

Summary
Calcium entry through CaV2.2 calcium channels clustered at the active zone (AZ) of the presynaptic nerve terminal gates synaptic vesicle (SV) fusion and the discharge of neurotransmitters, but the mechanism of channel scaffolding remains poorly understood. Recent studies have implicated the binding of a PDZ ligand domain (PDZ-LD) at the tip of the channel C terminal to a partner PDZ domain on RIM1/2, a synaptic vesicle-associated protein. To explore CaV2.2 scaffolding, we created intracellular region fusion proteins and used these to test for binding by ‘fishing’ for native CaV2.2 channels from cell lysates. Fusion proteins mimicking the distal half of the channel C terminal (C3strep) reliably captured CaV2.2 from whole brain crude membrane or purified synaptosome membrane lysates, whereas channel I–II loop or the distal half of the II–III loop proteins were negative. This capture could be replicated in a non-synaptic environment using CaV2.2 expressed in a cell line. The distal tip PDZ-LD, DDWC-COOH, was confirmed as the critical binding site by block of pull-down with mimetic peptides. Pull-down experiments using brain crude membrane lysates confirmed that RIM1/2 can bind to the DDWC PDZ-LD. However, robust CaV2.2 capture was observed from synaptosome membrane or in the cell line expression system with little or no RIM1/2 co-capture. Thus, we conclude that CaV2.2 channels can scaffold to each other via an interaction that involves the PDZ-LD by an inter-channel linkage bridged by an unknown protein.
Key words: CaV2.2, PDZ, Active zone, Calcium channel, Presynaptic, Scaffold
Introduction
Action potentials trigger synaptic vesicle (SV) fusion at the active zone of presynaptic terminals by gating the entry of Ca2+ through voltage gated calcium channels (CaVs) which binds to SV-associated calcium sensors (Llinás et al., 1981; Katz, 1969). Structural, functional and biochemical studies all support the conclusion that these CaVs are clustered within the active zone (AZ) presumably by interconnecting protein links. Further, it is now becoming generally accepted (Stanley, 1997; Mulligan et al., 2001; Eggermann et al., 2012) that the CaVs are intimately associated with the SVs to the extent that a single-channel calcium ion domain can gate a closely associated, attached SV (Stanley, 1993). These findings are consistent with a two-armed-scaffold model, an anchor maintaining the channels within the AZ, and a tether to attach the SVs to local CaV channels (Wong and Stanley, 2010). A release site sub-membrane structure that may correspond to the above structural elements has been imaged by EM tomography (Harlow et al., 2001).
The clustering of CaVs at release sites has been demonstrated by morphology (Pumplin et al., 1981; Robitaille et al., 1990; Haydon et al., 1994; Mirotznik et al., 2000), Ca2+ entry (Llinás et al., 1992; Smith et al., 1993), and direct cell-attached patch recording from the transmitter release face (Stanley, 1991; Sheng et al., 2012). Within this cluster the channels can be distributed in orderly arrays (Dreyer et al., 1973; Heuser et al., 1974; Pumplin et al., 1981) or looser short strings (Haydon et al., 1994) with an ∼15 nm inter-channel interval (Haydon et al., 1994; Stanley et al., 2003). CaV2.2 channels can exist in two main forms with either a short or long-splice C terminal (Maximov et al., 1999; Maximov and Bezprozvanny, 2002) and the latter is targeted to the release sites (Maximov and Bezprozvanny, 2002; Khanna et al., 2006b; Gardezi et al., 2010). This extended C terminal variant is of particular interest with respect to presynaptic scaffolding as it terminates in a PDZ ligand domain (PDZ-LD), E/DxWC/S-COOH (Maximov et al., 1999). Recent reports have suggested that channel scaffolding at the AZ involves binding of the PDZ-LD to a corresponding PDZ domain on RIM1/2 (Han et al., 2011; Kaeser et al., 2011).
In this study we reasoned that if CaV2.2s are maintained at the TRS by an interlinking scaffold it should be possible to detect inter channel binding by fishing for CaVs using fusion proteins of its intracellular regions. We therefore developed a direct in vitro biochemical assay for CaV2.2–CaV2.2 interaction. We generated CaV2.2 C terminal fusion protein constructs using a bacterial expression system and tested for intact channel capture from chick brain crude membrane, purified nerve terminal (synaptosome) membrane lysates, or cell line lysates after expression of CaV2.2. We also analyzed the role of CaV2.2 C terminal and its binding domains in channel–channel attachment and explored the involvement of bridging proteins.
Results
CaV2.2 cytoplasmic region fusion proteins
We generated four chick CaV2.2 bacterial expression constructs coding the I–II (I–II; GST and FLAG tagged), the distal half of the II–III loop (II–IIIdist, GST and FLAG tagged), and the distal half of the C-terminus (C3strep; Strep tagged) regions (Fig. 1). These fusion proteins were expressed successfully and their molecular mass (Mr) correspond to the predicted size by WB (Fig. 2A).
Fig. 1. Fusion protein amino acid sequences.

Chick CaV2.2 I–II loop (I–II; (A)) and II–III loop distal half (II–IIIdistal; (B)) fusion protein amino acid sequences aligned with the corresponding region of the native long splice-variant chick CaV2.2 (cdB1; accession number AAD51815) used for cloning. Both constructs have N terminal GST (not shown) and C terminal FLAG (grey highlight) tags. (C) Chick CaV2.2 distal C-terminus fusion protein (C3strep) amino acid sequence aligned with long splice CaV2.2 variant (cdB1, as above).
Fig. 2. Pull down of CaV2.2 by channel C terminal fusion protein.
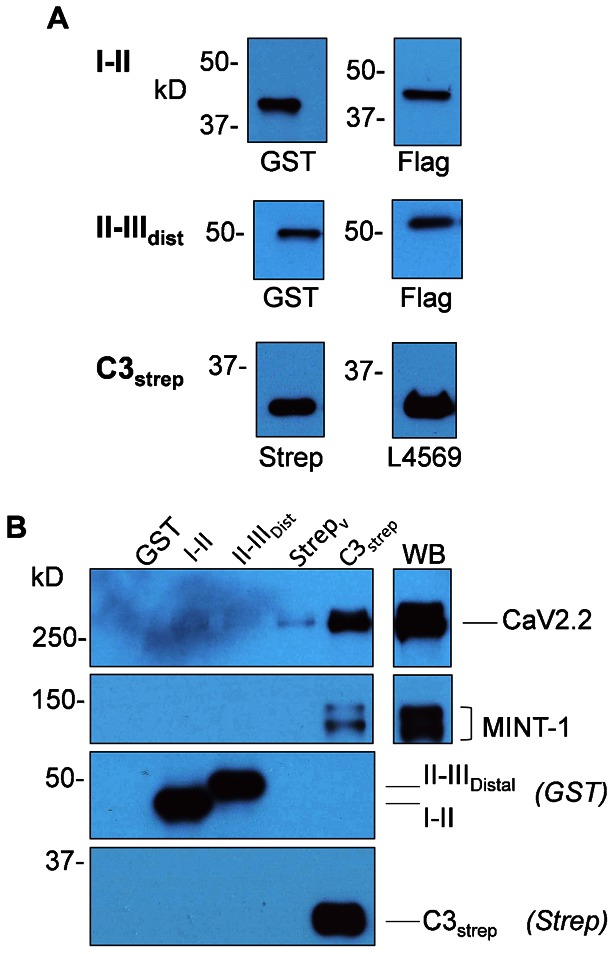
(A) I–II (as labeled), from bacterial protein lysate and purified protein II–IIIdistal were identified with anti-GST or anti-FLAG antibodies. Purified protein C3strep was identified with antibodies against the strep tag (left) and distal C-terminus (L4569; right). (B) Chick brain crude membrane lysate was incubated with I–II, II–IIIdist or Cstrep fusion proteins immobilized on the respective precipitation beads and captured proteins analyzed by WB. GST and strep vector were used as controls. The blots were probed for MINT-1 and CaV2.2. MINT-1 was only captured by C3strep. Negative or trace CaV2.2 capture was observed with I–II, II–IIIdistal or either of the two GST or strep vector (Strepv) controls, whereas a robust band was observed with C3strep. Fusion proteins were identified with anti-GST and anti-Strep respectively (bottom panels). In all blots 10% of lysate used for pull down assays was loaded in the WB lane.
Pull down assays
Fusion proteins immobilized on beads were incubated overnight with chick brain crude membrane lysate (see Materials and Methods). After washing, captured proteins were assayed by WB. Successful capture of MINT-1, which is known to bind the distal tip of the long-splice CaV2.2 C terminal (Maximov et al., 1999; see also below), confirmed that the C3strep fusion protein was both full length and functional (Fig. 2B).
We probed for pull down of chick CaV2.2 with Ab571, an antibody directed against the proximal II–III loop of the channel (Li et al., 2004). Pull down with I–II and II–IIIdist regions gave no, or very faint bands corresponding to CaV2.2 Mr (N = 4 and 3, respectively; Fig. 2B). However, protein pull down with C3strep fusion protein resulted in strong bands at the corresponding Mr (N = 20; Fig. 2B), indicating that this region of the channel can scaffold to CaV2.2 in brain crude membrane lysate.
The CaV2.2 binding region is in the distal sixth of the C-terminus
In order to further localize the CaV2.2-interacting region we generated two additional GST fusion proteins for the proximal and distal two thirds of the C3strep construct, and hence, with a one-third overlap region: C3prox and C3dist (Fig. 3A). These fusion proteins exhibited their appropriate Mr (Fig. 3B, left panel) and the latter was verified further by reactivity with L4569; an antibody directed against the CaV2.2 C terminal long-splice region (Khanna et al., 2006b) (Fig. 3B, right panel). These were used for GST pull down assays from purified synaptosome membrane lysates. C3dist, but not C3prox, pulled down MINT-1 from membrane lysate (Fig. 4, 2nd panel) and confirmed binding of MINT-1 to the PDZ-LD on the tip of the C terminal (Maximov et al., 1999).
Fig. 3. Fusion proteins subdividing the chick CaV2.2 C-terminus C3 region.
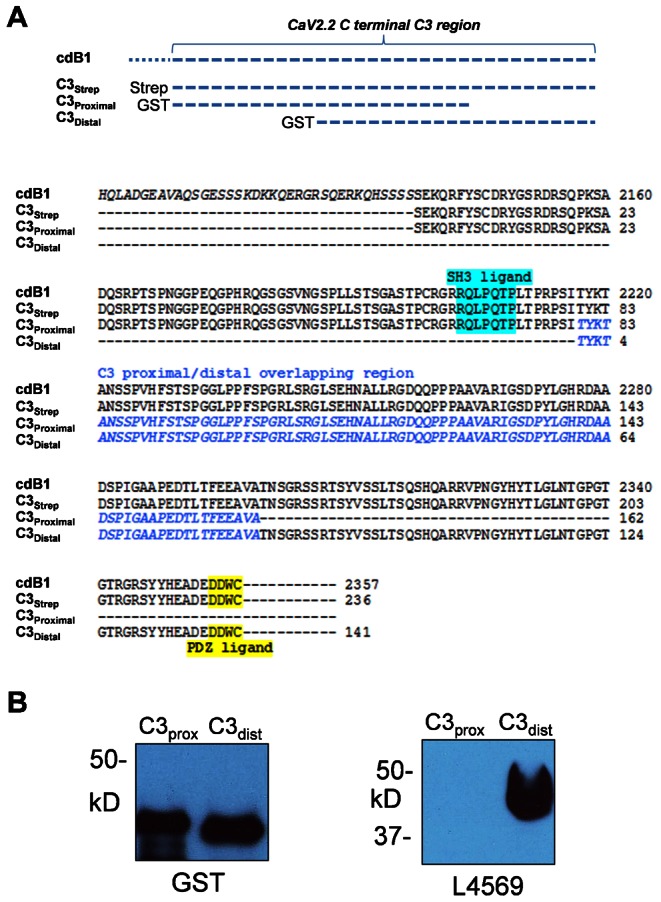
(A) Upper panel: diagram of the C3strep construct and two GST tagged constructs, C3prox and C3dist. C3prox and C3dist cover the first and last 2/3rds of the C3 region with a 1/3rd overlap. Amino acid sequences of all three constructs with that of the native channel are shown in the lower panel. Note that while C3strep contains both the SH3 ligand domain (blue) and PDZ-LD (yellow), C3prox includes only the former and C3dist only the latter. (B) C3 proximal (C3prox) and distal (C3dist) region purified fusion proteins are both identified with anti-GST while L4569, an antibody against the distal C-terminus, only detected C3dist.
Fig. 4. CaV2.2 is captured by the distal region of C terminal.
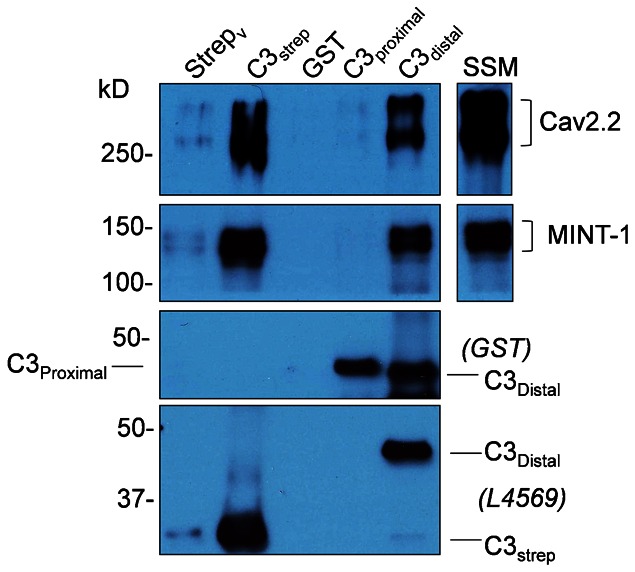
CaV2.2 capture from solubilized purified synaptosome membranes was compared with three C3 region fusion proteins using GST and strep vector as controls. Prominent CaV2.2 channel bands were observed with C3strep and C3dist, both of which include the PDZ-LD, but were negative with C3prox, which lacks that domain but includes the SH3 domain (top panel). A functional PDZ-LD on the C3strep and C3dist fusion proteins was confirmed by pull down of MINT-1 (middle panels). Fusion proteins are identified with their respective antibodies (bottom panels; the shorter C3dist fusion protein has a higher molecular weight due to the GST tag). The faint bands for MINT-1 and CaV2.2 observed in the Strepv lane are attributed to a trace contamination with the C3strep sample (see C3strep bands).
We compared the ability of C3prox and C3dist capture CaV2.2. C3prox exhibited a very weak or negligible pull down of CaV2.2 from solubilized synaptosome membrane (also used for all subsequent experiments) whereas that with C3dist was robust and similar to C3strep (N = 3; Fig. 4). Thus, the CaV2.2 binding region can also be localized to the distal sixth of the channel C terminal.
Role of the terminal PDZ ligand-domain in CaV2.2 capture
Previous studies have identified a highly conserved PDZ ligand motif, E/D-X-W-C-COOH at the tip of the C terminal and have demonstrated that this domain is responsible for capture of MINT-1 (Maximov et al., 1999). The amino acid sequence of this domain in the chick CaV2.2 is DDWC-COOH (herein DDWC; Fig. 3A). To test if the PDZ-ligand domain (PDZ-LD) plays a role in C-terminal pull down of CaV2.2 we synthesized competitive mimetic peptide blockers: DDWC by itself and also HEADEDDWC, which includes additional C terminal amino acids proximal to the PDZ-LD. Previous analysis of PDZ-LDs has shown that the identity of the terminal amino acid is crucial for its specificity (Tonikian et al., 2008). We therefore synthesized a very similar peptide but with the terminal amino acid mutated from C to A, DDWA, as a control. These peptides were added to chick synaptosome membrane lysate prior to C3strep pull down, as above. The mimetic DDWC and HEADEDDWC peptides effectively blocked PDZ binding, as evidenced by a marked reduction in MINT-1 pull down, whereas the control DDWA peptide had little effect (N = 4, Fig. 5A).
Fig. 5. CaV2.2 pull-down is reduced with distal C-terminus blocking peptides in synaptosome membrane lysate.
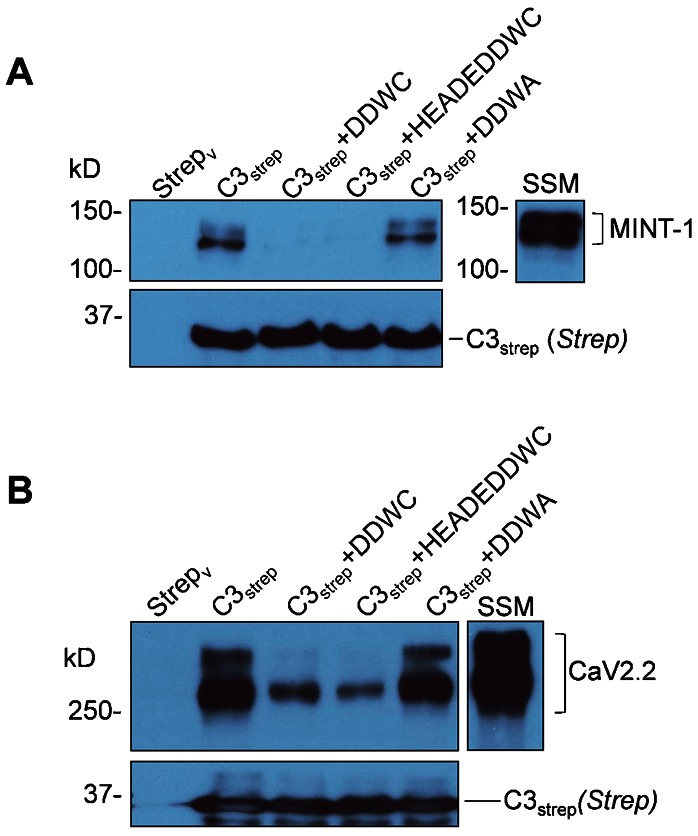
(A) The efficacy of the peptides to compete with PDZ-LD domain binding was tested using MINT-1 as the target protein. C3strep was used to pull down MINT-1 from chick synaptosome membrane lysate in the presence or absence of DDWC which mimics the PDZ-LD, HEADEDDWC which is also a PDZ-LD mimetic but with additional amino acids to increase specificity or control DDWA peptides. (B) DDWC and HEADEDDWC markedly inhibited CaV2.2 pull-down from chick brain synaptosome membrane lysates (SSM) lysate.
We next tested if the PDZ-LD mimetic peptides would affect CaV2.2 capture using the C3strep fusion protein. The usual robust channel pull down from synaptosome membrane lysate with this fusion protein was markedly inhibited in the presence of DDWC and HEADEDDWC (N = 4, Fig. 5B) where the latter didn't show a more obvious significant effect than the four amino acid PDZ-LD mimetic. The control DDWA had little detectable effect.
Can the C terminal capture expressed CaV2.2 channels?
To test if C terminal pull down of CaV2.2 was restricted to presynaptic tissue lysates, we expressed rat CaV2.2 channel in the mammalian tsA201 cell line (Fig. 6A) and repeated the above experiment using C3strep fusion protein. CaV2.2 recovery was probed with both Ab571 and anti-rat CaV2.2 (rCaV2.2, Table 1; Fig. 6A) as the former was raised against chick CaV2.2. Channel capture was observed in the cells with expressed CaV2.2 but not non-transfected controls (N = 5; Fig. 6B). This finding indicates that the C terminal can scaffold to CaV2.2 channels that did not originate from a presynaptic environment.
Fig. 6. Distal C-terminus pulls down CaV2.2 from channel transfected tsA201 cell lysates.
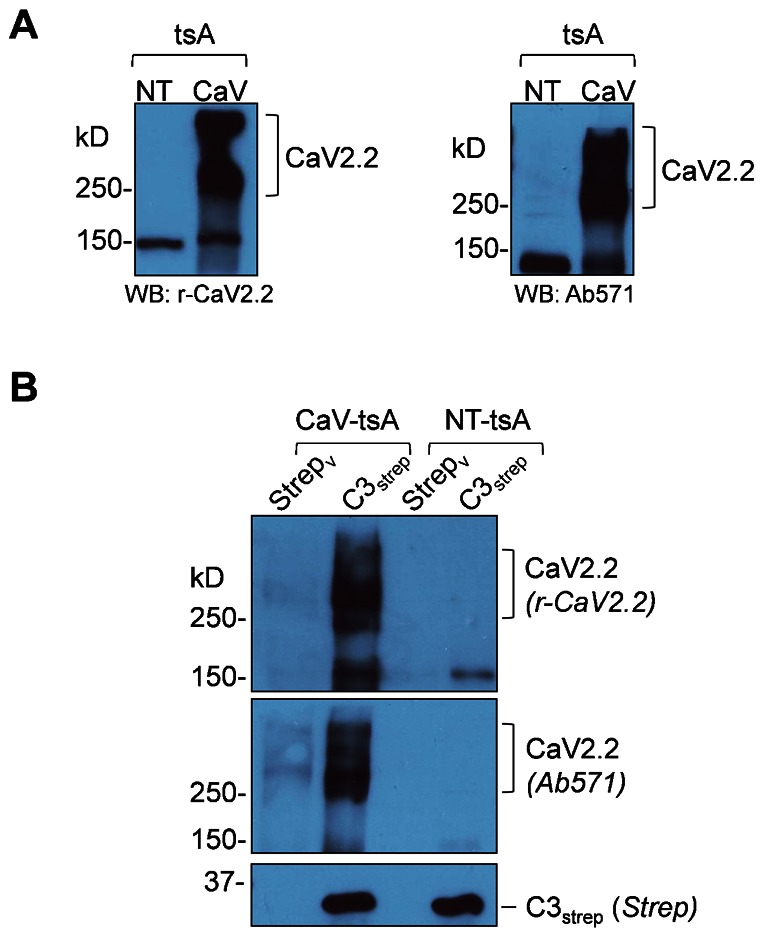
(A) Western blots of rat CaV2.2 in transfected (CaV lane) and non-transfected (NT lane) of tsA201 cell lysates with both r-CaV2.2 (left panel) and Ab571 (right panel). (B) C3strep pulls down the channel from CaV2.2 transfected (CaV-tsA) but not in non-transfected (NT) tsA201 cell lysates, as detected by both anti-CaV2.2 antibodies.
Table 1. Antibodies used in this study.

Does CaV2.2 scaffolding involve RIM1/2?
The PDZ binding protein RIM1/2 has been implicated as the scaffold protein that binds the CaV2.2 C terminal PDZ-LD. If this protein was responsible for channel–channel linkage, as assayed above, we would predict both that it should be co-captured by the C terminal fusion protein and also that CaV2.2 PD should fail in its absence. We used ‘polyRIM2’ (Synaptic Systems) because, despite its moniker, this antibody cross reacts with RIM1α and RIM2α proteins (Wong and Stanley, 2010) and hence, can be used to screen for both RIM proteins. As in our previous report, we term this antibody pRIM1,2 herein to avoid confusion. We did not attempt to differentiate between the closely related RIM1α and RIM2α variants and refer to the proteins collectively as RIM1/2.
RIM1/2 is readily identified in chick brain crude membrane lysate by Western blot and, as predicted above, was pulled down with C3strep and was blocked by DDWC peptide (N = 3, Fig. 7A). This result is consistent with previous reports suggesting that RIM1/2 binds to the C terminal via the DDWC PDZ ligand domain (Kaeser et al., 2011). However, this experiment does not differentiate whether RIM1/2 binds to the channel C terminal in conjunction, as it must if it is part of the scaffold, or independently of CaV2.2. Since the same channel binding domain is involved this question is difficult to address by biochemical means. Fortunately, a natural test was available because we found RIM1/2 pull down varied markedly between the three lysates tested: brain crude membrane, purified synaptosome membrane and the tsA201 expression cells. RIM1/2 was prominent in Western blots of brain crude membrane and purified synaptosome membrane (Fig. 7A,B) but whereas PD with C3strep captured a corresponding strong band of RIM1/2 in the former (Fig. 7A) only trace levels were detected from the synaptosome membrane lysate (N = 4, Fig. 7B). RIM1/2 protein bands were very faint or absent in tsA201 cell lysates, as assessed by Western blot (Fig. 7C, upper panel) and, not surprisingly RIM1/2 pull down with C3strep was also absent or negligible (N = 3, Fig. 7C, lower panel). In contrast, C3strep pull down of CaV2.2 was robust from all three lysates (Fig. 4, Fig. 5B, Fig. 6B and Fig. 7A,B). Thus, our results exhibit a stark dissociation between inter-CaV2.2 scaffolding, as indicated by C terminal pull down of channel, and RIM1/2 capture. We also tested tsA201 cell lysates for MINT-1 expression as this protein is captured by C3strep and blocked by PDZ-LD peptides (Fig. 5A). MINT-1 was virtually absent in tsA201 cells (Fig. 7D, upper panel) and failed to pull down with C3strep (Fig. 7D, bottom panels).
Fig. 7. PDZ-domain-dependent CaV2.2 pull down and the role of RIM1/2.
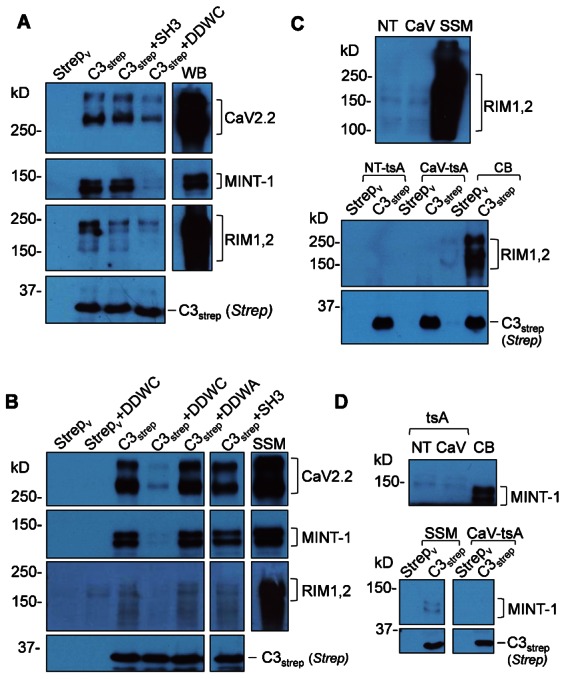
(A) CaV2.2 and MINT-1 are captured by C3strep pull down (Fig. 5) from chick brain crude membrane lysate by a PDZ domain-sensitive binding mechanism, as demonstrated by inhibition with DDWC. The figure also shows that channel and MINT-1 pull downs are not affected by a blocking peptide for the C terminal SH3 domain, consistent with the previous fusion protein analysis (Fig. 4). Western blot (right lane) demonstrates a prominent presence of RIM1/2 proteins in the lysate and C3strep captures a significant RIM1/2 band which was markedly inhibited by DDWC, consistent with binding to the PDZ ligand domain. (B) C3strep exhibits a robust, DDWC peptide-sensitive, but SH3 peptide insensitive, capture of CaV2.2 and MINT-1 from purified synaptosome membrane lysate. This lysate also contains a high concentration of RIM proteins, as evidenced by the Western blot. However, only trace amounts of RIM1/2 is detected in the C3strep pull down lanes although the little that is observed is blocked by DDWC, but not SH3 peptide. (C) C3strep pull downs in non-transfected (NT-tsA), CaV2.2 transfected (CaV-tsA) cell lysates failed to capture RIM1/2 although extremely faint bands were detected in the WB (upper panel). A chick brain crude membrane (CB) lysate pull down is shown as a positive control. (D) MINT-1 is absent in tsA201 cell lsyate WBs (upper panels) and fails to pull down with C3strep (bottom panels). Synaptosome membrane lysate (SSM) pull down is shown as a positive control (bottom panels).
A mimetic peptide blocker for the reported ‘RIM binding protein’ (RBP) SH3 binding site (Hibino et al., 2002) on the C terminal and blocked a fraction of RIM1/2 pull-down from brain crude membrane lysate (Fig. 7A) but had little consistent effect on CaV2.2 pull down by C3strep from either crude or purified synaptosome membrane lysates (N = 7, Fig. 7A,B), ruling out a significant contribution to the channel–channel linkage explored in this study.
Discussion
The main findings of this study are: we provide evidence for a mechanism of inter-CaV2.2 scaffolding; we show that this linkage involves the channel C terminal PDZ ligand domain; we demonstrate channel–channel linkage using purified native synaptosome membrane CaV2.2; and show this attachment can occur independently of RIM1/2. Our results identify an interlinking mechanism for CaV2.2 channels that may play a key role in their localization at presynaptic active zones.
We set out to test if the CaV2.2 calcium channels can scaffold to other CaV2.2 channels via its C terminal. Initial experiments using C terminal distal half fusion protein (C3strep) demonstrated robust CaV2.2 capture from solubilized chick brain crude membrane lysates, as assayed by Western blots and probing with a specific, and highly-characterized antibody directed against the channel II–III loop (Li et al., 2004; Gardezi et al., 2010). This pull down was specific as channel capture was minimal or absent with CaV2.2 I–II loop or the distal II–III loop fusion proteins.
We confirmed that the DxWC motif, DDWC in chick, functions as a PDZ-LD by demonstrating robust C terminal fusion protein pull down of MINT-1 (Figs 2, 4, 5, 7), its signature binding protein (Maximov et al., 1999). MINT-1 was captured by C3strep and C3dist but was not captured by C3prox (Fig. 4) which lacks the distal sixth of the channel C terminal. The finding that mimetic blockers, DDWC and HEADEDDWC, specifically and markedly inhibited CaV2.2 pull down (Fig. 5), provides compelling evidence for involvement of the C terminal PDZ-LD in channel binding.
Our initial data demonstrated channel capture by C terminal fusion proteins from brain crude membrane lystate. Robust capture of channels from purified synaptosome membrane lysate demonstrates that this scaffold mechanism is also present at the nerve terminal. The finding that CaV2.2 expressed in tsA201 cells pulls down with C3strep suggests that binding is not due simply to capture of a gross large presynaptic release site complex (Khanna et al., 2007) but involves a more intimate connection. However, as CaV2.2 lacks a PDZ domain, we must presume that some protein or protein complex serves as a scaffold bridge between the channels. An early study on presynaptic CaV scaffolding suggested that the modular adaptor proteins CASK and MINT-1 might serve such a role (Maximov et al., 1999) but a lack of co-variance with long-splice CaV2.2 at transmitter release sites, as assessed by quantitative immunocytochemistry (Khanna et al., 2006b), argued against this hypothesis. In agreement with that conclusion, our results in this study show that CaV2.2 can be captured from tsA201 cell lysates in the absence of MINT-1.
More recent reports have implicated RIM1/2, which contains a PDZ binding domain, (Kaeser et al., 2011), as a channel scaffold molecule (Han et al., 2011) and is essential for fast transmitter release. The present study identifies a robust channel–channel attachment mechanism and although we cannot as yet test whether this linkage plays a role at the release site itself, it can capture CaV2.2 derived from purified presynaptic membranes. We do not know which protein links the two channel PDZ-LDs but our results do not favor RIM1/2. While we can demonstrate RIM1/2 recovery using brain crude membrane lysate, trace RIM1/2 pull down using purified synaptosome membrane and its virtual absence in the tsA201 cell lysate (Fig. 7) in the face of robust CaV2.2 capture compels us to conclude that RIM1/2 binds to the C terminal independently of the channel. Further, it is hard to understand how RIM1/2 can bridge channels as it has only one PDZ domain and the possibility of linkage via the C terminal SH3 domain and RBP (Liu et al., 2011) was ruled out both by demonstrating channel capture using C3dist (Fig. 4) which lacks the SH3 domain (Fig. 3A), and by the negative effects with SH3 peptide (Fig. 7B). Lastly, in our previous studies on intact and native CaV2.2 we concluded that while RIM1/2 and CaV2.2 channels ‘covary’ at the release site, as assessed by quantitative immunocytochemistry (Li et al., 2004), they exhibit little evidence of biochemical binding as tested by immunoprecipitation in vitro (Wong and Stanley, 2010; Khanna et al., 2006a). We suggested that these two proteins are components of ‘two independent protein complexes that interact with each other with a fixed stoichiometric ratio’ (Khanna et al., 2006a) – consistent with a role in SV tethering but not as a channel release site anchor. We do not know the identity of the putative channel–channel bridge but predict either a multi-PDZ domain protein or a PDZ-binding domain protein that can form stable di- or multimers to support scaffolding. This protein(s) must be present in presynaptic nerve terminals but, since channel capture was patent in the tsA201 expression cells, it presumably has a wide cellular distribution.
This, and previous studies show that channel C terminal PDZ-LD can, as is common for these domains (Lee and Zheng, 2010), interact with multiple protein partners that include MINT-1, RIM1/2. There is also evidence for functional interactions including SV tethering (Kaeser et al., 2011), channel transport (Maximov and Bezprozvanny, 2002) and channel–channel binding, as in this report. It can be presumed that a PDZ domain cannot bind to two target proteins at the same time and hence, cannot both scaffold the channel and tether an SV. Since there is compelling evidence that CaV2.2 channels are tightly linked to the release site (as discussed above) a second, PDZ-independent, inter-channel link should be considered. An additional clue comes from structural analysis of release site channels. Freeze-fracture of the frog neuromuscular junction identifies orderly double arrays of large membrane particles that have been attributed to calcium channels (Pumplin et al., 1981) and quantitative analysis yielded an inter-particle distance 17 nm (inner row) or 14 nm (outer row) (Stanley et al., 2003). The distribution of tagged (using ω-conotoxin GVIA) CaV2.2 has also been analyzed by atomic force microscopy at the transmitter release face of the chick ciliary ganglion calyx where the channels were in short strings with a very similar, ∼16 nm interval (Haydon et al., 1994). These inter-channel estimates seem too short for a ∼350 amino acid C terminal (E.F.S., personal observations) and may indicate a second, and more direct, mechanism of channel–channel scaffolding. Our fusion protein analysis does not favor attachment via the I–II loop or the distal half of the II–III loop (Fig. 2) but further work is necessary to test the proximal II–III loop (Catterall, 1999) or proximal C terminal.
Materials and Methods
Antibodies
Antibodies used in this study are listed in Table 1.
Generation of fusion proteins
Chick E15 brain mRNA was used to synthesize cDNA using the reverse transcriptase II enzyme (Invitrogen). cDNA was used as a template for RT-PCR. PCR fragments of the CaV2.2 long splice (cdB1) variant was inserted into the TA cloning vector pCR2.1 (Invitrogen) and cut out at EcoRI and XhoI sites, then subcloned into pGEX-KG (GE healthcare), pPr-IBA (IBA; OneStrep tag), or pGEX-KG with a sub-cloned FLAG tag (to generate GST-FLAG tagged constructs) expression vectors. The DNA sequence in frame was confirmed by sequencing after transformation into DH5α competent cells (Invitrogen). Constructs were transformed into BL21 (DE3; Invitrogen) Escherichia coli cells for fusion protein production and purification.
Fusion protein purification
Fusion proteins were induced using isopropyl-β-D-thiogalactopyranoside (IPTG) and purified using standard protocols. I–II, II–IIIdistal GST-Flag and C3prox GST bacterial pellets were lysed using 1× PBS buffer (Gibco) supplemented with 0.1% Tween-20 and β-mercaptoethanol, 1 mM PMSF, and protease inhibitor cocktail (Sigma–Aldrich). Supernatants were incubated with glutathione sepharose beads (GE Healthcare) for 4–6 hrs at 4°C and washed 2× with ice-cold PBS (supplemented with 0.1% Tween-20, 1 mM PMSF and protease inhibitor cocktail; PBS-T), 3× with lysis buffer followed by 3× again with PBS-T. GST fusion proteins with the exception of I–II, which was used directly on bead, were eluted with a reduced glutathione buffer (50 mM Tris-HCl pH 8.0, 20 mM reduced glutathione). Eluted proteins were concentrated and elution buffer replaced using a 0.5 ml 10 K microcon (Millipore). C3dist GST fusion protein was prepared as described above with the following modifications. Bacterial pellets were lysed using 1× PBS supplemented with 1 mM EDTA, 0.1% Triton X-100, 1 mM PMSF, and protease inhibitors. Supernatants were incubated with glutathione sepharose beads for 2 h on ice and washed 3× with ice-cold PBS with 1 mM PMSF. C3distal GST fusion protein was eluted/concentrated as described above. C3Strep bacteria pellets were resuspended in lysis buffer (0.1 M Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 1 mM PMSF and a protease inhibitor cocktail). The supernatant was incubated with Strep-tactin superflow beads (IBA) for 2 h on ice and washed 3× with lysis buffer. The C3Strep protein was not eluted and used directly on bead. Fusion proteins were characterized using SDS-PAGE followed by Western blotting before using for experiments. Protein yield was determined using Coomassie blue gel staining by comparison with BSA standards.
Brain and purified synaptosome membrane lysate preparation
Whole brains from embryonic day 15–17 chicks were used for both membrane preparations. Brain crude membrane pellet was prepared as described in Gardezi et al. as well as Wong and Stanley (Gardezi et al., 2010; Wong and Stanley, 2010). Purified synaptosomes were prepared as described in Gardezi et al. (Gardezi et al., 2010) but with an additional discontinuous sucrose gradient step for separation of purified synaptosome membrane (SSM). Briefly, purified synaptosomes were collected from the 0.8/1.2 M gradient interphase and diluted with homogenization buffer (0.32 M sucrose, 2 mM EDTA, 10 mM HEPES pH 7.4, 1 mM PMSF and protease inhibitor cocktail from Sigma–Aldrich) followed by centrifugation at 22,000 g for 30 min. The pelleted synaptosomes were lysed using HEPES lysis buffer (2 mM EDTA, 50 mM HEPES pH 7.4, 1 mM PMSF and protease inhibitor cocktail) and centrifuged for 147,000 g for 4 hours. The lysed synaptosomal pellet was resuspended in 0.2 M buffered sucrose and layered onto a discontinuous sucrose gradient (0.2 (sample)/0.4/0.6/0.8/1.0 M sucrose) followed by centrifugation at 100,000 g for 1.5 hour in a swinging bucket rotor (SW41 TI). The gradient interphase (0.8/1.0 M) containing SSM was diluted with homogenization buffer and centrifuged at 215,000 g for 3 hours in a swinging bucket rotor (SW60TI) yielding a purified SSM pellet. Both brain crude membrane and purified synaptosome membrane pellets were solubilized in RIPA lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% NP-40, 0.5% Na deoxycholate, 1 mM PMSF and protease inhibitor cocktail) and stored at −80°C until use. Protein concentrations were measured using Bradford reagent and a Beckman spectrophotometer. All the high speed spins were carried out in a Beckman (L-80) ultra-centrifuge using a 70TI rotor or swinging bucket rotors as described above.
tsA201 cell transfection and lysate preparation
tsA201 cells were maintained in 1× high glucose Dulbecco's modified Eagle's medium (DMEM; Gibco) with 5% penicillin–streptomycin (Gibco) at 37°C. Cells were transfected with expression vectors containing cDNA encoding rat CaV2.2 α1B, β1b and α2δ subunits using lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. CaV2.2 expression constructs were a gift from Dr T. Snutch (University of British Columbia). Cells were harvested after 48 hours of transfection using RIPA lysis buffer and were centrifuged at 5000 g for 15 min to pellet cell debris. Cell lysates were stored at −80°C until use and protein concentration was determined using a Bradford assay.
Fusion protein pull-down assays
Lysates were pre-cleared with Strep-Tactin or glutathione sepharose bead 50% slurry (40 µl of bead slurry per 1 ml of sample) for 1 hour at 4°C. The pre-cleared lysates were incubated with immobilized fusion proteins overnight at 4°C. Fusion proteins were either used on bead (not eluted) or were immobilized prior to incubation with lysates. Pull-down beads were washed 5× with RIPA lysis buffer and proteins were denatured by the addition of Laemmli sample buffer (Biorad) supplemented with 5% β-mercaptoethanol. Protein complexes were analyzed by SDS-PAGE followed by Western blotting.
CaV2.2 C-terminal peptide synthesis and treatment
CaV2.2 distal C-terminus PDZ ligand domain mimetic peptide DDWC, HEADEDDWC, its mutated control DDWA, and SH3 ligand mimetic peptide RQLPQTPL (referred to as “SH3” herein) was synthesized at the SickKids Advanced Protein Technology Centre. Peptides were dissolved in 1× PBS (Gibco) and used at a final concentration of 1 mM. Untreated control lysates were incubated with a comparable volume of buffer used to reconstitute the peptides. Pre-cleared membrane lysates were incubated with peptides for 4 hours at 4°C prior to incubation with immobilized C3strep protein.
Western blots
Western blotting was carried out as described previously (Wong and Stanley, 2010).
Acknowledgments
We would like to thank Fiona Wong, Drs Bill Trimble, Christine Bear and Shuzo Sugita for helpful suggestions and Ali Farsi and Fenghao Xu for technical assistance. This project was funded by CIHR award MOP 86599 and CRC to E.F.S. and an Ontario Graduate Scholarship award to S.R.G.
Footnotes
Competing interests: The authors have no competing interests to declare.
References
- Catterall W. A. (1999). Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann. N. Y. Acad. Sci. 868, 144–159 10.1111/j.1749-6632.1999.tb11284.x [DOI] [PubMed] [Google Scholar]
- Dreyer F., Peper K., Akert K., Sandri C., Moor H. (1973). Ultrastructure of the “active zone” in the frog neuromuscular junction. Brain Res. 62, 373–380 10.1016/0006-8993(73)90699-9 [DOI] [PubMed] [Google Scholar]
- Eggermann E., Bucurenciu I., Goswami S. P., Jonas P. (2012). Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 13, 7–21 10.1038/nrn3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardezi S. R., Taylor P., Stanley E. F. (2010). Long C terminal splice variant CaV2.2 identified in presynaptic membrane by mass spectrometric analysis. Channels (Austin) 4, 58–62 10.4161/chan.4.1.10364 [DOI] [PubMed] [Google Scholar]
- Han Y., Kaeser P. S., Südhof T. C., Schneggenburger R. (2011). RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 69, 304–316 10.1016/j.neuron.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow M. L., Ress D., Stoschek A., Marshall R. M., McMahan U. J. (2001). The architecture of active zone material at the frog's neuromuscular junction. Nature 409, 479–484 10.1038/35054000 [DOI] [PubMed] [Google Scholar]
- Haydon P. G., Henderson E., Stanley E. F. (1994). Localization of individual calcium channels at the release face of a presynaptic nerve terminal. Neuron 13, 1275–1280 10.1016/0896-6273(94)90414-6 [DOI] [PubMed] [Google Scholar]
- Heuser J. E., Reese T. S., Landis D. M. D. (1974). Functional changes in frog neuromuscular junctions studied with freeze-fracture. J. Neurocytol. 3, 109–131 10.1007/BF01111936 [DOI] [PubMed] [Google Scholar]
- Hibino H., Pironkova R., Onwumere O., Vologodskaia M., Hudspeth A. J., Lesage F. (2002). RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca2+ channels. Neuron 34, 411–423 10.1016/S0896-6273(02)00667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser P. S., Deng L., Wang Y., Dulubova I., Liu X., Rizo J., Südhof T. C. (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295 10.1016/j.cell.2010.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. (1969). The Release Of Neural Transmitter Substances. Liverpool: Liverpool University Press. [Google Scholar]
- Khanna R., Li Q., Sun L., Collins T. J., Stanley E. F. (2006a). N type Ca2+ channels and RIM scaffold protein covary at the presynaptic transmitter release face but are components of independent protein complexes. Neuroscience 140, 1201–1208 10.1016/j.neuroscience.2006.04.053 [DOI] [PubMed] [Google Scholar]
- Khanna R., Sun L., Li Q., Guo L., Stanley E. F. (2006b). Long splice variant N type calcium channels are clustered at presynaptic transmitter release sites without modular adaptor proteins. Neuroscience 138, 1115–1125 10.1016/j.neuroscience.2005.12.050 [DOI] [PubMed] [Google Scholar]
- Khanna R., Li Q., Bewersdorf J., Stanley E. F. (2007). The presynaptic CaV2.2 channel-transmitter release site core complex. Eur. J. Neurosci. 26, 547–559 10.1111/j.1460-9568.2007.05680.x [DOI] [PubMed] [Google Scholar]
- Lee H. J., Zheng J. J. (2010). PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun. Signal. 8, 8 10.1186/1478-811X-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Lau A., Morris T. J., Guo L., Fordyce C. B., Stanley E. F. (2004). A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J. Neurosci. 24, 4070–4081 10.1523/JNEUROSCI.0346-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. S., Siebert M., Mertel S., Knoche E., Wegener S., Wichmann C., Matkovic T., Muhammad K., Depner H., Mettke C.et al. (2011). RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 334, 1565–1569 10.1126/science.1212991 [DOI] [PubMed] [Google Scholar]
- Llinás R. R., Steinberg I. Z., Walton K. (1981). Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys. J. 33, 323–351 10.1016/S0006-3495(81)84899-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R., Sugimori M., Silver R. B. (1992). Microdomains of high calcium concentration in a presynaptic terminal. Science 256, 677–679 10.1126/science.1350109 [DOI] [PubMed] [Google Scholar]
- Maximov A., Bezprozvanny I. (2002). Synaptic targeting of N-type calcium channels in hippocampal neurons. J. Neurosci. 22, 6939–6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A., Südhof T. C., Bezprozvanny I. (1999). Association of neuronal calcium channels with modular adaptor proteins. J. Biol. Chem. 274, 24453–24456 10.1074/jbc.274.35.24453 [DOI] [PubMed] [Google Scholar]
- Mirotznik R. R., Zheng X., Stanley E. F. (2000). G-Protein types involved in calcium channel inhibition at a presynaptic nerve terminal. J. Neurosci. 20, 7614–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan S. J., Davison I., Delaney K. R. (2001). Mitral cell presynaptic Ca2+ influx and synaptic transmission in frog amygdala. Neuroscience 104, 137–151 10.1016/S0306-4522(01)00057-4 [DOI] [PubMed] [Google Scholar]
- Pumplin D. W., Reese T. S., Llinás R. (1981). Are the presynaptic membrane particles the calcium channels? Proc. Natl. Acad. Sci. USA 78, 7210–7213 10.1073/pnas.78.11.7210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R., Adler E. M., Charlton M. P. (1990). Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron 5, 773–779 10.1016/0896-6273(90)90336-E [DOI] [PubMed] [Google Scholar]
- Sheng J., He L., Zheng H., Xue L., Luo F., Shin W., Sun T., Kuner T., Yue D. T., Wu L. G. (2012). Calcium-channel number critically influences synaptic strength and plasticity at the active zone. Nat. Neurosci. 15, 998–1006 10.1038/nn.3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. J., Buchanan J., Osses L. R., Charlton M. P., Augustine G. J. (1993). The spatial distribution of calcium signals in squid presynaptic terminals. J. Physiol. 472, 573–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley E. F. (1991). Single calcium channels on a cholinergic presynaptic nerve terminal. Neuron 7, 585–591 10.1016/0896-6273(91)90371-6 [DOI] [PubMed] [Google Scholar]
- Stanley E. F. (1993). Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron 11, 1007–1011 10.1016/0896-6273(93)90214-C [DOI] [PubMed] [Google Scholar]
- Stanley E. F. (1997). The calcium channel and the organization of the presynaptic transmitter release face. Trends Neurosci. 20, 404–409 10.1016/S0166-2236(97)01091-6 [DOI] [PubMed] [Google Scholar]
- Stanley E. F., Reese T. S., Wang G. Z. (2003). Molecular scaffold reorganization at the transmitter release site with vesicle exocytosis or botulinum toxin C1. Eur. J. Neurosci. 18, 2403–2407 10.1046/j.1460-9568.2003.02948.x [DOI] [PubMed] [Google Scholar]
- Tonikian R., Zhang Y., Sazinsky S. L., Currell B., Yeh J. H., Reva B., Held H. A., Appleton B. A., Evangelista M., Wu Y.et al. (2008). A specificity map for the PDZ domain family. PLoS Biol. 6, e239 10.1371/journal.pbio.0060239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F. K., Stanley E. F. (2010). Rab3a interacting molecule (RIM) and the tethering of pre-synaptic transmitter release site-associated CaV2.2 calcium channels. J. Neurochem. 112, 463–473 10.1111/j.1471-4159.2009.06466.x [DOI] [PubMed] [Google Scholar]