMed] [Google Scholar]
- Franke K, Harder T, Aerts L, Melchior K, Fahrenkrog S, Rodekamp E, Ziska T, Van Assche FA, Dudenhausen JW, Plagemann A. ‘Programming’ of orexigenic and anorexigenic hypothalamic neurons in offspring of treated and untreated diabetic mother rats. Brain Res. 2005;1031:276–283. doi: 10.1016/j.brainres.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Garza J, Guo M, Zhang W, Lu XY. Leptin Increases Adult Hippocampal Neurogenesis in Vivo and in Vitro. J Biol Chem. 2008;283:18238–18247. doi: 10.1074/jbc.M800053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind S, Kozma R, Monfries C, Lim L, Ahmed S. Cdc42hs Facilitates Cytoskeletal Reorganization and Neurite Outgrowth by Localizing the 58-Kd Insulin Receptor Substrate to Filamentous Actin. The Journal of Cell Biology. 2001;152:579–594. doi: 10.1083/jcb.152.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove KL, Allen S, Grayson BE, Smith MS. Postnatal development of the hypothalamic neuropeptide Y system. Neuroscience. 2003;116:393–406. doi: 10.1016/s0306-4522(02)00668-1. [DOI] [PubMed] [Google Scholar]
- Havrankova J, Roth J, Brownstein MJ. Concentrations of Insulin and of Insulin Receptors in the Brain are Independent of Peripheral Insulin Levels. J Clin Invest. 1979;64:636–642. doi: 10.1172/JCI109504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida T, Nakahara K, Mondal MS, Date Y, Nakazato M, Kojima M, Kangawa K, Murakami N. Ghrelin in neonatal rats: distribution in stomach and its possible role. J Endocrinol. 2002;173:239–245. doi: 10.1677/joe.0.1730239. [DOI] [PubMed] [Google Scholar]
- Hileman SM, Tornoe J, Flier JS, Bjorbak C. Transcellular Transport of Leptin by the Short Leptin Receptor Isoform ObRa in Madin-Darby Canine Kidney Cells. Endocrinology. 2000;141:1955–1961. doi: 10.1210/endo.141.6.7450. [DOI] [PubMed] [Google Scholar]
- Hoggard N, Hunter L, Duncan JS, Williams LM, Trayhurn P, Mercer JG. Leptin and leptin receptor mRNA and protein expression in the murine fetus and placenta. PNAS. 1997;94:11073–11078. doi: 10.1073/pnas.94.20.11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Nakahara K, Kangawa K, Murakami N. Transitional change in rat fetal cell proliferation in response to ghrelin and des-acyl ghrelin during the last stage of pregnancy. Biochemical and Biophysical Research Communications. 2010;393:455–460. doi: 10.1016/j.bbrc.2010.02.022. [DOI] [PubMed] [Google Scholar]
- Ishii Y, Bouret SG. Embryonic Birthdate of Hypothalamic Leptin-Activated Neurons in Mice. Endocrinology. 2012;153:3657–3667. doi: 10.1210/en.2012-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A, Olster D, States B. Maternal insulin manipulations in rats organize body weight and noradrenergic innervation of the hypothalamus in gonadally intact male offspring. Developmental Brain Research. 1996;97:16–21. doi: 10.1016/s0165-3806(96)00128-9. [DOI] [PubMed] [Google Scholar]
- Jones AP, Pothos EN, Rada P, Olster DH, Hoebel BG. Maternal hormonal manipulations in rats cause obesity and increase medial hypothalamic norepinephrine release in male offspring. Developmental Brain Research. 1995;88:127–131. doi: 10.1016/0165-3806(95)00078-r. [DOI] [PubMed] [Google Scholar]
- Kar S, Chabot JG, Quirion R. Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol. 1993;333:375–397. doi: 10.1002/cne.903330306. [DOI] [PubMed] [Google Scholar]
- Kastin AJ, Pan W, Maness LM, Koletsky RJ, Ernsberger P. Decreased transport of leptin across the blood-brain barrier in rats lacking the short form of the leptin receptor*. Peptides. 1999;20:1449–1453. doi: 10.1016/s0196-9781(99)00156-4. [DOI] [PubMed] [Google Scholar]
- Kokoeva MV, Yin H, Flier JS. Neurogenesis in the Hypothalamus of Adult Mice: Potential Role in Energy Balance. Science. 2005;310:679–683. doi: 10.1126/science.1115360. [DOI] [PubMed] [Google Scholar]
- Kokoeva MV, Yin H, Flier JS. Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. The Journal of Comparative Neurology. 2007;505:209–220. doi: 10.1002/cne.21492. [DOI] [PubMed] [Google Scholar]
- Lee DA, Bedont JL, Pak T, Wang H, Song J, Miranda-Angulo A, Takiar V, Charubhumi V, Balordi F, Takebayashi H, Aja S, Ford E, Fishell G, Blackshaw S. Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nat Neurosci. 2012;15:700–702. doi: 10.1038/nn.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis GW, Greenwald-Yarnell M, Phillips R, Coolen LM, Lehman MN, Myers MG. Molecular Mapping of the Neural Pathways Linking Leptin to the Neuroendocrine Reproductive Axis. Endocrinology. 2011;152:2302–2310. doi: 10.1210/en.2011-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik SA, Marino G, BenYounes A, Shen S, Harper F, Maiuri MC, Kroemer G. Neuroendocrine regulation of autophagy by leptin. Cell Cycle. 2011;10:2917–2923. doi: 10.4161/cc.10.17.17067. [DOI] [PubMed] [Google Scholar]
- Markakis EA. Development of the neuroendocrine hypothalamus. Front Neuroendocrinol. 2002;23:257–291. doi: 10.1016/s0091-3022(02)00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Gronert MS, Ozanne SE. Programming of appetite and type 2 diabetes. Early Human Development. 2005;81:981–988. doi: 10.1016/j.earlhumdev.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Arai Y. Developmental changes in synaptic formation in the hypothalamic arcuate nucleus of female rats. Cell Tissue Res. 1976;14:143–156. doi: 10.1007/BF00214204. [DOI] [PubMed] [Google Scholar]
- McCarthy MM. Estradiol and the Developing Brain. Physiol Rev. 2008;88:91–134. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellana KM, Parker KL, Tobet SA. Development of the ventromedial nucleus of the hypothalamus. Frontiers in Neuroendocrinology. 2006;27:193–209. doi: 10.1016/j.yfrne.2006.02.002. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Steroid hormones: effect on brain development and function. Horm Res. 1992;37:1–10. doi: 10.1159/000182393. [DOI] [PubMed] [Google Scholar]
- Melnick I, Pronchuck N, Cowley MA, Grove KL, Colmers WF. Developmental switch in neuropeptide Y and melanocortin effects in the paraventricular nucleus of the hypothalamus. Neuron. 2007;56:1103–1115. doi: 10.1016/j.neuron.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Miki H, Takenawa T. WAVE2 serves a functional partner of IRSp53 by regulating its interaction with Rac. Biochemical and Biophysical Research Communications. 2002;293:93–99. doi: 10.1016/S0006-291X(02)00218-8. [DOI] [PubMed] [Google Scholar]
- Mitchell V, Bouret S, B J, Schilling A, Perret M, Kordon C, Epelbaum J. Comparative distribution of mRNA encoding the growth hormone secretagogue-receptor (GHS-R) in Microcebus murinus (Primate, lemurian) and rat forebrain and pituitary. J Comp Neurol. 2001;429:469–489. doi: 10.1002/1096-9861(20010115)429:3<469::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Nakahara K, Nakagawa M, Baba Y, Sato M, Toshinai K, Date Y, Nakazato M, Kojima M, Miyazato M, Kaiya H, Hosoda H, Kangawa K, Murakami N. Maternal Ghrelin Plays an Important Role in Rat Fetal Development during Pregnancy. Endocrinology. 2006;147:1333–1342. doi: 10.1210/en.2005-0708. [DOI] [PubMed] [Google Scholar]
- Nilsson I, Johansen JE, Schalling M, Hokfelt T, Fetissov SO. Maturation of the hypothalamic arcuate agouti-related protein system during postnatal development in the mouse. Developmental Brain Research. 2005;155:147–154. doi: 10.1016/j.devbrainres.2005.01.009. [DOI] [PubMed] [Google Scholar]
- O'Malley D, MacDonald N, Mizielinska S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Molecular and Cellular Neuroscience. 2007;35:559–572. doi: 10.1016/j.mcn.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paderson J. Diabetes mellitus and pregnancy: present status of the hyperglycaemia--hyperinsulinism theory and the weight of the newborn baby. Postgrad Med J. 1971;(Suppl):66–67. [PubMed] [Google Scholar]
- Paderson J, Osler M. Hyperglycemia as the cause of characteristic features of the foetus and newborn of diabetic mothers. Dan Med Bull. 1961:78–83. [PubMed] [Google Scholar]
- Padilla SL, Carmody JS, Zeltser LM. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nature Medicine. 2010;16:403–405. doi: 10.1038/nm.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Hsuchou H, Hong T, Kastin A. Developmental changes of leptin receptors in cerebral microvessels: unexpected relation to leptin transport. Endocrinology. 2008;149:877–885. doi: 10.1210/en.2007-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao H, Hosoda H, Kangawa K, Murata T, Narita K, Higuchi T. Ghrelin Stimulates Milk Intake by Affecting Adult Type Feeding Behaviour in Postnatal Rats. Journal of Neuroendocrinology. 2008;20:330–334. doi: 10.1111/j.1365-2826.2007.01644.x. [DOI] [PubMed] [Google Scholar]
- Pierce AA, Xu AW. De Novo Neurogenesis in Adult Hypothalamus as a Compensatory Mechanism to Regulate Energy Balance. J Neurosci. 2010;30:723–730. doi: 10.1523/JNEUROSCI.2479-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid Rewiring of Arcuate Nucleus Feeding Circuits by Leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Heidrich I, Götz F, Rohde W, D G. Lifelong enhanced diabetes susceptibility and obesity after temporary intrahypothalamic hyperinsulinism during brain organization. Exp Clin Endocrinol. 1992;99:91–95. doi: 10.1055/s-0029-1211143. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Janert U, Rake A, Rittel F, Rohde W, Dorner G. Malformations of Hypothalamic Nuclei in Hyperinsulinemic Offspring of Rats with Gestational Diabetes. Developmental Neuroscience. 1999a;21:58–67. doi: 10.1159/000017367. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Melchior K, Rake A, Rohde W, Dörner G. Elevation of hypothalamic neuropeptide Y-neurons in adult offspring of diabetic mother rats. Neuroreport. 1998;10:3211–3216. doi: 10.1097/00001756-199910190-00016. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Rake A, Janert U, Melchior K, Rohde W, Dorner G. Morphological alterations of hypothalamic nuclei due to intrahypothalamic hyperinsulinism in newborn rats. International Journal of Developmental Neuroscience. 1999b;17:37–44. doi: 10.1016/s0736-5748(98)00064-1. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Rake A, Melchior K, Rohde W, Dorner Gn. Hypothalamic Nuclei Are Malformed in Weanling Offspring of Low Protein Malnourished Rat Dams. The Journal of Nutrition. 2000;130:2582–2589. doi: 10.1093/jn/130.10.2582. [DOI] [PubMed] [Google Scholar]
- Plum L, Lin HV, Aizawa KS, Liu Y, Accili D. InsR/FoxO1 Signaling Curtails Hypothalamic POMC Neuron Number. PLoS ONE. 2012;7:e31487. doi: 10.1371/journal.pone.0031487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Nakahara K, Goto S, Kaiya H, Miyazato M, Date Y, Nakazato M, Kangawa K, Murakami N. Effects of ghrelin and des-acyl ghrelin on neurogenesis of the rat fetal spinal cord. Biochemical and Biophysical Research Communications. 2006;350:598–603. doi: 10.1016/j.bbrc.2006.09.088. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, OMalley D, Irving AJ, Ashford ML, Harvey J. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J Physiol. 2002;545:933–944. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Nakamura T. Time of neuron origin in mouse hypothalamic nuclei. Experimental Neurology. 1973;41:163–173. doi: 10.1016/0014-4886(73)90187-8. [DOI] [PubMed] [Google Scholar]
- Simerly RB. Wired for reproduction: organization and development of sexually dimorphic circuits in the mammalian forebrain. Annual Review of Neuroscience. 2002;25:507–536. doi: 10.1146/annurev.neuro.25.112701.142745. [DOI] [PubMed] [Google Scholar]
- Spaventi R, Antica M, Pavelic K. Insulin and insulin-like growth factor I (IGF I) in early mouse embryogenesis. Development. 1990;108:491–495. doi: 10.1242/dev.108.3.491. [DOI] [PubMed] [Google Scholar]
- Steculorum SM, Bouret SG. Developmental effects of ghrelin. Peptides. 2011a;32:2362–2366. doi: 10.1016/j.peptides.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steculorum SM, Bouret SG. Maternal Diabetes Compromises the Organization of Hypothalamic Feeding Circuits and Impairs Leptin Sensitivity in Offspring. Endocrinology. 2011b;152:4171–4179. doi: 10.1210/en.2011-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steppan C, Swick A. A role for leptin in brain development. Biochemical and Biophysical Research Communications. 1999;256:600–602. doi: 10.1006/bbrc.1999.0382. [DOI] [PubMed] [Google Scholar]
- Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4679–4684. doi: 10.1073/pnas.0305930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toran-Allerand CD, Bentham W, Miranda RC, Anderson JP. Insulin influences astroglial morphology and glial fibrillary acidic protein (GFAP) expression in organotypic cultures. Brain Res. 1991;558:296–304. doi: 10.1016/0006-8993(91)90781-p. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Ellis L, Pfenninger KH. Estrogen and insulin synergism in neurite growth enhancement in vitro: mediation of steroid effects by interactions with growth factors? Dev Brain Res. 1988;41:87–100. doi: 10.1016/0165-3806(88)90172-1. [DOI] [PubMed] [Google Scholar]
- Torsello A, Scibona B, Leo G, Bresciani E, Avallone R, Bulgarelli I, Luoni M, Zoli M, Rindi G, Cocchi D, Locatelli V. Ontogeny and tissue-specific regulation of ghrelin mRNA expression suggest that ghrelin is primarily involved in the control of extraendocrine functions in the rat. Neuroendocrinology. 2003;77:91–99. doi: 10.1159/000068653. [DOI] [PubMed] [Google Scholar]
- Udagawa J, Hashimoto R, Hioki K, Otani H. The role of leptin in the development of the cortical neuron in mouse embryos. Brain Research. 2006a;1120:74–82. doi: 10.1016/j.brainres.2006.08.116. [DOI] [PubMed] [Google Scholar]
- Udagawa J, Hashimoto R, Suzuki H, Hatta T, Sotomaru Y, Hioki K, Kagohashi Y, Nomura T, Minami Y, Otani H. The Role of Leptin in the Development of the Cerebral Cortex in Mouse Embryos. Endocrinology. 2006b;147:647–658. doi: 10.1210/en.2005-0791. [DOI] [PubMed] [Google Scholar]
- Udagawa J, Hatta T, Naora H, Otani H. Expression of the long form of leptin receptor (Ob-Rb) mRNA in the brain of mouse embryos and newborn mice. Brain Research. 2000;868:251–258. doi: 10.1016/s0006-8993(00)02334-9. [DOI] [PubMed] [Google Scholar]
- Valerio A, Ghisi V, Dossena M, Tonello C, Giordano A, Frontini A, Ferrario M, Pizzi M, Spano P, Carruba MO, Nisoli E. Leptin Increases Axonal Growth Cone Size in Developing Mouse Cortical Neurons by Convergent Signals Inactivating Glycogen Synthase Kinase-3beta. J Biol Chem. 2006;281:12950–12958. doi: 10.1074/jbc.M508691200. [DOI] [PubMed] [Google Scholar]
- Wierup N, Svensson H, Mulder H, Sundler F. The ghrelin cell: a novel developmentally regulated islet cell in the human pancreas. Regulatory Peptides. 2002;107:63–69. doi: 10.1016/s0167-0115(02)00067-8. [DOI] [PubMed] [Google Scholar]
- Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nature Neuroscience. 2012;15:1350–1355. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Hu Y, Lin TR, Fan Y, Mulholland MW. Stimulation of neurogenesis in rat nucleus of the solitary tract by ghrelin. Peptides. 2005;26:2280–2288. doi: 10.1016/j.peptides.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Zhang W, Lin TR, Hu Y, Fan Y, Zhao L, Stuenkel EL, Mulholland MW. Ghrelin stimulates neurogenesis in the dorsal motor nucleus of the vagus. The Journal of Physiology. 2004;559:729–737. doi: 10.1113/jphysiol.2004.064121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Zigman JM, ones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. The Journal of Comparative Neurology. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]