

Abstract
Posttraumatic stress disorder (PTSD) develops in approximately one-quarter of trauma-exposed individuals, leading us and others to question the mechanisms underlying this heterogeneous response to trauma. We suggest that the reasons for the heterogeneity relate to a complex interaction between genes and the environment, shaping each individual's recovery trajectory based on both historical and trauma-specific variables. Epigenetic modifications provide a unique opportunity to elucidate how preexisting risk factors may contribute to PTSD risk through changes in the methylation of DNA. Preexisting risks for PTSD, including depression, stress, and trauma, result in differential DNA methylation of endocrine genes, which may then result in a different biological responses to trauma and subsequently a greater risk for PTSD onset. Although these relationships are complex and currently inadequately described, we provide a critical review of recent studies to examine how differences in genetic and proteomic biomarkers shape an individual's vulnerability to PTSD development, thereby contributing to a heterogeneous response to trauma.
1. Introduction
Up to 90% of individuals experience a traumatic event at some time during their lives, yet most recover and suffer from no long-term ramifications; however, a subset of individuals develop posttraumatic stress disorder (PTSD) and are at high risk for health decline [1–4]. This heterogeneous response suggests that preexisting factors might influence how people respond to traumatic situations. We suggest that a more comprehensive examination of biomarkers that approximate PTSD risk would be of great value. Biomarker discovery in PTSD has been hindered by the lack of prospective studies in traumatized individuals, resulting in an insufficient understanding of the preexisting risk factors for PTSD onset as well as the mechanistic pathways that underlie this risk. Without this knowledge, we are unable to identify traumatized individuals at risk for PTSD development and, therefore, unable to implement effective, preventive interventions. Yet, additional biological analysis methods have become available, and these new strategies will be useful in addressing this critical issue. Specifically, the use of whole-genome gene expression and epigenetic modifications (DNA methylation) provide an opportunity to explore more than candidate biomarkers, to identify novel mechanisms related to PTSD risk, and to pinpoint genetic pathways that may be implicated in both risk and resilience to trauma.
A traumatic event places individuals at high risk for developing PTSD, resulting in rates of PTSD between 18% and 36% in trauma-exposed patients [1–4]; as mentioned above, most trauma-exposed individuals recover and do not develop PTSD. PTSD is viewed as a disorder of dysregulation of fear and processing of stimuli associated with trauma, and it is characterized by three main clusters of symptoms: reexperiencing, avoidance, and hyperarousal. Reexperiencing symptoms include distressing recollections or dreams of the event, flashbacks, and intense psychological and physiological reactivity to internal and external cues; avoidance symptoms include evasion of any thoughts or feelings of people and places that remind the individual of the traumatic event, decreased interest in participating in activities, and emotional numbing; and hyperarousal symptoms include problems falling or staying asleep, irritability or anger, hypervigilance, and exaggerated startle reflex. It has become increasingly clear that environmental influences early in development remain pervasive into adulthood, a relationship that is attributed to an interaction of gene function and environment. Both genetic and environmental factors are critical to developmental processes, and even minor changes in either type of factor can result in trajectories of resilience or vulnerability [5]; however, it is the interaction between these factors that may provide the most vital information to understanding the heterogeneous response to trauma. Epigenetic modifications occur in response to an environmental factor and include DNA methylation, acetylation, and histone modification which alter DNA accessibility and chromatin structure, thereby regulating activity of the gene in a long-lasting manner. Because the risk for PTSD onset is influenced by environmental factors that predate trauma exposure, epigenetics may provide novel insights into the heterogeneous response to a trauma. Large epidemiological studies link pretrauma risks including previous depression, stressors, and traumatic events to a greater risk for PTSD onset following trauma exposure [6], suggesting that these factors may contribute to variability of individuals in response to a trauma.
The support for the mediating link of epigenetic modifications and PTSD vulnerability is reported in preclinical studies. Specifically, preclinical models illustrate these complex relationships and link epigenetic modifications in neurons to psychological vulnerability following stressors. In rats, the offspring of the high caring mothers (i.e., high licking) exhibit the reduced methylation of the glucocorticoid receptor gene [7] and endocrine regulation of a subsequent stressor [8]. In contrast, in non-human primates, offspring that face early adversity exhibits endocrine dysregulation [9] as well as reductions in neuronal plasticity in the prefrontal cortex that persist into adulthood [10]. Early stressors in humans have also been linked to epigenetic modifications, including increased methylation of the glucocorticoid promoter in hippocampal neurons of suicide completers with histories of early childhood maltreatment [11]. Cord blood collected from mothers with high levels of depression and anxiety during the third trimester displays similar glucocorticoid methylation differences, which are linked to a dysregulation of the endocrine system [12]. In studies of rats who exhibit PTSD-like behavior, there is evidence of increased methylation of stress response genes including brain-derived neurotrophic factor [13] and nuclear protein phosphate-1 [14] in neurons, which result in the onset of PTSD-like behavior [15]. These preclinical studies provide insights into the heterogeneous response to trauma and stress and suggest that epigenetic modifications in neurons result in the onset of PTSD [16, 17].
We suggest that the heterogeneous response to trauma relates to complex interactions between genes and the environment, shaping each individual's recovery trajectory based on both historical and trauma specific variables (see Figure 1). One of the major environmental factors linked to epigenetic changes is stress, which is a critical factor in the pathogenesis of PTSD. There is robust evidence linking PTSD onset to epigenetic changes following stress; yet this evidence is only starting to accumulate from the clinical studies. Recent advances in laboratory analyses and biostatistical methods provide new opportunities to determine the mechanisms of PTSD onset; however, many of these advances are not yet applied. Additionally, current studies are limited by our inability to determine pretrauma epigenetic status as well as our restriction to peripheral blood samples in clinical samples. Here, we provide a critical review of recent studies to examine how differences in genetic and proteomic biomarkers may underlie the vulnerability of the individual to develop PTSD, and how this field of study may provide fundamental insights into the identification of individuals at risk for PTSD development following trauma and the development of novel, interventional strategies.
Figure 1.
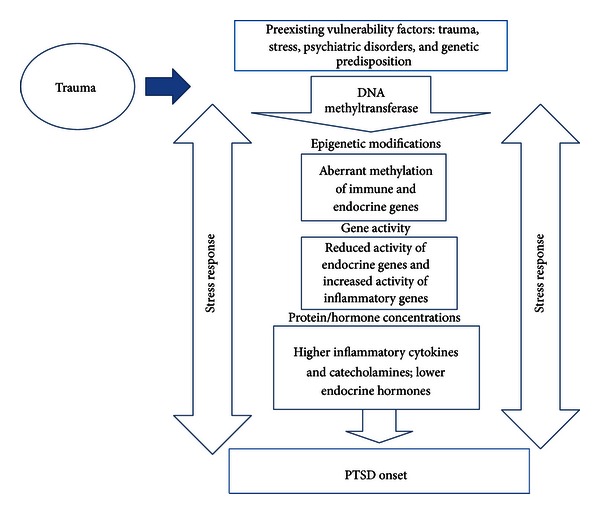
Epigenetic mechanisms of PTSD onset.
2. Methods
In order to obtain a comprehensive pool of prospective studies investigating peripheral biomarkers of PTSD following trauma, PubMed, OVID/MEDLINE, the Cochran Database, Embase, Scopus, CINAHL, and PsychInfo were systematically searched using the following National Library of Medicine Medical Subject Headings (MeSH) “posttraumatic stress disorder,” “genes,” “cytokines,” “neuropeptides,” and “inflammation.” Additionally, each key word was cross-referenced with each other in each of the various databases. The search extended to the literature published between 2000 and 2013, and studies were included in the review if they (1) were primary research articles, (2) were published in English, (3) were conducted in humans, and (4) examined epigenetic modifications (i.e., DNA methylation), gene expression, or a proteomic biomarker that was linked to PTSD risk following a trauma. The following components and variables of interest were appraised to provide an overall synthesis of the available literature: study purpose, design, methods, sample size, demographics, type of trauma, severity of injuries sustained (mild, moderate, and severe), type of biological sample collected, and times of collection following trauma. We were able to locate 6 clinical studies that examined epigenetic modifications (i.e., DNA methylation), 9 studies of gene expression, and 14 studies that used a proteomic biomarker to examine the risk for PTSD onset (3 pediatric and 9 adult studies). All studies were reviewed and are presented in Tables 1, 2, and 3.
Table 1.
Epigenetics studies.
| Study | Analysis method | Sample | Findings |
|---|---|---|---|
| Uddin et al., 2010 [18] | DNA methylation; >14,000 genes |
100 individuals from DNHS (PTSD = 23) | PTSD had greater methylation of toll-like receptors 1 and 3, IL-8, and others compared to controls and had a greater overall number of uniquely methylated genes. |
|
| |||
| Koenen et al., 2011 [22] | DNA methylation and genotype; SLC6A4 | 100 individuals from DNHS (PTSD = 23) | Neither genotype nor methylation of SLC6A4 was associated with PTSD; however, when controlling with genotype, lower methylation levels were associated with increased risk for developing PTSD among individuals with more traumatic events. |
|
| |||
| Ressler et al., 2011 [19] | DNA methylation; 44 SNPs of PACAP and PAC1 |
64 individuals, primarily African American (PTSD = 24) |
An SNP in ADCYAP1R1, rs2267735, predicted PTSD diagnosis and symptoms in women; methylation of this gene was also associated with PTSD. |
|
| |||
| Smith et al., 2011 [23] | DNA methylation; global and site specific | 110 African Americans (PTSD = 50) |
Global methylation was increased in subjects with PTSD, as compared to control subjects or subjects with a history of childhood trauma; CpG sites in TPR, CLEC9A, APC5, ANXA2, and TLR8 were differentially methylated in subjects with PTSD. |
|
| |||
| Uddin et al., 2011 [24] |
DNA methylation; 33 candidate genes |
100 individuals from DNHS (PTSD = 23) | One candidate gene, MAN2C1, showed significantly higher methylation in subjects with lifetime PTSD. |
|
| |||
| Rusiecki et al., 2012 [25] | DNA methylation; LINE-1 and Alu | 150 service members (PTSD = 75) |
LINE-1 was hypomethylated in PTSD versus controls postdeployment and hypermethylated postdeployment versus predeployment in controls; Alu was hypermethylated in PTSD versus controls predeployment. |
Table 2.
Gene expression studies.
| Study | Analysis method | Sample | Findings |
|---|---|---|---|
| Zieker et al., 2007 [26] | Pre-selected stress-immune genes, whole-blood | 16 individuals (PTSD = 8) |
In PTSD, upregulated (4): glutamate transported, IGF-2; downregulated (14): IL-18, IL-16, colony stimulating factor. |
|
| |||
| Yehuda et al., 2009 [27] | Whole blood gene expression | 35 individuals exposed to 9/11 (PTSD = 15) |
FKBP5, STAT5B, and MHC class II showed reduced expression in individuals with PTSD. |
|
| |||
| Neylan et al., 2011 [28] | CD14+ monocyte; gene expression | 67 ± trauma-exposed individuals (PTSD = 34) |
In PTSD patients, three monocyte genes were underexpressed in men but not in women. |
|
| |||
| Sarapas et al., 2011 [29] | Genome-wide gene expression | 40 individuals exposed to 9/11 (PTSD = 20) | PTSD patients showed a reduction in gene expression of STAT5B and nuclear factor I/A. |
|
| |||
| Mehta et al. 2011 [30] | Whole-blood gene expression and SNP of FKBP5 | 211 low income (PTSD = 75) |
With FKBP5 SNP added to interaction with PTSD, there was a reduction in 32 genes including IL-18 and STAT pathway. |
|
| |||
| Pace et al., 2012 [31] | Nuclear factor-κB activity in peripheral blood | 36 women (PTSD = 12) |
Increased nuclear factor-κB activity was associated with women with PTSD as compared to controls. |
|
| |||
| van Zuiden et al., 2012 [32] | GR number; FKBP5, GILZ, and SGK1 mRNA expression | 448 military personnel (PTSD = 35) |
Predeployment high GR number, low FKBP5 mRNA expression, and high GILZ expression predicted PTSD development. |
|
| |||
| van Zuiden et al., 2012 [33] | GC sensitivity of leukocytes | 526 military personnel (PTSD = 46) |
Predeployment sensitivity of GCRs on leukocytes predicted development of PTSD. |
|
| |||
| Matić et al., 2013 [34] | GR function and expression using PCR | 347 ± war trauma-exposed individuals (PTSD = 113) |
Lower GR sensitivity in PBMCs and low gene-expression of GR were found in PTSD patients. |
Table 3.
Proteomic studies.
| Pediatric studies | Sample | Collection; followup | Outcome | Results |
|---|---|---|---|---|
| Delahanty et al., 2005 [35] | 58 ED patients | ED; 6 wks | Diagnosis | High cortisol and epinephrine predicted acute PTSD symptoms. |
| Ostrowski et al., 2007 [36] | 54 ED patients | ED; 6 wks, 7 mos | Diagnosis | High cortisol predicted acute PTSD symptoms and PTSD onset in boys. |
| Pervanidou et al., 2007 [37] | 56 MVA patients (9 = PTSD) | ED; 1, 6 mos | Diagnosis | High cortisol and IL-6 predicted PTSD onset. |
|
| ||||
| Adult studies | Sample | Collection; followup | Outcome | Results |
|
| ||||
| Resnick et al., 1995 [38] | 37 rape survivors (19 = PTSD) | ED; 17–157 days | Diagnosis | Low cortisol in previously assaulted women predicted PTSD onset. |
| Yehuda et al., 1998 [39] | 20 rape survivors (11 = PTSD) | ED; 27–157 days | Diagnosis | Cortisol and MHPG did not predict PTSD onset. |
| Delahanty et al., 2000 [40] | 99 MVA patients (9 = ASD) | ED; 1 mo | Diagnosis | Low cortisol predicted acute PTSD symptoms. |
| Bonne et al., 2003 [41] | 21 ED patients (8 = PTSD) | 1 wk; 6 mos | Diagnosis | Cortisol did not predict PTSD onset. |
| Heinrichs et al., 2005 [42] | 43 firefighters (7 = PTSD at 2 yrs) | Training; 6, 9, 12, 24 mos | Symptom report | Cortisol and CA did not predict PTSD onset. |
| McFarlane et al., 1997 [43] | 40 MVA patients (7 = PTSD) | ED; 2, 10 days and 6 mos | Diagnosis | Low cortisol predicted PTSD onset. |
| Ehring et al., 2008 [44] | 53 MVA patients (5 = PTSD) | ED; 2 wks, 6 mos | Diagnosis | Low cortisol predicted PTSD onset. |
| Shalev et al., 2008 [45]; Videlock et al., 2008 [46] | 155 ED patients (31 = PTSD) | ED; 10 days, and 1, 5 mos | Diagnosis | Cortisol, ACTH, GR, and NE did not predict PTSD onset. |
| Cohen et al., 2011 [47] | 48 orthopedic patients, 13 HC | At hospitalization; 1 mo | Symptom report | High IL-8 and low TGF-β predicted acute PTSD symptoms. |
| van Zuiden et al., 2011 [48] | 68 service members, (34 = PTSD) | Prior; following deployment | Symptom report | High GR predicted PTSD onset. |
2.1. Epigenetics
Clinical studies are restricted to examining epigenetic modifications in samples of peripheral blood, but these studies do provide some key insights into how these molecular changes relate to PTSD risk (see Table 1). Specifically, these studies implicate insufficient regulation of inflammatory activity and reduced neurotransmitter activity in PTSD risk. In support of this, studies in chronic PTSD-affected patients report hypermethylation of inflammatory initiator genes (toll-like receptors 1 & 3, IL-8, chemokine ligand 1, and others) and demethylation of inflammatory regulatory genes (FK506 binding protein-5 [FKBP5]) [18]. In a subsequent study, Ressler et al. (2011) linked these molecular-genetic mechanisms to higher concentrations of inflammatory cytokines in patients with chronic PTSD [19]. Together, these association studies suggest that observations by us and others of excessive inflammation in chronic PTSD [20, 21] likely relate to these DNA methylation differences; however, their cross- sectional designs prohibit linking these molecular differences to PTSD vulnerability.
In contrast, a recent study measured DNA methylation and reported that postdeployment hypomethylation of LINE-1 was associated with PTSD onset following deployment [25]. The authors of this report postulate that LINE-1, a regulator of stress response in the immune system, may represent an adaptive response to combat stress that protects against PTSD; yet additional studies are needed in more generalized samples that include women and various traumatic events. In addition, this study did not determine predeployment factors such as previous PTSD, depression, or trauma that contributed to precombat methylation differences, resulting in an inability to understand how the interactions between genes and environment prior to trauma underlies PTSD risk.
Other studies suggest that differential methylation of neurotransmitter genes is linked to PTSD onset. In two studies using samples of civilians from the Detroit Neighborhood Health Study, neurotransmitter genes were implicated in the risk for PTSD development. In one study that examined the serotonin transporter gene (SLC6A4), methylation levels were modified by the effect of the number of traumatic events on PTSD after controlling for SLC6A4 genotype, such that persons with more traumatic events were at increased risk for PTSD, but only at lower methylation levels. At higher methylation levels, individuals who reported more traumatic events were protected from this disorder, suggesting that the serotonin transporter gene may also be important in trauma-related resilience [22]. In a study using the same patient sample, the candidate gene MAN2C1 showed a significant methylation × trauma experience interaction such that those with both higher MAN2C1 methylation and greater exposure to traumatic events showed an increase in risk of lifetime PTSD [24]. Although these studies provide unique insights into risks for PTSD based on DNA methylation, trauma, and psychiatric-related symptoms, these studies used cross-sectional design and did not include definitive diagnoses determined through a clinical interview.
Although not epigenetic in nature, previous studies that examine genetic inheritance provide further evidence of genetic underpinnings in endocrine modulating genes in the risk for PTSD. In brief, previous studies link single nucleotide polymorphisms (SNPs) of FKBP5, a negative regulator of GCR sensitivity to a greater risk for PTSD onset [29, 49, 50]. Current findings indicate that genetic predisposition differences in another key inflammatory gene, corticotrophin- releasing hormone type 1 receptor gene (CRHR1), increase PTSD onset in children who were abused at an early age [51]. With pediatric injury patients, a longitudinal study identified one SNP significantly related to acute PTSD symptoms as well as trajectory of symptoms over time [52]. These findings are meaningful in terms of genetic and environmental interplay producing risk for PTSD; however, current studies do not explicate the link between baseline epigenetics in immune regulatory genes and the risk for PTSD onset following a trauma. Yet, taking these studies together with studies that examine gene function, there is evidence that a combination of genetic and environmental factors contributes to psychological vulnerability following a trauma by shaping an individual's neuronal and biological stress response, warranting future prospective studies to determine these important temporal relationships [53].
2.2. Gene Expression
Nine studies have also used gene expression analysis of peripheral blood to examine the mechanisms of PTSD, providing additional insights into the complex biological processes underlying PTSD onset. Gene expression involves processes that alter the ultimate product of the gene, which is most often the production of proteins. Current studies using gene expression analysis methods also support the role of insufficient inflammatory regulation in the risk for PTSD onset, by implicating higher expression of inflammatory genes and lower expression of genes that regulate inflammatory processes (see Table 2). In a recent cross-sectional study of PTSD patients, a reduction in the transcriptional activity of genes that regulate inflammation including FKBP5 and IL18 and STAT pathways was reported in an urban sample of primarily African American participants [30]. Reductions in gene expression of STAT5B and MHC II class receptor genes in participants who developed PTSD following the 9-11 terrorist attacks were also reported [29]. Also, in a sample of participants who developed PTSD following 9-11, reduced FKBP5 gene expression was most related to PTSD severity in regression analysis as well as a reduction in STAT5B, a direct inhibitor of GR, and a major histocompatibility complex (MHC) class II gene expression; however, significant differences were related to PTSD symptom severity and not a definitive diagnosis of PTSD [27]. Lastly, in a recent study of 12 women with PTSD related to child abuse, increased glucocorticoid receptor sensitivity in monocytes was linked to increased NF-κB activity; however, this study did not use a whole-genome analysis like the other reviewed studies, but instead a DNA binding ELISA in mononuclear cells [31]. Thus, together, these studies suggest that reduced gene expression of immune regulatory genes is associated with PTSD but is limited to small sample sizes and cross-sectional design.
In contrast, two recent studies provide vital insights into how gene expression differences contribute to PTSD onset through the use of a prospective design in male service members who are evaluated prior to and then following deployment. In a study of 448 male soldiers, predeployment high glucocorticoid receptor numbers and low FKBP5 mRNA expression were associated with increased risk for a high level of PTSD symptoms [32]. In a similar sample, a high level of PTSD symptoms after deployment was independently associated with a high DEX sensitivity of T-cell proliferation before deployment, but only in individuals who reported PTSD symptoms without depressive symptoms [33]. These studies link alterations in gene expression to functional immune cell changes. Importantly, these studies also link preexisting biological differences in cell function and gene expression to the risk for PTSD onset following a trauma. These studies are limited by their use of gene expression analysis of only predetermined target genes, restricting our ability to identify novel biological targets for PTSD risk and to understand how known risks relate to less well known genes related to PTSD risk. Although these studies have some limitations, they provide fundamental information regarding the mechanisms of PTSD onset. Specifically, these studies report that it is likely an accumulation of preexisting factors that result in differential gene activity and vulnerability to develop PTSD following trauma exposure.
2.3. Proteomics
Studies of proteomic biomarkers of PTSD risk have been undertaken for far longer than studies that use genetic analyses; yet they have been limited by many methodological issues. These issues include an inability to control for timing of the sample collection, with most proteins exhibiting substantial circadian variation, the impact of the physical consequences of trauma, and differences in the timing between the occurrence of the trauma and the biological sample. Despite these limitations, these studies provide some vital insights into the biological mechanisms underlying PTSD vulnerability. Proteins play important roles in both intracellular processes and intercellular communication, thereby contributing to the orchestration of responses to traumatic events. Thus, proteins should be considered as possible PTSD biomarkers. Studies of acutely traumatized individuals suggest that high concentrations of inflammatory proteins are linked to PTSD onset. Here, we review 14 studies that collect a biological sample within the acute period following a trauma and use this biomarker to approximate PTSD risk (see Table 3).
In children following a motor vehicle accident (MVA), high concentrations of IL-6 twenty four hours after the MVA predicted PTSD onset at six months [37]; however, these elevations were no longer evident in those children who developed PTSD. Similar findings are reported in a study of adults hospitalized for orthopedic injuries, reporting that high concentrations of IL-6 as well as IL-8 related to the risk acute stress disorders symptoms; however, this study did not link these biomarkers to the risk for PTSD onset [47]. IL-8 is a mediator of the innate immune system, with higher concentrations indicating a greater immune challenge. In contrast, low concentrations of IL-8 predicted high PTSD symptoms following the earthquakes in China, whereas high IL-6 predicted anxiety and depression symptoms but was not specific to PTSD [54]. The Song et al. [54] study differs from the other two studies as biomarker collection was within months, not hours of the trauma, and many of the participants did not sustain physical injuries. A benefit of the Song study was the evaluation of cooccurring depression with PTSD, which is a limitation of the other prospective studies. In studies of chronic PTSD, cooccurring depression is associated with higher concentrations of IL-6 [20, 55]. Therefore, inflammation in the acute recovery period may be very detrimental to psychological recovery as high concentrations of inflammatory cytokines exert central effects including the induction of “sickness behavior” [56–60]. In addition, inflammatory cytokines increase blood brain barrier disruption [61–63] and alter metabolism of serotonin [64–67], all of which contributes to neuronal vulnerability despite of the excessive central inflammatory actions, including overactivation of microglia [68–70]. Together, these studies suggest that increased concentrations of inflammatory cytokines relate to the risk for PTSD onset.
There is also evidence that endocrine dysregulation relates to PTSD risk; however, this risk may relate to previous trauma. In three studies of children recruited from emergency departments, high cortisol measured in the urine predicted acute stress symptoms at six weeks following the trauma; however, in the Ostrowski et al. (2007) study high cortisol predicted PTSD onset in boys only [36]. This is of interest as studies of adult women report that it is low cortisol concentrations that predict PTSD onset [39]. Resnick et al. (1995) support this finding in a similar sample but reports that this relationship only relates to those women who report a trauma prior to the rape [38]. In samples of men and women following an MVA, low cortisol concentrations are also linked to PTSD risk [40, 44]; however, the role of previous trauma is not reported in these studies. Other studies do not link cortisol concentrations to PTSD risk [39, 41, 42, 45, 46]. In a unique study of service members prior to and following deployment, concentrations of cortisol were not related to PTSD risk; however, greater glucocorticoid binding in mononuclear cells prior to deployment predicted PTSD onset, suggesting that systems that regulate cortisol activity relate to PTSD risk [48].
ANS dysregulation contributes to PTSD vulnerability [71, 72]; however, current biomarker studies that measure catecholamines or metabolites have not supported this relationship. Studies that examined catecholamine concentrations from blood samples did not significantly differ in studies of adults [39, 46] or children [35, 37]. Studies of concentrations of catecholamines using urine samples also did not show significant findings in adults [35, 40, 42, 46] or in children [35]. Therefore, ANS dysregulation may relate to PTSD risk yet this is not supported in biomarkers studies; this discrepancy may be related to the high degree of turnover of catecholamines in circulation, in which current studies may not be able to capture. Future studies that are able to approximate ANS dysregulation will be important as catecholamines influence immune and endocrine function, and also neuronal circuitry required to psychologically cope with trauma [73].
3. Conclusions
In this review, we provide a comprehensive examination of the biomarkers that contribute to PTSD risk. We suggest that epigenetic modifications shape the resulting proteomic response of the individual through differential gene activity, thereby contributing to the heterogeneous response to trauma and differential risk for PTSD onset. Additional studies are needed to elucidate these relationships and to design technological methods to use these biomarkers to identify trauma patients at the highest risk for PTSD onset. Preventative interventions would be of great value in patients at the highest risk for PTSD to prevent chronic symptomatology. In addition, these studies may inform the development of novel interventions including pharmacological agents that are better able to prevent the onset of PTSD.
Addressing this issue is critical, as delivering effective, preventive interventions for PTSD could save the USA up to 180 million dollars each year in health care costs [74–79] and the lives of up to 9,000 individuals from suicide [80–83]. Health care providers have an opportunity to reduce the risk for PTSD onset as up to 3 million adults seek immediate medical care for traumatic injuries each year [84], resulting in the onset of PTSD in almost 1 million of Americans annually [1, 4, 85–87]. However, even in this group, there is a high level of interindividual response variability to traumatic injuries, suggesting that a biomarker that is able to approximate PTSD risk would be of great value in directing preventive measures. Preventing PTSD is paramount and would reduce the substantial morbidity and health-related mortality associated with this devastating disorder [2, 85].
References
- 1.Davydow DS, Katon WJ, Zatzick DF. Psychiatric morbidity and functional impairments in survivors of burns, traumatic injuries, and ICU stays for other critical illnesses: a review of the literature. International Review of Psychiatry. 2009;21(6):531–538. doi: 10.3109/09540260903343877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zatzick D. Posttraumatic stress, functional impairment, and service utilization after injury: a public health approach. Seminars in Clinical Neuropsychiatry. 2003;8(3):149–157. doi: 10.1016/s1084-3612(03)00017-0. [DOI] [PubMed] [Google Scholar]
- 3.Coplan JA. Rotational motion of the knee: a comparison of normal and pronating subjects. Journal of Orthopaedic and Sports Physical Therapy. 1989;10(9):366–369. doi: 10.2519/jospt.1989.10.9.366. [DOI] [PubMed] [Google Scholar]
- 4.Zatzick DF, Grossman DC, Russo J, et al. Predicting posttraumatic stress symptoms longitudinally in a representative sample of hospitalized injured adolescents. Journal of the American Academy of Child and Adolescent Psychiatry. 2006;45(10):1188–1195. doi: 10.1097/01.chi.0000231975.21096.45. [DOI] [PubMed] [Google Scholar]
- 5.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics. 2003;33, supplement:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 6.Breslau N. Epidemiologic studies of trauma, posttraumatic stress disorder, and other psychiatric disorders. Canadian Journal of Psychiatry. 2002;47(10):923–929. doi: 10.1177/070674370204701003. [DOI] [PubMed] [Google Scholar]
- 7.Weaver ICG, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nature Neuroscience. 2004;7(8):847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 8.Liu D, Diorio J, Tannenbaum B, et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277(5332):1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez MM. The impact of early adverse care on HPA axis development: nonhuman primate models. Hormones and Behavior. 2006;50(4):623–631. doi: 10.1016/j.yhbeh.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Fumagalli F, Molteni R, Racagni G, Riva MA. Stress during development: impact on neuroplasticity and relevance to psychopathology. Progress in Neurobiology. 2007;81(4):197–217. doi: 10.1016/j.pneurobio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 11.McGowan PO, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience. 2009;12(3):342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3(2):97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- 13.Roth TL, Zoladz PR, Sweatt JD, Diamond DM. Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. Journal of Psychiatric Research. 2011;45(7):919–926. doi: 10.1016/j.jpsychires.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koshibu K, Gräff J, Mansuy IM. Nuclear protein phosphatase-1: an epigenetic regulator of fear memory and amygdala long-term potentiation. Neuroscience. 2011;173:30–36. doi: 10.1016/j.neuroscience.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 15.Tian F, Marini AM, Lipsky RH. Effects of histone deacetylase inhibitor Trichostatin A on epigenetic changes and transcriptional activation of Bdnf promoter 1 by rat hippocampal neurons. Annals of the New York Academy of Sciences. 2010;1199:186–193. doi: 10.1111/j.1749-6632.2009.05175.x. [DOI] [PubMed] [Google Scholar]
- 16.Vanderbilt-Adriance E, Shaw DS. Neighborhood risk and the development of resilience. Annals of the New York Academy of Sciences. 2006;1094:359–362. doi: 10.1196/annals.1376.050. [DOI] [PubMed] [Google Scholar]
- 17.Vanderbilt-Adriance E, Shaw DS. Conceptualizing and re-evaluating resilience across levels of risk, time, and domains of competence. Clinical Child and Family Psychology Review. 2008;11(1-2):30–58. doi: 10.1007/s10567-008-0031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uddin M, Aiello AE, Wildman DE, et al. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(20):9470–9475. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ressler KJ, Mercer KB, Bradley B, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470(7335):492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill J, Luckenbaugh D, Charney D, Vythilingam M. Sustained elevation of serum interleukin-6 and relative insensitivity to hydrocortisone differentiates posttraumatic stress disorder with and without depression. Biological Psychiatry. 2010;68(11):999–1006. doi: 10.1016/j.biopsych.2010.07.033. [DOI] [PubMed] [Google Scholar]
- 21.de Kloet CS, Vermetten E, Rademaker AR, Geuze E, Westenberg HGM. Neuroendocrine and immune responses to a cognitive stress challenge in veterans with and without PTSD. European Journal of Psychotraumatology. 2012;2012(article 3) doi: 10.3402/ejpt.v3i0.16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koenen KC, Uddin M, Chang SC, et al. SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder. Depression and Anxiety. 2011;28(8):639–647. doi: 10.1002/da.20825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith AK, Conneely KN, Kilaru V, et al. Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. American Journal of Medical Genetics B. 2011;156(6):700–708. doi: 10.1002/ajmg.b.31212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uddin M, Galea S, Chang SC, et al. Gene expression and methylation signatures of MAN2C1 are associated with PTSD. Disease Markers. 2011;30(2-3):111–121. doi: 10.3233/DMA-2011-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rusiecki JA, Chen L, Srikantan V, et al. DNA methylation in repetitive elements and post-traumatic stress disorder: a case-control study of US military service members. Epigenomics. 2012;4(1):29–40. doi: 10.2217/epi.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zieker J, Zieker D, Jatzko A, et al. Differential gene expression in peripheral blood of patients suffering from post-traumatic stress disorder. Molecular Psychiatry. 2007;12(2):116–118. doi: 10.1038/sj.mp.4001905. [DOI] [PubMed] [Google Scholar]
- 27.Yehuda R, Cai G, Golier JA, et al. Gene expression patterns associated with posttraumatic stress disorder following exposure to the world trade center attacks. Biological Psychiatry. 2009;66(7):708–711. doi: 10.1016/j.biopsych.2009.02.034. [DOI] [PubMed] [Google Scholar]
- 28.Neylan TC, Sun B, Rempel H, et al. Suppressed monocyte gene expression profile in men versus women with PTSD. Brain, Behavior, and Immunity. 2011;25(3):524–531. doi: 10.1016/j.bbi.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarapas C, Cai G, Bierer LM, et al. Genetic markers for PTSD risk and resilience among survivors of the world trade center attacks. Disease Markers. 2011;30(2-3):101–110. doi: 10.3233/DMA-2011-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta D, Gonik M, Klengel T, et al. Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from endocrine and gene expression studies. Archives of General Psychiatry. 2011;68(9):901–910. doi: 10.1001/archgenpsychiatry.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pace TW, Wingenfeld K, Schmidt I, Meinlschmidt G, Hellhammer DH, Heim CM. Increased peripheral NF-κB pathway activity in women with childhood abuse-related posttraumatic stress disorder. Brain, Behavior, and Immunity. 2012;26(1):13–17. doi: 10.1016/j.bbi.2011.07.232. [DOI] [PubMed] [Google Scholar]
- 32.van Zuiden M, Geuze E, Willemen HL, et al. Glucocorticoid receptor pathway components predict posttraumatic stress disorder symptom development: a prospective study. Biological Psychiatry. 2012;71(4):309–316. doi: 10.1016/j.biopsych.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 33.van Zuiden M, Heijnen CJ, Maas M, et al. Glucocorticoid sensitivity of leukocytes predicts PTSD, depressive and fatigue symptoms after military deployment: a prospective study. Psychoneuroendocrinology. 2012;37(11):1822–1836. doi: 10.1016/j.psyneuen.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Matić G, Milutinović DV, Nestorov J, et al. Lymphocyte glucocorticoid receptor expression level and hormone-binding properties differ between war trauma-exposed men with and without PTSD. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2013;43:238–245. doi: 10.1016/j.pnpbp.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Delahanty DL, Nugent NR, Christopher NC, Walsh M. Initial urinary epinephrine and cortisol levels predict acute PTSD symptoms in child trauma victims. Psychoneuroendocrinology. 2005;30(2):121–128. doi: 10.1016/j.psyneuen.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Ostrowski SA, Christopher NC, Van Dulmen MHM, Delahanty DL. Acute child and mother psychophysiological responses and subsequent PTSD symptoms following a child’s traumatic event. Journal of Traumatic Stress. 2007;20(5):677–687. doi: 10.1002/jts.20286. [DOI] [PubMed] [Google Scholar]
- 37.Pervanidou P, Kolaitis G, Charitaki S, et al. Elevated morning serum interleukin (IL)-6 or evening salivary cortisol concentrations predict posttraumatic stress disorder in children and adolescents six months after a motor vehicle accident. Psychoneuroendocrinology. 2007;32(8–10):991–999. doi: 10.1016/j.psyneuen.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Resnick HS, Yehuda R, Pitman RK, Foy DW. Effect of previous trauma on acute plasma cortisol level following rape. The American Journal of Psychiatry. 1995;152(11):1675–1677. doi: 10.1176/ajp.152.11.1675. [DOI] [PubMed] [Google Scholar]
- 39.Yehuda R, Resnick HS, Schmeidler J, Yang RK, Pitman RK. Predictors of cortisol and 3-methoxy-4-hydroxyphenylglycol responses in the acute aftermath of rape. Biological Psychiatry. 1998;43(11):855–859. doi: 10.1016/s0006-3223(97)00554-4. [DOI] [PubMed] [Google Scholar]
- 40.Delahanty DL, Raimonde AJ, Spoonster E. Initial posttraumatic urinary cortisol levels predict subsequent PTSD symptoms in motor vehicle accident victims. Biological Psychiatry. 2000;48(9):940–947. doi: 10.1016/s0006-3223(00)00896-9. [DOI] [PubMed] [Google Scholar]
- 41.Bonne O, Brandes D, Segman R, Pitman RK, Yehuda R, Shalev AY. Prospective evaluation of plasma cortisol in recent trauma survivors with posttraumatic stress disorder. Psychiatry Research. 2003;119(1-2):171–175. doi: 10.1016/s0165-1781(03)00098-2. [DOI] [PubMed] [Google Scholar]
- 42.Heinrichs M, Wagner D, Schoch W, Soravia LM, Hellhammer DH, Ehlert U. Predicting posttraumatic stress symptoms from pretraumatic risk factors: a 2-year prospective follow-up study in firefighters. The American Journal of Psychiatry. 2005;162(12):2276–2286. doi: 10.1176/appi.ajp.162.12.2276. [DOI] [PubMed] [Google Scholar]
- 43.McFarlane AC, Atchison M, Yehuda R. The acute stress response following motor vehicle accidents and its relation to PTSD. Annals of the New York Academy of Sciences. 1997;821:437–441. doi: 10.1111/j.1749-6632.1997.tb48299.x. [DOI] [PubMed] [Google Scholar]
- 44.Ehring T, Ehlers A, Cleare AJ, Glucksman E. Do acute psychological and psychobiological responses to trauma predict subsequent symptom severities of PTSD and depression? Psychiatry Research. 2008;161(1):67–75. doi: 10.1016/j.psychres.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shalev AY, Videlock EJ, Peleg T, Segman R, Pitman RK, Yehuda R. Stress hormones and post-traumatic stress disorder in civilian trauma victims: a longitudinal study—part I: HPA axis responses. International Journal of Neuropsychopharmacology. 2008;11(3):365–372. doi: 10.1017/S1461145707008127. [DOI] [PubMed] [Google Scholar]
- 46.Videlock EJ, Peleg T, Segman R, Yehuda R, Pitman RK, Shalev AY. Stress hormones and post-traumatic stress disorder in civilian trauma victims: a longitudinal study—part II: the adrenergic response. International Journal of Neuropsychopharmacology. 2008;11(3):373–380. doi: 10.1017/S1461145707008139. [DOI] [PubMed] [Google Scholar]
- 47.Cohen M, Meir T, Klein E, Volpin G, Assaf M, Pollack S. Cytokine levels as potential biomarkers for predicting the development of posttraumatic stress symptoms in casualties of accidents. International Journal of Psychiatry in Medicine. 2011;42(2):117–131. doi: 10.2190/PM.42.2.b. [DOI] [PubMed] [Google Scholar]
- 48.van Zuiden M, Geuze E, Willemen HLDM, et al. Pre-existing high glucocorticoid receptor number predicting development of posttraumatic stress symptoms after military deployment. The American Journal of Psychiatry. 2011;168(1):89–96. doi: 10.1176/appi.ajp.2010.10050706. [DOI] [PubMed] [Google Scholar]
- 49.Mehta D, Binder EB. Gene x environment vulnerability factors for PTSD: the HPA-axis. Neuropharmacology. 2012;62(2):654–662. doi: 10.1016/j.neuropharm.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 50.Boscarino JA, Erlich PM, Hoffman SN, Rukstalis M, Stewart WF. Association of FKBP5, COMT and CHRNA5 polymorphisms with PTSD among outpatients at risk for PTSD. Psychiatry Research. 2011;188(1):173–174. doi: 10.1016/j.psychres.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gillespie CF, Phifer J, Bradley B, Ressler KJ. Risk and resilience: genetic and environmental influences on development of the stress response. Depression and Anxiety. 2009;26(11):984–992. doi: 10.1002/da.20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amstadter AB, Nugent NR, Yang BZ, et al. Corticotrophin-releasing hormone type 1 receptor gene (CRHR1) variants predict posttraumatic stress disorder onset and course in pediatric injury patients. Disease Markers. 2011;30(2-3):89–99. doi: 10.3233/DMA-2011-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szanton SL, Gill JM. Facilitating resilience using a society-to-cells framework: a theory of nursing essentials applied to research and practice. Advances in Nursing Science. 2010;33(4):329–343. doi: 10.1097/ANS.0b013e3181fb2ea2. [DOI] [PubMed] [Google Scholar]
- 54.Song Y, Zhou D, Guan Z, Wang X. Disturbance of serum interleukin-2 and interleukin-8 levels in posttraumatic and non-posttraumatic stress disorder earthquake survivors in Northern China. NeuroImmunoModulation. 2007;14(5):248–254. doi: 10.1159/000112050. [DOI] [PubMed] [Google Scholar]
- 55.Gill J, Vythilingam M, Page GG. Low cortisol, high DHEA, and high levels of stimulated TNF-α, and IL-6 in women with PTSD. Journal of Traumatic Stress. 2008;21(6):530–539. doi: 10.1002/jts.20372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonaccorso S, Marino V, Puzella A, et al. Increased depressive ratings in patients with hepatitis C receiving interferon-α-based immunotherapy are related to interferon-α-induced changes in the serotonergic system. Journal of Clinical Psychopharmacology. 2002;22(1):86–90. doi: 10.1097/00004714-200202000-00014. [DOI] [PubMed] [Google Scholar]
- 57.Bonanno GA, Mancini AD. The human capacity to thrive in the face of potential trauma. Pediatrics. 2008;121(2):369–375. doi: 10.1542/peds.2007-1648. [DOI] [PubMed] [Google Scholar]
- 58.Capuron L, Gumnick JF, Musselman DL, et al. Neurobehavioral effects of interferon-α in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002;26(5):643–652. doi: 10.1016/S0893-133X(01)00407-9. [DOI] [PubMed] [Google Scholar]
- 59.Capuron L, Neurauter G, Musselman DL, et al. Interferon-α-induced changes in tryptophan metabolism: relationship to depression and paroxetine treatment. Biological Psychiatry. 2003;54(9):906–914. doi: 10.1016/s0006-3223(03)00173-2. [DOI] [PubMed] [Google Scholar]
- 60.Wichers MC, Kenis G, Koek GH, Robaeys G, Nicolson NA, Maes M. Interferon-α-induced depressive symptoms are related to changes in the cytokine network but not to cortisol. Journal of Psychosomatic Research. 2007;62(2):207–214. doi: 10.1016/j.jpsychores.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 61.Wu TH, Lin CH. IL-6 mediated alterations on immobile behavior of rats in the forced swim test via ERK1/2 activation in specific brain regions. Behavioural Brain Research. 2008;193(2):183–191. doi: 10.1016/j.bbr.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 62.Maes M, Kubera M, Leunis JC. The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro endocrinology letters. 2008;29(1):117–124. [PubMed] [Google Scholar]
- 63.Campbell SJ, Deacon RMJ, Jiang Y, Ferrari C, Pitossi FJ, Anthony DC. Overexpression of IL-1β by adenoviral-mediated gene transfer in the rat brain causes a prolonged hepatic chemokine response, axonal injury and the suppression of spontaneous behaviour. Neurobiology of Disease. 2007;27(2):151–163. doi: 10.1016/j.nbd.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 64.Pierucci-Lagha A, Covault J, Bonkovsky HL, et al. A functional serotonin transporter gene polymorphism and depressive effects associated with interferon-α treatment. Psychosomatics. 2010;51(2):137–148. doi: 10.1176/appi.psy.51.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Su S, Zhao J, Bremner JD, et al. Serotonin transporter gene, depressive symptoms,and interleukin-6. Circulation: Cardiovascular Genetics. 2009;2(6):614–620. doi: 10.1161/CIRCGENETICS.109.870386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schäfer A, Scheurlen M, Seufert J, et al. Platelet serotonin (5-HT) levels in interferon-treated patients with hepatitis C and its possible association with interferon-induced depression. Journal of Hepatology. 2010;52(1):10–15. doi: 10.1016/j.jhep.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 67.Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpé S, Maes M. IDO and interferon-α-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Molecular Psychiatry. 2005;10(6):538–544. doi: 10.1038/sj.mp.4001600. [DOI] [PubMed] [Google Scholar]
- 68.Järvelä JT, Ruohonen S, Kukko-Lukjanov TK, Plysjuk A, Lopez-Picon FR, Holopainen IE. Kainic acid-induced neurodegeneration and activation of inflammatory processes in organotypic hippocampal slice cultures: treatment with cyclooxygenase-2 inhibitor does not prevent neuronal death. Neuropharmacology. 2011;60(7-8):1116–1125. doi: 10.1016/j.neuropharm.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 69.Yu JT, Lee CH, Yoo KY, et al. Maintenance of anti-inflammatory cytokines and reduction of glial activation in the ischemic hippocampal CA1 region preconditioned with lipopolysaccharide. Journal of the Neurological Sciences. 2010;296(1-2):69–78. doi: 10.1016/j.jns.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 70.Munhoz CD, García-Bueno B, Madrigal JL, Lepsch LB, Scavone C, Leza JC. Stress-induced neuroinflammation: mechanisms and new pharmacological targets. Brazilian Journal of Medical and Biological Research. 2008;41(12):1037–1046. doi: 10.1590/s0100-879x2008001200001. [DOI] [PubMed] [Google Scholar]
- 71.Bryant RA, Marosszeky JE, Crooks J, Gurka JA. Elevated resting heart rate as a predictor of posttraumatic stress disorder after severe traumatic brain injury. Psychosomatic Medicine. 2004;66(5):760–761. doi: 10.1097/01.psy.0000138121.13198.84. [DOI] [PubMed] [Google Scholar]
- 72.Bryant RA, Harvey AG, Guthrie RM, Moulds ML. A prospective study of psychophysiological arousal, acute stress disorder, and posttraumatic stress disorder. Journal of Abnormal Psychology. 2000;109(2):341–344. [PubMed] [Google Scholar]
- 73.Krystal JH, Neumeister A. Noradrenergic and serotonergic mechanisms in the neurobiology of posttraumatic stress disorder and resilience. Brain Research. 2009;1293:13–23. doi: 10.1016/j.brainres.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Walker EA, Katon W, Russo J, Ciechanowski P, Newman E, Wagner AW. Health care costs associated with posttraumatic stress disorder symptoms in women. Archives of General Psychiatry. 2003;60(4):369–374. doi: 10.1001/archpsyc.60.4.369. [DOI] [PubMed] [Google Scholar]
- 75.Marciniak MD, Lage MJ, Dunayevich E, et al. The cost of treating anxiety: the medical and demographic correlates that impact total medical costs. Depression and Anxiety. 2005;21(4):178–184. doi: 10.1002/da.20074. [DOI] [PubMed] [Google Scholar]
- 76.Priebe S, Gavrilovic JJ, Matanov A, et al. Treatment outcomes and costs at specialized centers for the treatment of PTSD after the war in former Yugoslavia. Psychiatric Services. 2010;61(6):598–604. doi: 10.1176/ps.2010.61.6.598. [DOI] [PubMed] [Google Scholar]
- 77.O’Donnell ML, Creamer M, Elliott P, Atkin C. Health costs following motor vehicle accidents: the role of posttraumatic stress disorder. Journal of Traumatic Stress. 2005;18(5):557–561. doi: 10.1002/jts.20064. [DOI] [PubMed] [Google Scholar]
- 78.Kimerling R, Alvarez J, Pavao J, MacK KP, Smith MW, Baumrind N. Unemployment among women: examining the relationship of physical and psychological intimate partner violence and posttraumatic stress disorder. Journal of Interpersonal Violence. 2009;24(3):450–463. doi: 10.1177/0886260508317191. [DOI] [PubMed] [Google Scholar]
- 79.Nandi A, Galea S, Tracy M, et al. Job loss, unemployment, work stress, job satisfaction, and the persistence of posttraumatic stress disorder one year after the september 11 attacks. Journal of Occupational and Environmental Medicine. 2004;46(10):1057–1064. doi: 10.1097/01.jom.0000141663.22902.0a. [DOI] [PubMed] [Google Scholar]
- 80.Wilcox HC, Storr CL, Breslau N. Posttraumatic stress disorder and suicide attempts in a community sample of urban American young adults. Archives of General Psychiatry. 2009;66(3):305–311. doi: 10.1001/archgenpsychiatry.2008.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stein DJ, Chiu WT, Hwang I, et al. Cross-national analysis of the associations between traumatic events and suicidal behavior: findings from the WHO world mental health surveys. PloS ONE. 2010;5(5) doi: 10.1371/journal.pone.0010574.e10574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gradus JL, Qin P, Lincoln AK, et al. Posttraumatic stress disorder and completed suicide. The American Journal of Epidemiology. 2010;171(6):721–727. doi: 10.1093/aje/kwp456. [DOI] [PubMed] [Google Scholar]
- 83.Cougle JR, Resnick H, Kilpatrick DG. PTSD, depression, and their comorbidity in relation to suicidality: cross-sectional and prospective analyses of a national probability sample of women. Depression and Anxiety. 2009;26(12):1151–1157. doi: 10.1002/da.20621. [DOI] [PubMed] [Google Scholar]
- 84.Vyrostek SB, Annest JL, Ryan GW. Surveillance for fatal and nonfatal injuries—United States, 2001. Morbidity and Mortality Weekly Report: Surveillance Summaries. 2004;53(7):1–57. [PubMed] [Google Scholar]
- 85.Zatzick D, Jurkovich GJ, Rivara FP, et al. A national US study of posttraumatic stress disorder, depression, and work and functional outcomes after hospitalization for traumatic injury. Annals of Surgery. 2008;248(3):429–437. doi: 10.1097/SLA.0b013e318185a6b8. [DOI] [PubMed] [Google Scholar]
- 86.Breslau N. Trauma and mental health in US inner-city populations. General Hospital Psychiatry. 2009;31(6):501–502. doi: 10.1016/j.genhosppsych.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 87.Zatzick D, Russo J, Grossman DC, et al. Posttraumatic stress and depressive symptoms, alcohol use, and recurrent traumatic life events in a representative sample of hospitalized injured adolescents and their parents. Journal of Pediatric Psychology. 2006;31(4):377–387. doi: 10.1093/jpepsy/jsj056. [DOI] [PubMed] [Google Scholar]