

Abstract
More than 65 loci, encoding up to 500 different genes, have been implicated by genome-wide association studies (GWAS) as conferring an increased risk of developing type 2 diabetes (T2D). Whilst mouse models have in the past been central to understanding the mechanisms through which more penetrant risk genes for T2D, for example, those responsible for neonatal or maturity-onset diabetes of the young, only a few of those identified by GWAS, notably TCF7L2 and ZnT8/SLC30A8, have to date been examined in mouse models. We discuss here the animal models available for the latter genes and provide perspectives for future, higher throughput approaches towards efficiently mining the information provided by human genetics.
1. Introduction
The estimated global prevalence for diabetes in 2011 was 366 million, and the disease is expected to affect 552 million people by 2030 (Diabetes U.K. figures; [1] accessed 09/01/13). Type 2 diabetes (T2D) is a complex and multifactorial disease characterised by impaired insulin secretion and insulin resistance. Disease risk/progression is determined by a combination of genetic and environmental factors. It has been consistently demonstrated that lifestyle factors are associated with risk of T2D across populations [2–8], with increased adiposity being the greatest modifiable risk factor for the disease [9, 10]. Inactivity [3, 11], “bad” diet [2, 6, 8, 12–14], smoking, and other vices [8, 15, 16] and the nutritional environment during pre- and postnatal life [17] also contribute to the risk for developing diabetes.
It has been estimated that 30–70% of T2D risk may be due to genetics [18]. Whilst pedigree-based linkage analysis and the candidate gene approach led to the discovery of highly penetrant genetic defects which account for the development of diabetes [19–24], it is the advent of large scale genome-wide association studies (GWAS) which have led to the accelerated discovery of risk-variants associated with T2D [25–34]. Currently, over 60 common risk variants have been identified [30–34], with a combined disease risk of 5–10% [34, 35], suggesting the existence of many more as yet undiscovered loci [34, 36, 37]. Most of the GWAS-identified associations for T2D have high linkage disequilibrium with a causal variant with a small effect size; the largest common variant-signal identified to date is that for TCF7L2, which has a per allelle odds ratio of 1.35 [27–29].
Most of the common variant signals identified by GWAS are associated with defective pancreatic islet function, indicating that this is the primary driver for the development of T2D [34, 38]. However, most of the GWAS signals map to noncoding regions of the genome, making it difficult to establish functional links to specific transcripts. As a result, determination both of (a) the identity of the likely transcript(s) involved and (b) the mechanisms of actions on disease risk, require the use of genetically tractable organisms where the expression of candidate genes can be manipulated at will in a cell type-specific manner. Of the available models (which include lower organisms such as C. elegans, D. melanogaster, etc.), mice arguably represent the best compromise between ease of genetic manipulation and similarity to man, in terms of both genome structure and physiology. In this review, we discuss the use of mouse models to study the contribution of genetic variations, identified by GWAS, in the TCF7L2 and SLC30A8 genes to the development of T2D via their effects on pancreatic islet function.
2. TCF7L2
2.1. Background
The gene-encoding Transcription 7 Like-2 (TCF7L2, previously called TCF4) is the most important T2D susceptibility gene identified to date, with genetic variants strongly associated with diabetes in all major racial groups [27–29, 39–59]. Signals in this locus are the most consistently identified across various GWAS and are associated with the highest elevation of risk of developing adult-onset T2D. Each copy of the risk T-allele at rs7903146 has an increased odds ratio for T2D of 1.4-1.5 [60]. Inheritance of the risk allele is also a useful predictor for the likelihood of conversion from a state of prediabetes to T2D [61, 62]. Additionally, results from a small number of studies also indicate that TCF7L2 variation may play an important role in cases of early onset T2D [63, 64].
TCF7L2 is a member of the TCF family of transcription factors involved in the control of cell growth and signalling downstream of wingless-type MMTV integration site family (Wnt) receptors [65]. Activation of the Wnt pathway leads to release of β-catenin from an inhibitory complex and its translocation to the nucleus, where it binds TCF7L2 and other related TCF factors [66]. The function of this transcriptional complex is context dependent; that is it may act as either a transcriptional activator or repressor [66].
In recent years, the product of the TCF7L2 gene has been associated with dysregulated pancreatic β cell function and T2D [25, 27, 28]. Enhanced Wnt signalling has been shown to lead to proliferation of islets [67] and the pancreatic epithelium [68]. Whilst loss of β-catenin signalling has been shown to lead to pancreatic hypoplasia [69], stabilisation of β-catenin has been shown to result in the formation of large pancreatic tumours [70].
Individuals carrying the risk alleles of rs7903146 in the TCF7L2 gene display lowered insulin secretion [61, 71, 72], impaired insulin processing [71], and decreased sensitivity to the incretin glucagon-like peptide 1 (GLP-1) [72, 73] compared to controls. TCF7L2 message levels were elevated in T2D patients [72, 74], whilst TCF7L2 protein content was depressed [75]. The decrease in protein content was associated with downregulation of GLP-1 and gastric inhibitory peptide (GIP) receptor expression and impaired pancreatic β cell function [74, 76, 77]. Studies have shown that silencing of Tcf7l2 gene expression in clonal mouse β cell lines [76] and primary islets [75] leads to increased apoptosis [75] and impaired β cell function [19, 20]. Gene expression analysis following Tcf7l2 silencing revealed changes in the expression of a number of genes in mouse pancreatic islets [76], one of which was Glp1r [73, 78]. TCF7L2 may mediate GLP-1-induced β cell proliferation through activation of the Wnt signalling pathway [79]. Since GLP-1 is implicated in β cell survival, the increased incidence of apoptosis in TCF7L2-silenced islets [74, 75] and in individuals carrying the variants of TCF7L2 [73] is consistent with lowered GLP-1 signalling [73, 78]. Correspondingly, the diminished insulinotropic effect of GLP-1 in Tcf7l2-silenced islets may be due, at least in part, to the lack of cognate receptors on the cell surface [74].
2.2. Mouse Models for TCF7L2
2.2.1. Whole Body Knockout Model
Prior to its association with T2D, TCF7L2 was previously best known for its association with cancer development [80–82]. Homozygous Tcf7l2 knockout (Tcf7l2 −/−) mice die shortly after birth, with a lack of stem cells in their intestinal crypts [83]. Newborn Tcf7l2 −/− mice have reduced body weight with significantly lower blood glucose 3 h postpartum than control littermates, which is not caused by excessive insulin release but by impaired carbohydrate and lipid metabolism in the newborn liver [84].
Heterozygote Tcf7l2 +/− mice display >20% decrease in body weight compared to wild-type littermates, with decreased glucose, insulin, fatty acid, triglyceride, and cholesterol in adult mice [84]. Tcf7l2 +/− mice displayed increased insulin sensitivity, improved glucose tolerance, and reduced hepatic glucose output [84, 85]. Improved glucose tolerance was also observed in heterozygote null mice generated using zinc finger nucleases [85] and insertion of a loxP site and FRT-flanked neomycin selection cassette within intron 4 and a loxP site within intron 5 [86], with data from the latter study also pointing to reduced lipogenesis and hepatic triglyceride levels and decreased peripheral fat deposition following exposure to a high fat diet in heterozygote mice compared to control littermates.
Pancreatic development is grossly normal in Tcf7l2 −/− mice [83, 84]. This observation and a report suggesting that TCF7L2 was not expressed in the pancreas [87] led to the proposal that the principle defect underlying decreased insulin production in TC- or TT-bearing individuals may be inadequate production of GLP-1, from gut L-cells [88]. However, evidence for differences in GLP-1 level in individuals with the common versus the at-risk TCF7L2 allele is currently absent [89], and patient studies have indicated that the primary defect lies in pancreatic β cells [71, 72, 75]. For this reason, mouse models which allow Tcf7l2 gene expression to be selectively ablated in the islet were required.
2.2.2. Pancreas Knockout Model
We used the Pdx1 promoter-driven Cre recombinase (PDX1.Cre) deleter strain [90] to effect deletion in all cells of pancreatic lineage in transgenic mice with a floxed Tcf7l2 exon 1 to address the question whether selective deletion of Tcf7l2 in pancreas impairs or improves glucose homeostasis and insulin secretion [77]. This approach allowed us to detect the potential effects of Tcf7l2 deletion early in pancreatic development, as TCF7L2 has previously been shown to regulate cell proliferation during development: the Tcf7l2 −/− mouse exhibited defects in the accumulation of stem cells in the intestinal crypt [83]. This approach also offered an advantage over the use of the commonly deployed rat insulin 2-promoter-driven Cre recombinase (RIP2.Cre) deleter strain since the latter also leads to deletion in the central nervous system [91–93]. Pancreas-specific Tcf7l2 −/− (pTcf7l2) mice showed age-dependent glucose intolerance by 20 weeks of age when challenged with an intraperitoneal glucose bolus [77]. Glucose intolerance was detected from 12 weeks of age when glucose was administered by the oral route, indicating that the incretin response was impaired [77]. Tolerance to glucose introduced by both the oral and intraperitoneal route was exacerbated in pTcf7l2 mice that were exposed to a high fat diet, with a concomitant decrease in β cell mass [77]. The latter observation is consistent with observations by Shu and colleagues in high-fat-fed rats [94], where the authors found a correlation of Tcf7l2 expression and β cell regeneration from pancreatic ductal cells and may reflect the inability of β cells to proliferate or regenerate from progenitor cells in the absence of functional Tcf7l2. The decreased expression of the cyclin D1 gene [77] from islets of Langerhans extracted from 20-week-old pTcf7l2 mice may contribute towards the lack of cell proliferation.
pTcf7l2 −/− islets displayed impaired glucose and GLP-1-stimulated insulin secretion and decreased expression of the gene encoding for the GLP-1 receptor [77], consistent with in vitro human and mouse islet and cell line siRNA-mediated-silencing experiments [74–76]. Whilst the PDX1.Cre strain is likely to result in deletion in other (non-β) cell types [95], we observed no difference in plasma glucagon and GLP-1 levels and in insulin sensitivity in pTcf7l2 mice [77]. Our preliminary data obtained using a more β cell selective deleter strain (Ins1.Cre; J. Ferrer, B. Thorens, unpublished) also indicate deficiencies in insulin secretion and glucose tolerance, suggesting that TCF7L2 plays a critical and cell autonomous role in the β cell compartment.
2.2.3. β Cell Knockout Model
Recently, Boj and colleagues generated a β cell Tcf7l2 knockout (βTCF4KO) mouse using the tamoxifen inducible RIP2.Cre-ERT2 deleter strain [96] bred against a conditional mouse-bearing Tcf7l2 alleles with a floxed exon 10 [84]. Although the use of the RIP2.Cre-ERT2 may affect metabolic phenotype through the expression of Cre recombinase in islet cells as well as in hypothalamic neurons [95], it is unclear whether Tcf7l2 expression was affected in the hypothalamus of βTCF4KO mice.
βTCF4KO mice on normal or high fat diet displayed normal glucose tolerance when glucose was introduced by the intraperitoneal route [84]. There was no difference in plasma insulin and insulin release from isolated islets of βTCF4KO mice, versus control littermates, in response to glucose challenge [84]. Importantly, however, mice were not examined beyond 12 weeks of age, and oral glucose tolerances were not reported in this later study.
2.2.4. Transgenic Models
In the three previous mouse models described in this section, Tcf7l2 gene expression was ablated either constitutively [83] or specifically in islet cells [77, 84]. Savic and colleagues took a different approach whereby they engineered mice that expressed LacZ under the control of human bacterial artificial chromosomes (BACs) containing the genomic interval encompassing the diabetes associated SNPs (which are intronic) for TCF7L2 [85]. Using this technique, they demonstrated the presence of enhancer function in the SNPs-containing region which drives expression in, for example, intestine and pancreas, but not in adult islets [85]. Transgenic mice with Tcf7l2 overexpression driven by the human BAC sequence exhibited glucose intolerance when placed on a high fat diet [85]. These data are consistent with that presented by Gaulton and colleagues [103] indicating that the chromatin of the TCF7L2 intronic variant is in an islet-specific “open” conformation, and reporter assays demonstrated increased enhancer activity of the at-risk T-allelle compared with the C-allelle in β cell lines.
The discrepancy in data between the various mouse models could be partly due to the involvement of TCF7L2 in glucose homeostasis in more than one tissue, and at different times during development. The Tcf7l2 gene was manipulated in different ways in the various experimental models and this may alter the tissue-specific splicing of the gene [104–107]. The expression of different variants may lead to different outcomes in different tissue types [104–107]. Analysis of glucose homeostasis at different time points during the life time of the animals and exposure to differing amounts of time to diets with different fat composition could all contribute to the differences in observations.
3. ZnT8 (SLC30A8)
3.1. Background
ZnT8 (encoding by the SLC30A8 gene) is a member of the zinc transporter family (ZnTs) important for extruding zinc from the cytosol into either the extracellular space or intracellular organelles [108]. In particular, the expression of ZnT8 is largely (but not exclusively) restricted to α and β cells of the islets of Langerhans, where the mature protein resides chiefly on the limiting membrane of dense core secretory granule [97, 109, 110]. Its function thus, appears to be chiefly to transport Zn2+ from the cytosol into the granules where, in beta cells, this is required for insulin crystallisation [111]. By contrast, the role of Zn2+ in glucagon storage in the pancreatic alpha cell granule is not fully understood.
From the discovery that a single nucleotide polymorphism in the SLC30A8 gene leads to an increased risk of developing T2D [27, 29], much work has been done to elucidate the function of the encoded protein and the role that ZnT8 plays in the pathogenesis of the disease. In contrast with the majority of GWAS-identified polymorphisms, rs13266634 in the SLC30A8 gene encodes the replacement of Trp for Arg at position 325 (R325W) at the C-terminus of the protein and is associated with a ~20% increased risk of developing T2D per allele [28]. Given the highly restricted expression pattern of the transporter, hopes have been raised that ZnT8 may provide an exciting new drug target to enhance insulin release in diabetic patients.
3.2. Mouse Models Exploring ZnT8/SLC30A8 Function
A number of mouse models have been generated in order to elucidate the function of this molecule and its role in the pathogenesis of diabetes. These include whole body [97, 99–101] and cell type-specific (α or β cell) [102] ZnT8 knockout animals. Systemic ZnT8 knockout models have up to now been investigated by three different groups [97, 99–101]. These studies have revealed gross abnormalities (albeit age and gender dependent) in insulin crystallisation and storage [97, 99] (Figure 1(a)), confirming the importance of ZnT8 in granular zinc accumulation. Nonetheless, significant differences were apparent both in terms of the regulation of insulin secretion and whole body glucose homeostasis. These differences are likely the result of subtle differences in genetic background, gender, and the age of the animals. Local environmental factors including diet and gut microbiome may also play a role [112]. Thus, glucose tolerance was found to be impaired at an early age (4–6 weeks of age) in three of these studies but not at an older age (>18 weeks) [97, 99, 100], suggesting that the penetrance of the phenotype decreases with age. While insulin sensitivity was unaltered in all of the studies, defects in insulin secretion were reported in two of the studies [97, 100]. None of these changes was associated with altered beta cell mass (Figures 1(b) and 1(c)) These data support the view that decreased ZnT8 activity is likely to influence glucose homeostasis in man and may underlie the defects which increase the risk of developing T2D. Differences in the phenotype are summarised in Table 1.
Figure 1.
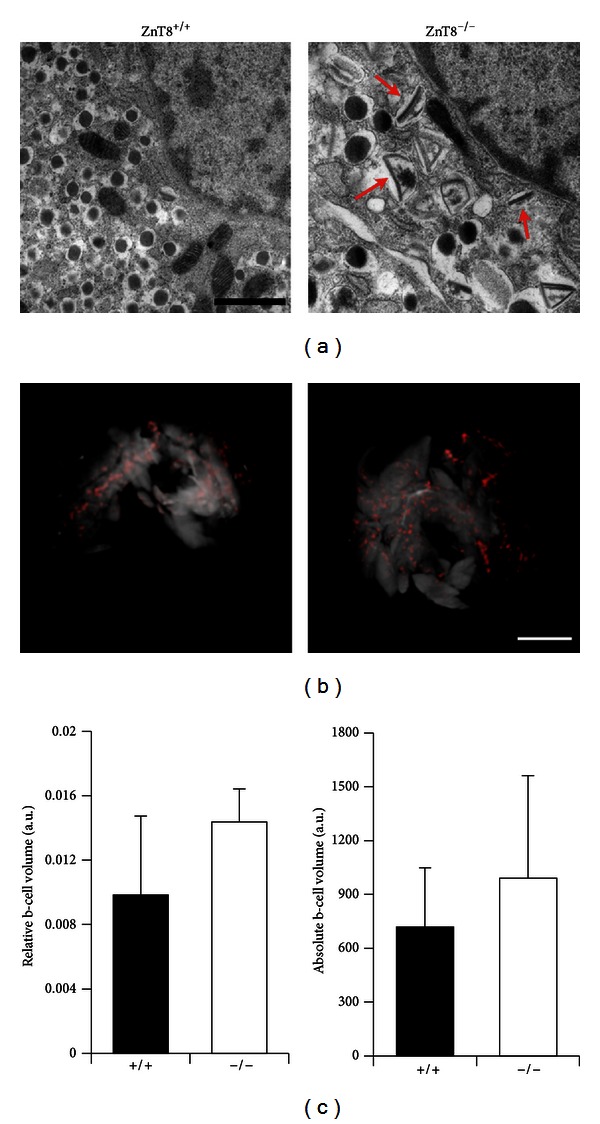
Electron Micrographs and Optical Projection Tomography (OPT) in ZnT8+/+ and ZnT8−/− mice. (a) Transmission electron microscopy images of isolated islets from ZnT8+/+ and ZnT8−/− male mice at high magnification (scale bar 1 mm) reveals the appearance of rod-shaped core granules in ZnT8−/− cells, indicated by red arrows (n = 3 mice). Sections were cut and images were acquired by Dr. Raffaella Carzaniga and Ms. Katrin Kronenberger. (b) Representative three-dimensional OPT projections of whole fixed and permeabilised pancreas from ZnT8−/− and ZnT8+/+ mice. In red are the insulin positive structures (β cells). The overall shape of the whole pancreas was visualized as autofluorescence and is apparent as white/grey shading. Scale bar = 1 cm. (c) Relative (right panel) and absolute (left panel) β-cell volume (n = 2 pancreata per genotype).
Table 1.
Summary of the major phenotype of the different colonies of ZnT8 KO mice. ZnT8-αKO and ZnT8-βKO for α and β-cell-specific knockout mice, respectively; GSIS for glucose-stimulated insulin secretion.
| Phenotype/model | ZnT8KO-London [97] |
ZnT8KO- Toronto [98] |
ZnT8KO- Leuven [99] |
ZnT8KO- Vanderbilt [100] (129SvEvBrd × C57BL/6J) |
ZnT8KO-Vanderbilt [101] (C57BL/6J) | ZnT8-αKO | ZnT8-βKO [102] |
|---|---|---|---|---|---|---|---|
| Glucose tolerance | |||||||
| ≤6 weeks | ♂ intolerant | ♂ intolerant ♀ intolerant |
Normal | ♂ intolerant | Normal | Intolerant | |
| 12 weeks | ♂ intolerant ♀ normal |
♂ normal ♀ intolerant |
Normal | ||||
| ≥18 weeks | Normal | Normal | Normal | ||||
|
| |||||||
| Insulin sensitivity | Normal | Normal | Normal | Normal | Normal | ||
|
| |||||||
| Plasma glucose | ♂: Elevated (fasting) at 6 wks, normal afterwards. ♀: Normal |
Normal | Normal | Normal (fasting) | |||
|
| |||||||
| Plasma insulin | Decreased | Normal | Decreased | Normal (fasting) | Normal. (Plasma glucagon normal.) |
Normal | |
|
| |||||||
| Islet insulin content | Normal (glucagon content was normal.) |
Normal | Normal | ||||
|
| |||||||
| Insulin secretion | |||||||
| In vivo | Reduced | ||||||
| In vitro | Basal secretion enhanced GSIS normal |
GSIS enhanced | GSIS normal | GSIS reduced | GSIS normal | Reduced first phase | |
|
| |||||||
| Glucagon secretion | Unaffected | ||||||
|
| |||||||
| Insulin processing | Normal | Normal | |||||
|
| |||||||
| Granule morphology | Abnormal | Abnormal | Abnormal | Normal | Normal | Abnormal | |
Of note, the two recent studies of Pound et al. stress the importance of the genetic background. In the first [100], the genetically modified animals were maintained on a mixed background, while in the second [101], the mice were backcrossed onto a pure C57BL/6 background. Strikingly, whereas glucose-stimulated insulin secretion was unaltered in islets from ZnT8 knockout mice on a pure C57BL/6 background, islets from mice on the mixed background showed clear abnormalities in this parameter. Again, plasma insulin was decreased in the mixed background animals, while it was found normal in mice on a pure background. Since these mice were generated and kept in the same animal facility, it seems reasonable to exclude environmental differences as playing a role. Instead, these data support the view that background is a critical determinant of the penetrance of null ZnT8 alleles. Whether this impacts the preservation of functional β cell mass in the face of differing insulin sensitivities between the strains, altered intracellular Zn2+ handling, or defective auto/paracrine Zn2+ signalling between islets cells remains to be elucidated.
As mentioned above, ZnT8 is present in both α and β cells such that the systemic knockout model reflects the impact of deletion from both cell types (and perhaps others where ZnT8 is expressed at low but detectable levels). The generation of cell-specific knockout models has therefore helped in understanding the contribution of each cell type to the overall phenotype observed. Wijesekara et al. described both the animal models in a recent paper [102]. Deletion of ZnT8 selectively from β cells (βZnT8 null mice; using the RIP2 promoter) led to similar effects on glucose homeostasis as those observed in the systemic knockout developed by the same group and by ourselves [97], confirming that the transporter is required for proper insulin processing, crystallization, and packaging. However, βZnT8 null mice displayed additional abnormalities in the expression of key genes required for normal glucose sensing in β cells. Whilst the underlying reasons for this greater penetrance are unclear, it is possible that changes in intracellular free Zn2+ levels are more marked in the β cell selective model since the α cell complement remains as an efficient sink for Zn2+ release, thus, more efficiently depleting β cell Zn2+. Nonetheless, the broad similarities between ZnT8 whole body knockout and the β-cell-specific mouse model suggest that the phenotype of the former is primarily a consequence of ZnT8 deletion in β and not α cells; α cell-selective ZnT8 null mouse displayed unaltered glucose tolerance. However, glucagon secretion was not measured in these animals under conditions where the latter is likely to be physiologically important that is hypoglycaemia. It therefore remains possible that ZnT8 plays a significant role in the α cell, a possibility which awaits more detailed examination of α cell-selective null mice in the future.
Because T2D is a polygenic disease that is also influenced by environmental factors, it is important to mention new studies where ZnT8 knockout animals were maintained on a high fat content diet (HFD) [98, 99]. In both of these studies, ZnT8 null mice displayed an increase in body weight as well as fasting blood glucose and insulin levels compared to wild type controls. In particular, in one of these studies, 50% of the ZnT8 knockout animals became hyperglycemic after exposure to HFD, while none of the controls did so [99]. Each of these studies was performed using animals on a mixed (sv129/C57BL6) background. On the other hand, a further recent study using animals backcrossed onto a C57BL6 background [101] revealed that ZnT8 null animals were protected against the effects of high fat, again stressing the likely importance of modifier genes in determining the final penetrance of the effect. Although it is difficult to provide a straightforward rationalisation for these differences, it is noteworthy that sv129 mice are more insulin sensitive than C57BL6 animals [113], with the latter producing more insulin in hyperglycemic clamps. It is possible, therefore, that C57BL6 mice are better equipped to tolerate perturbations in insulin storage and secretion following ZnT8 deletion.
These data reinforce the idea that mice, at least, are able to adapt metabolically to the loss of ZnT8 alleles under many circumstances. However, under metabolic stress, such as in the case of a diet rich in fat, the impact of defective insulin storage and or secretion are more apparent at least for mice on a mixed genetic background.
Whilst complete inactivation of ZnT8 in the mouse has been useful as a means of understanding the function of this protein, it is clear that more work needs to be done in order to elucidate the significance of the diabetes-associated polymorphism in vivo. At present, knock-in models for either the protective (W325) or risk (R325) forms of ZnT8 are missing and may be revealing, provided that the impact on transporter activity is sufficiently large [97]. Of note, such models would more closely mimic the situation in humans and help us to better understand the metabolic, signaling, and other pathways that are altered in tissues which express the transporter.
4. Perspectives
4.1. Better Mouse Models
A key point to bear in mind in assessing the usefulness of mouse models is the relative plasticity displayed by rodents faced with gene deletions. Thus, differences between the penetrance of mutations in human genes linked to monogenic forms of diabetes, including maturity onset diabetes of the young (MODY), between humans and mice, are usually observed [114] with the mouse equivalents showing far less marked disturbances in glycemia or changes which are seen only after deletion of both alleles. This clearly reflects the limitations of the use of mice (weight ~25 g, life expectancy ~3 years) for comparisons with human subjects. Nonetheless, and although the phenotypes of the above murine models are thus often more subtle than the human counterparts, they remain useful models for the study of diabetes, allowing single-targeted gene deletions which are impossible in man. For example, human populations with different genetic backgrounds have different susceptibility to the R235W ZnT8 polymorphism. We should not, therefore, find surprising the results that different genetic backgrounds and different diet reveal different phenotypes in ZnT8 knockout models.
The study of knockout mouse models is most useful if the likely target gene is clearly defined, as is the case when a SNP lies in an exon and encodes a nonsense or missense mutation (as for SLC30A8). One of the difficulties in studying the contribution of the SNPs identified for increased risk of T2D is that many of the SNPs identified to date mainly reside in intronic regions. This may be due to the technical limitations of identifying the disease-causing gene using current methods for GWAS, or that the disease-inducing variation may indeed reside in the intronic region of the gene, which may have regulatory function, as may be the case for TCF7L2 [85]. Frequently, the sequences within the SNP regions are poorly conserved between mouse and man, for example, the sequences spanning SNP rs7903146 for TCF7L2 lies within a repetitive element that is absent in mice. One possibility is to conduct physiological studies in “humanized” mice [85], but it is difficult to fully replicate the human genetic environment in mouse models. Additionally, it is technically difficult to introduce targeted changes at high efficiency at precise locations. The emergence of genome modification technologies such as transcription activator-like effector nucleases (TALENs) [115–117] can substantially speed up the making of a tailored mutant animal model for whole system approaches to study the contribution of risk variants identified by GWAS to disease progression and may be useful in those instances where the region containing the variation is sufficiently similar to that found in humans. Importantly, such gene-editing approaches may also facilitate the use of alternative species (such as the rat or even the pig) whose physiology more closely resembles that of man.
An additional complication is that disease-causing SNPs do not exist in isolation. The genetic landscape of each individual may play a part in an individual's risk of developing a certain disease. For example, the risk of T2D is additive: the larger the number of risk SNPs present in an individual's genome, the higher the risk for the development of T2D [37, 118–121]. Thus, future animal models may require careful mapping of the genetic variations present in the model animal and the introduction of more than one genetic variation to model the diabetic phenotype conferred by these combined genetic variations.
4.2. Gene-Environment Interactions
An individual's risk of developing T2D is the product of interaction between the individual's genetic constitution and the environment inhabited by the individual. Whilst the contribution of genetic factors to disease risk is relatively easy to quantify, the impact of environmental exposure is less easily measured in a clinical setting. Nevertheless, efforts have been made to study the interactions between some of the known susceptibility loci for T2D and the environment, and these findings may be useful for the development of prediction models and tailoring clinical treatment for T2D [122, 123]. For example, for carriers of the risk allele for TCF7L2, diets of low glycaemic load [124, 125] and a more intensive lifestyle modification regime (versus that recommended for nonrisk carriers) [61, 62, 126, 127] have been shown to reduce the risk of T2D. Meaningful studies for gene-environment interactions will require samples of sufficient size to increase statistical power [128] and accurate methods for measuring environmental exposure, for example, the use of metabolomics to identify and assess metabolic characteristics, changes, and phenotypes in response to the environment, diet, lifestyle, and pathophysiological states. This information will allow the generation of better risk prediction models and personalisation/stratification of treatment, the holy grail of GWAS.
4.3. Cancer versus Diabetes (Opposing Mechanisms Hypothesis)
One other observation from GWAS that should be mentioned, as it may have implications on treatment, is the link between cancer and T2D. There is epidemiological evidence that links T2D and cancer [129]. A large number of T2D genes found via GWAS are involved in cell cycle regulation [34], for example, the T2D association SNP mapping to chromosome 9p21 in the vicinity of the tumour suppressor genes CDKN2A and CDKN2B [130–132] and the CDKN2B regulator ANRIL [133–135]. Recent genetic data suggest that common genetic variants influence cancer and diabetes in opposite directions [136, 137].
4.4. Which Genes Do We Study?
A fundamental challenge facing those wishing to determine which of the genes in a particular locus is responsible for affecting disease risk, and dissect how this/these act, is the very scale of the problem (currently more than 500 genes in total to interrogate, with others emerging) [35] (and McCarthy M, personal communication). Clearly, new strategies will be required both to prioritise genes and thus develop models for those most likely to be involved: assessment of the impact of a particular variant (odds ratio) as well as expression profile (notably expression in β cells for those genes affecting insulin secretion), and finally, the likely biological impact of variations in a particular gene based on published knowledge are all essential to this process. Further, “experimental filtration” through higher throughput approaches (e.g., siRNA in β cell lines, including novel human lines [138]) are likely to be needed. Finally, more high throughput means to inactivate or overexpress genes in specific tissues in living mice without the need to engineer the latter via conventional recombination-based engineering of embryonic stem cells (e.g., through virus-mediated delivery) [139] and are likely to be increasingly important. A further challenge is that of understanding how the identified genes affect disease risk work via different tissues; systems and computational biology are likely to be highly important here.
Acknowledgments
This work is supported by Wellcome Trust Senior Investigator (WT098424AIA), Royal Society Wolfson Research Merit, MRC Programme (MR/J0003042/1), and Diabetes UK Studentship grants to Guy A. Rutter. Gabriela da Silva Xavier and Guy A. Rutter thank the European Foundation for the Study of Diabetes (EFSD) for Project grants. The work leading to this publication has also received support from the Innovative Medicines Initiative Joint Undertaking under Grant Agreement no. 155005 (IMIDIA), resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies' in kind contribution.
References
- 1.Diabetes UK. Diabetes in the UK 2012: key statistics on diabetes. 2013.
- 2.Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes Care. 2011;34:1249–1257. doi: 10.2337/dc11-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu FB, Li TY, Colditz GA, Willett WC, Manson JE. Television watching and other sedentary behaviors in relation to risk of obesity and type 2 diabetes mellitus in women. Journal of the American Medical Association. 2003;289(14):1785–1791. doi: 10.1001/jama.289.14.1785. [DOI] [PubMed] [Google Scholar]
- 4.Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. New England Journal of Medicine. 2002;346(6):393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramachandran A, Snehalatha C, Mary S, Mukesh B, Bhaskar AD, Vijay V. The Indian Diabetes Prevention Programme shows that lifestyle modification and metformin prevent type 2 diabetes in Asian Indian subjects with impaired glucose tolerance (IDPP-1) Diabetologia. 2006;49(2):289–297. doi: 10.1007/s00125-005-0097-z. [DOI] [PubMed] [Google Scholar]
- 6.Salas-Salvadó J, Bulló M, Babio N, et al. Reduction in the incidence of type 2 diabetes with the mediterranean diet: results of the PREDIMED-Reus nutrition intervention randomized trial. Diabetes Care. 2011;34(1):14–19. doi: 10.2337/dc10-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuomilehto J, Lindström J, Eriksson JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. New England Journal of Medicine. 2001;344(18):1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 8.Van Dam RM. The epidemiology of lifestyle and risk for type 2 diabetes. European Journal of Epidemiology. 2003;18(12):1115–1125. doi: 10.1023/b:ejep.0000006612.70245.24. [DOI] [PubMed] [Google Scholar]
- 9.Yach D, Stuckler D, Brownell KD. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nature Medicine. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- 10.Wilson PWF, Meigs JB, Sullivan L, Fox CS, Nathan DM, D’Agostino RB., Sr. Prediction of incident diabetes mellitus in middle-aged adults: the framingham offspring study. Archives of Internal Medicine. 2007;167(10):1068–1074. doi: 10.1001/archinte.167.10.1068. [DOI] [PubMed] [Google Scholar]
- 11.Rana JS, Li TY, Manson JE, Hu FB. Adiposity compared with physical inactivity and risk of type 2 diabetes in women. Diabetes Care. 2007;30(1):53–58. doi: 10.2337/dc06-1456. [DOI] [PubMed] [Google Scholar]
- 12.Malik VS, Popkin BM, Bray GA, Després JP, Willett WC, Hu FB. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care. 2010;33(11):2477–2483. doi: 10.2337/dc10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Risérus U, Willett WC, Hu FB. Dietary fats and prevention of type 2 diabetes. Progress in Lipid Research. 2009;48(1):44–51. doi: 10.1016/j.plipres.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barclay AW, Petocz P, McMillan-Price J, et al. Glycemic index, glycemic load, and chronic disease risk—a metaanalysis of observational studies. American Journal of Clinical Nutrition. 2008;87(3):627–637. doi: 10.1093/ajcn/87.3.627. [DOI] [PubMed] [Google Scholar]
- 15.Christakis NA, Fowler JH. The spread of obesity in a large social network over 32 years. New England Journal of Medicine. 2007;357(4):370–379. doi: 10.1056/NEJMsa066082. [DOI] [PubMed] [Google Scholar]
- 16.Willi C, Bodenmann P, Ghali WA, Faris PD, Cornuz J. Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. Journal of the American Medical Association. 2007;298(22):2654–2664. doi: 10.1001/jama.298.22.2654. [DOI] [PubMed] [Google Scholar]
- 17.Burdge GC, Lillycrop KA. Nutrition, epigenetics, and developmental plasticity: implications for understanding human disease. Annual Review of Nutrition. 2010;30:315–339. doi: 10.1146/annurev.nutr.012809.104751. [DOI] [PubMed] [Google Scholar]
- 18.Poulsen P, Ohm Kyvik K, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—a population-based twin study. Diabetologia. 1999;42(2):139–145. doi: 10.1007/s001250051131. [DOI] [PubMed] [Google Scholar]
- 19.Owen K, Hattersley AT. Maturity-onset diabetes of the young: from clinical description to molecular genetic characterization. Best Practice and Research. 2001;15(3):309–323. doi: 10.1053/beem.2001.0148. [DOI] [PubMed] [Google Scholar]
- 20.Farooqi IS, O’Rahilly S. Genetics of obesity in humans. Endocrine Reviews. 2006;27(7):710–718. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 21.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273(5281):1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 22.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genetics. 2000;26(1):76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 23.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ) Journal of Biological Chemistry. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 24.Gloyn AL, Weedon MN, Owen KR, et al. Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52(2):568–572. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 25.Grant SFA, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics. 2006;38(3):320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 26.Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 27.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in finns detects multiple susceptibility variants. Science. 2007;316(5829):1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445(7130):881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 29.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho YS, Chen CH, Hu C, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nature Genetics. 2012;44:67–72. doi: 10.1038/ng.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nature Genetics. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kooner JS, Saleheen D, Sim X, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nature Genetics. 2011;43:984–989. doi: 10.1038/ng.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer ND, McDonough CW, Hicks PJ, et al. A genome-wide association search for type 2 diabetes genes in African Americans. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0029202.e29202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nature Genetics. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nature Genetics. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornelis MC, Qi L, Zhang C, et al. Joint effects of common genetic variants on the risk for type 2 diabetes in U.S. men and women of European ancestry. Annals of Internal Medicine. 2009;150(8):541–550. doi: 10.7326/0003-4819-150-8-200904210-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Miguel-Yanes JM, Shrader P, Pencina MJ, et al. Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care. 2011;34(1):121–125. doi: 10.2337/dc10-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Florez JC. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia. 2008;51(7):1100–1110. doi: 10.1007/s00125-008-1025-9. [DOI] [PubMed] [Google Scholar]
- 39.Cauchi S, Meyre D, Dina C, et al. Transcription factor TCF7L2 genetic study in the French population: expression in human β-cells and adipose tissue and strong association with type 2 diabetes. Diabetes. 2006;55(10):2903–2908. doi: 10.2337/db06-0474. [DOI] [PubMed] [Google Scholar]
- 40.Maeda S, Osawa N, Hayashi T, Tsukada S, Kobayashi M, Kikkawa R. Genetic variations associated with diabetic nephropathy and type II diabetes in a Japanese population. Kidney International. 2007;72(106, supplement):S43–S48. doi: 10.1038/sj.ki.5002385. [DOI] [PubMed] [Google Scholar]
- 41.Meigs JB, Rutter MK, Sullivan LM, Fox CS, D’Agostino RB, Wilson PWF. Impact of insulin resistance on risk of type 2 diabetes and cardiovascular disease in people with metabolic syndrome. Diabetes Care. 2007;30(5):1219–1225. doi: 10.2337/dc06-2484. [DOI] [PubMed] [Google Scholar]
- 42.Herder C, Rathmann W, Strassburger K, et al. Variants of the PPARG, IGF2BP2, CDKAL1, HHEX, and TCF7L2 genes confer risk of type 2 diabetes independently of BMI in the German KORA studies. Hormone and Metabolic Research. 2008;40(10):722–726. doi: 10.1055/s-2008-1078730. [DOI] [PubMed] [Google Scholar]
- 43.Sanghera DK, Ortega L, Han S, et al. Impact of nine common type 2 diabetes risk polymorphisms in Asian Indian Sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTO variants confer a significant risk. BMC Medical Genetics. 2008;9, article 59 doi: 10.1186/1471-2350-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho YM, Kim TH, Lim S, et al. Type 2 diabetes-associated genetic variants discovered in the recent genome-wide association studies are related to gestational diabetes mellitus in the Korean population. Diabetologia. 2009;52(2):253–261. doi: 10.1007/s00125-008-1196-4. [DOI] [PubMed] [Google Scholar]
- 45.Tabara Y, Osawa H, Kawamoto R, et al. Replication study of candidate genes associated with type 2 diabetes based on genome-wide screening. Diabetes. 2009;58(2):493–498. doi: 10.2337/db07-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marquezine GF, Pereira AC, Sousa AGP, Mill JG, Hueb WA, Krieger JE. TCF7L2 variant genotypes and type 2 diabetes risk in Brazil: significant association, but not a significant tool for risk stratification in the general population. BMC Medical Genetics. 2008;9, article 106 doi: 10.1186/1471-2350-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ezzidi I, Mtiraoui N, Cauchi S, et al. Contribution of type 2 diabetes associated loci in the Arabic population from Tunisia: a case-control study. BMC Medical Genetics. 2009;10, article 33 doi: 10.1186/1471-2350-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takeuchi F, Serizawa M, Yamamoto K, et al. Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes. 2009;58(7):1690–1699. doi: 10.2337/db08-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ereqat S, Nasereddin A, Cauchi S, Azmi K, Abdeen Z, Amin R. Association of a common variant in TCF7L2 gene with type 2 diabetes mellitus in the Palestinian population. Acta Diabetologica. 2010;47(1, supplement):S195–S198. doi: 10.1007/s00592-009-0161-0. [DOI] [PubMed] [Google Scholar]
- 50.Wen J, Rönn T, Olsson A, et al. Investigation of type 2 diabetes risk alleles support CDKN2A/B, CDKAL1, and TCF7L2 as susceptibility genes in a Han Chinese cohort. PLoS ONE. 2010;5(2) doi: 10.1371/journal.pone.0009153.e9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chauhan G, Spurgeon CJ, Tabassum R, et al. Impact of common variants of PPARG, KCNJ11, TCF7L2, SLC30A8, HHEX, CDKN2A, IGF2BP2, and CDKAL1 on the risk of type 2 diabetes in 5,164 Indians. Diabetes. 2010;59(8):2068–2074. doi: 10.2337/db09-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karns R, Zhang G, Jeran N, et al. Replication of genetic variants from genome-wide association studies with metabolic traits in an island population of the Adriatic coast of Croatia. European Journal of Human Genetics. 2011;19(3):341–346. doi: 10.1038/ejhg.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramos E, Chen G, Shriner D, et al. Replication of genome-wide association studies (GWAS) loci for fasting plasma glucose in African-Americans. Diabetologia. 2011;54(4):783–788. doi: 10.1007/s00125-010-2002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rees SD, Hydrie MZ, Shera AS, et al. Replication of 13 genome-wide association (GWA)-validated risk variants for type 2 diabetes in Pakistani populations. Diabetologia. 2011;54:1368–1374. doi: 10.1007/s00125-011-2063-2. [DOI] [PubMed] [Google Scholar]
- 55.Saxena R, Elbers CC, Guo Y, et al. Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. American Journal of Human Genetics. 2012;90:410–425. doi: 10.1016/j.ajhg.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cauchi S, Ezzidi I, El AY, et al. European genetic variants associated with type 2 diabetes in North African Arabs. Diabetes & Metabolism. 2012;38:316–323. doi: 10.1016/j.diabet.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 57.Mtiraoui N, Turki A, Nemr R, et al. Contribution of common variants of ENPP1, IGF2BP2, KCNJ11, MLXIPL, PPARgamma, SLC30A8 and TCF7L2 to the risk of type 2 diabetes in Lebanese and Tunisian Arabs. Diabetes & Metabolism. 2012;38:444–449. doi: 10.1016/j.diabet.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 58.Turki A, Al-Zaben GS, Mtiraoui N, Marmmuoch H, Mahjoub T, Almawi WY. Transcription factor-7-like 2 gene variants are strongly associated with type 2 diabetes in Tunisian Arab subjects. Gene. 2013;513:244–248. doi: 10.1016/j.gene.2012.10.086. [DOI] [PubMed] [Google Scholar]
- 59.Long J, Edwards T, Signorello LB, et al. Evaluation of genome-wide association study-identified type 2 diabetes loci in African Americans. American Journal of Epidemiology. 2012;176:995–1001. doi: 10.1093/aje/kws176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Helgason A, Pálsson S, Thorleifsson G, et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nature Genetics. 2007;39(2):218–225. doi: 10.1038/ng1960. [DOI] [PubMed] [Google Scholar]
- 61.Florez JC, Jablonski KA, Bayley N, et al. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. New England Journal of Medicine. 2006;355(3):241–250. doi: 10.1056/NEJMoa062418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Kuusisto J, Vänttinen M, et al. Variants of transcription factor 7-like 2 (TCF7L2) gene predict conversion to type 2 diabetes in the Finnish Diabetes Prevention Study and are associated with impaired glucose regulation and impaired insulin secretion. Diabetologia. 2007;50(6):1192–1200. doi: 10.1007/s00125-007-0656-6. [DOI] [PubMed] [Google Scholar]
- 63.Dabelea D, Dolan LM, D’Agostino R, et al. Association testing of TCF7L2 polymorphisms with type 2 diabetes in multi-ethnic youth. Diabetologia. 2011;54(3):535–539. doi: 10.1007/s00125-010-1982-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raitakari OT, Rönnemaa T, Huupponen R, et al. Variation of the transcription factor 7-like 2 (TCF7L2) gene predicts impaired fasting glucose in healthy young adults. The Cardiovascular Risk in Young Finns Study. Diabetes Care. 2007;30:2299–2301. doi: 10.2337/dc07-0539. [DOI] [PubMed] [Google Scholar]
- 65.Jin T, Liu L. The Wnt signaling pathway effector TCF7L2 and type 2 diabetes mellitus. Molecular Endocrinology. 2008;22(11):2383–2392. doi: 10.1210/me.2008-0135. [DOI] [PubMed] [Google Scholar]
- 66.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 67.Rulifson IC, Karnik SK, Heiser PW, et al. Wnt signaling regulates pancreatic β cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(15):6247–6252. doi: 10.1073/pnas.0701509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Papadopoulou S, Edlund H. Attenuated Wnt signaling perturbs pancreatic growth but not pancreatic function. Diabetes. 2005;54(10):2844–2851. doi: 10.2337/diabetes.54.10.2844. [DOI] [PubMed] [Google Scholar]
- 69.Wells JM, Esni F, Boivin GP, et al. Wnt/β-catenin signaling is required for development of the exocrine pancreas. BMC Developmental Biology. 2007;7, article 4 doi: 10.1186/1471-213X-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heiser PW, Lau J, Taketo MM, Herrera PL, Hebrok M. Stabilization of β-catenin impacts pancreas growth. Development. 2006;133(10):2023–2032. doi: 10.1242/dev.02366. [DOI] [PubMed] [Google Scholar]
- 71.Loos RJF, Franks PW, Francis RW, et al. TCF7L2 polymorphisms modulate proinsulin levels and β-cell function in a British europid population. Diabetes. 2007;56(7):1943–1947. doi: 10.2337/db07-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lyssenko V, Lupi R, Marchetti P, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. Journal of Clinical Investigation. 2007;117(8):2155–2163. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Villareal DT, Robertson H, Bell GI, et al. TCF7L2 variant rs7903146 affects the risk of type 2 diabetes by modulating incretin action. Diabetes. 2010;59(2):479–485. doi: 10.2337/db09-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CHS, Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Human Molecular Genetics. 2009;18(13):2388–2399. doi: 10.1093/hmg/ddp178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shu L, Sauter NS, Schulthess FT, Matveyenko AV, Oberholzer J, Maedler K. Transcription factor 7-like 2 regulates β-cell survival and function in human pancreatic islets. Diabetes. 2008;57(3):645–653. doi: 10.2337/db07-0847. [DOI] [PubMed] [Google Scholar]
- 76.Da Silva Xavier G, Loder MK, McDonald A, et al. TCF7L2 regulates late events in insulin secretion from pancreatic islet β-cells. Diabetes. 2009;58(4):894–905. doi: 10.2337/db08-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.da Silva Xavier G, Mondragon A, Sun G, et al. Abnormal glucose tolerance and insulin secretion in pancreas-specific Tcf7l2-null mice. Diabetologia. 2012;55:2667–2676. doi: 10.1007/s00125-012-2600-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Prokunina-Olsson L, Welch C, Hansson O, et al. Tissue-specific alternative splicing of TCF7L2. Human Molecular Genetics. 2009;18(20):3795–3804. doi: 10.1093/hmg/ddp321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Z, Habener JF. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. Journal of Biological Chemistry. 2008;283(13):8723–8735. doi: 10.1074/jbc.M706105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Slattery ML, Folsom AR, Wolff R, Herrick J, Caan BJ, Potter JD. Transcription factor 7-like 2 polymorphism and colon cancer. Cancer Epidemiology Biomarkers and Prevention. 2008;17(4):978–982. doi: 10.1158/1055-9965.EPI-07-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saadeddin A, Babaei-Jadidi R, Spencer-Dene B, Nateri AS. The links between transcription, β-catenin/JNK signaling, and carcinogenesis. Molecular Cancer Research. 2009;7(8):1189–1196. doi: 10.1158/1541-7786.MCR-09-0027. [DOI] [PubMed] [Google Scholar]
- 82.Roose J, Clevers H. TCF transcription factors: molecular switches in carcinogenesis. Biochimica et Biophysica Acta. 1999;1424(2-3):M23–M37. doi: 10.1016/s0304-419x(99)00026-8. [DOI] [PubMed] [Google Scholar]
- 83.Korinek V, Barker N, Moerer P, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nature Genetics. 1998;19(4):379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 84.Boj SF, van Es JH, Huch M, et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell. 2012;151(7):1595–1607. doi: 10.1016/j.cell.2012.10.053. [DOI] [PubMed] [Google Scholar]
- 85.Savic D, Ye H, Aneas I, Park SY, Bell GI, Nobrega MA. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Research. 2011;21:1417–1425. doi: 10.1101/gr.123745.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang H, Li Q, Lee JH, Shu Y. Reduction in Tcf7l2 expression decreases diabetic susceptibility in mice. International Journal of Biological Sciences. 2012;8:791–801. doi: 10.7150/ijbs.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barker N, Huls G, Korinek V, Clevers H. Restricted high level expression of Tcf-4 protein in intestinal and mammary gland epithelium. American Journal of Pathology. 1999;154(1):29–35. doi: 10.1016/S0002-9440(10)65247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horikoshi M, Hara K, Ito C, Nagai R, Froguel P, Kadowaki T. A genetic variation of the transcription factor 7-like 2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia. 2007;50(4):747–751. doi: 10.1007/s00125-006-0588-6. [DOI] [PubMed] [Google Scholar]
- 89.Schäfer SA, Tschritter O, Machicao F, et al. Impaired glucagon-like peptide-1-induced insulin secretion in carriers of transcription factor 7-like 2 (TCF7L2) gene polymorphisms. Diabetologia. 2007;50(12):2443–2450. doi: 10.1007/s00125-007-0753-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 91.Hisadome K, Smith MA, Choudhury AI, Claret M, Withers DJ, Ashford MLJ. 5-HT inhibition of rat insulin 2 promoter Cre recombinase transgene and proopiomelanocortin neuron excitability in the mouse arcuate nucleus. Neuroscience. 2009;159(1):83–93. doi: 10.1016/j.neuroscience.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun G, Tarasov AI, McGinty JA, et al. LKB1 deletion with the RIP2.Cre transgene modifies pancreatic β-cell morphology and enhances insulin secretion in vivo. American Journal of Physiology: Endocrinology and Metabolism. 2010;298(6):E1261–E1273. doi: 10.1152/ajpendo.00100.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun G, Reynolds R, Leclerc I, Rutter GA. RIP2-mediated LKB1 deletion causes axon degeneration in the spinal cord and hind-limb paralysis. DMM Disease Models and Mechanisms. 2011;4(2):193–202. doi: 10.1242/dmm.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shu L, Zien K, Gutjahr G, et al. TCF7L2 promotes beta cell regeneration in human and mouse pancreas. Diabetologia. 2012;55:3296–3307. doi: 10.1007/s00125-012-2693-z. [DOI] [PubMed] [Google Scholar]
- 95.Wicksteed B, Brissova M, Yan W, et al. Conditional gene targeting in mouse pancreatic β-cells: analysis of ectopic cre transgene expression in the brain. Diabetes. 2010;59(12):3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 97.Nicolson TJ, Bellomo EA, Wijesekara N, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes. 2009;58(9):2070–2083. doi: 10.2337/db09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hardy AB, Wijesekara N, Genkin I, et al. Effects of high-fat diet feeding on Znt8-null mice: differences between beta-cell and global knockout of Znt8. American Journal of Physiology: Endocrinology and Metabolism. 2012;302:E1084–E1096. doi: 10.1152/ajpendo.00448.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lemaire K, Ravier MA, Schraenen A, et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(35):14872–14877. doi: 10.1073/pnas.0906587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pound LD, Sarkar SA, Benninger RKP, et al. Deletion of the mouse Slc30a8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochemical Journal. 2009;421(3):371–376. doi: 10.1042/BJ20090530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pound LD, Sarkar SA, Ustione A, et al. The physiological effects of deleting the mouse SLC30A8 gene encoding zinc transporter-8 are influenced by gender and genetic background. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0040972.e40972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wijesekara N, Dai FF, Hardy AB, et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia. 2010;53(8):1656–1668. doi: 10.1007/s00125-010-1733-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gaulton KJ, Nammo T, Pasquali L, et al. A map of open chromatin in human pancreatic islets. Nature Genetics. 2010;42(3):255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duval A, Rolland S, Tubacher E, Bui H, Thomas G, Hamelin R. The human T-cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Research. 2000;60(14):3872–3879. [PubMed] [Google Scholar]
- 105.Le Bacquer O, Shu L, Marchand M, et al. TCF7L2 splice variants have distinct effects on β-cell turnover and function. Human Molecular Genetics. 2011;20(10):1906–1915. doi: 10.1093/hmg/ddr072. [DOI] [PubMed] [Google Scholar]
- 106.Mondal AK, Das SK, Baldini G, et al. Genotype and tissue-specific effects on alternative splicing of the transcription factor 7-like 2 gene in humans. Journal of Clinical Endocrinology and Metabolism. 2010;95(3):1450–1457. doi: 10.1210/jc.2009-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weise A, Bruser K, Elfert S, et al. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/β-catenin targets. Nucleic Acids Research. 2009;38(6):1964–1981. doi: 10.1093/nar/gkp1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lichten LA, Cousins RJ. Mammalian zinc transporters: nutritional and physiologic regulation. Annual Review of Nutrition. 2009;29:153–176. doi: 10.1146/annurev-nutr-033009-083312. [DOI] [PubMed] [Google Scholar]
- 109.Chimienti F, Devergnas S, Favier A, Seve M. Identification and cloning of a β-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes. 2004;53(9):2330–2337. doi: 10.2337/diabetes.53.9.2330. [DOI] [PubMed] [Google Scholar]
- 110.Chimienti F, Favier A, Seve M. ZnT-8, a pancreatic beta-cell-specific zinc transporter. BioMetals. 2005;18(4):313–317. doi: 10.1007/s10534-005-3687-9. [DOI] [PubMed] [Google Scholar]
- 111.Emdin SO, Dodson GG, Cutfield JM, Cutfield SM. Role of zinc in insulin biosynthesis. Some possible zinc-insulin interactions in the pancreatic B-cell. Diabetologia. 1980;19(3):174–182. doi: 10.1007/BF00275265. [DOI] [PubMed] [Google Scholar]
- 112.Rutter GA. Think zinc: new roles for zinc in the control of insulin secretion. Islets. 2010;2(1):49–50. doi: 10.4161/isl.2.1.10259. [DOI] [PubMed] [Google Scholar]
- 113.Berglund ED, Li CY, Poffenberger G, et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes. 2008;57(7):1790–1799. doi: 10.2337/db07-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hattersley AT. Unlocking the secrets of the pancreatic β cell: man and mouse provide the key. Journal of Clinical Investigation. 2004;114(3):314–316. doi: 10.1172/JCI22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nature Biotechnology. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mussolino C, Cathomen T. TALE nucleases: tailored genome engineering made easy. Current Opinion in Biotechnology. 2012;23:644–650. doi: 10.1016/j.copbio.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 117.Hockemeyer D, Wang H, Kiani S, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 2011;29(8):731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. New England Journal of Medicine. 2008;359(21):2208–2219. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lyssenko V, Jonsson A, Almgren P, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. New England Journal of Medicine. 2008;359(21):2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 120.Talmud PJ, Hingorani AD, Cooper JA, et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: whitehall II prospective cohort study. British Medical Journal. 2010;340, article b4838 doi: 10.1136/bmj.b4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Langothe H, Palmer CNA, Morris AD, et al. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes. 2008;57(11):3129–3135. doi: 10.2337/db08-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Simmons RK, Harding AH, Wareham NJ, Griffin SJ. Do simple questions about diet and physical activity help to identify those at risk of Type 2 diabetes? Diabetic Medicine. 2007;24(8):830–835. doi: 10.1111/j.1464-5491.2007.02173.x. [DOI] [PubMed] [Google Scholar]
- 123.Lindström J, Tuomilehto J. The diabetes risk score: a practical tool to predict type 2 diabetes risk. Diabetes Care. 2003;26(3):725–731. doi: 10.2337/diacare.26.3.725. [DOI] [PubMed] [Google Scholar]
- 124.Cornelis MC, Qi L, Kraft P, Hu FB. TCF7L2, dietary carbohydrate, and risk of type 2 diabetes in US women. American Journal of Clinical Nutrition. 2009;89(4):1256–1262. doi: 10.3945/ajcn.2008.27058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fisher E, Boeing H, Fritsche A, Doering F, Joost HG, Schulze MB. Whole-grain consumption and transcription factor-7-like 2 (TCF7L2) rs7903146: gene-diet interaction in modulating type 2 diabetes risk. British Journal of Nutrition. 2009;101(4):478–481. doi: 10.1017/S0007114508020369. [DOI] [PubMed] [Google Scholar]
- 126.Haupt A, Thamer C, Heni M, et al. Gene variants of TCF7L2 influence weight loss and body composition during lifestyle intervention in a population at risk for type 2 diabetes. Diabetes. 2010;59(3):747–750. doi: 10.2337/db09-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reinehr T, Friedel S, Mueller TD, Toschke AM, Hebebrand J, Hinney A. Evidence for an influence of TCF7L2 polymorphism rs7903146 on insulin resistance and sensitivity indices in overweight children and adolescents during a lifestyle intervention. International Journal of Obesity. 2008;32(10):1521–1524. doi: 10.1038/ijo.2008.146. [DOI] [PubMed] [Google Scholar]
- 128.Thomas D. Gene—environment-wide association studies: emerging approaches. Nature Reviews Genetics. 2010;11(4):259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smith U, Gale EAM. Cancer and diabetes: are we ready for prime time? Diabetologia. 2010;53(8):1541–1544. doi: 10.1007/s00125-010-1815-8. [DOI] [PubMed] [Google Scholar]
- 130.Kim WY, Sharpless NE. The Regulation of INK4/ARF in Cancer and Aging. Cell. 2006;127(2):265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 131.Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 132.Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nature Genetics. 1999;22(1):44–54. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 133.Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:196–206. doi: 10.1161/ATVBAHA.111.232678. [DOI] [PubMed] [Google Scholar]
- 134.Pasmant E, Laurendeau I, Héron D, Vidaud M, Vidaud D, Bièche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Research. 2007;67(8):3963–3969. doi: 10.1158/0008-5472.CAN-06-2004. [DOI] [PubMed] [Google Scholar]
- 135.Visel A, Zhu Y, May D, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464(7287):409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gudmundsson J, Sulem P, Steinthorsdottir V, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nature Genetics. 2007;39(8):977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 137.Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nature Genetics. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 138.Ravassard P, Hazhouz Y, Pechberty S, et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. Journal of Clinical Investigation. 2011;121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Seidler B, Schmidt A, Mayr U, et al. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(29):10137–10142. doi: 10.1073/pnas.0800487105. [DOI] [PMC free article] [PubMed] [Google Scholar]