

Abstract
Progression-free survival (PFS) is increasingly used as an important and even a primary endpoint in randomized cancer clinical trials in the evaluation of patients with solid tumors, because of both practical and clinical considerations. Although in its simplest form PFS is the time from randomization to a pre-defined endpoint, there are many factors that can influence the exact moment of when disease progression is recorded. In this overview, we review the circumstances that can devalue the use of PFS as a primary endpoint, and attempt to provide a pathway for a future desired state when PFS will become not just a secondary alternative to overall survival but rather an endpoint of choice.
Introduction
Progression-free survival (PFS) is increasingly used as an important and even a primary endpoint in cancer clinical trials for patients with solid tumors. The reasons for this increase in the use of PFS are many, and include the practical (shorter time to a given number of events compared to other endpoints) and the clinical (less influenced by subsequent therapy than overall survival and more relevant with targeted agents than response). However, use of PFS raises many issues of definition, measurement and measurement error, possible observer bias, assessment schedule, and missing or incomplete data of various kinds. These issues are addressed in the several papers in this CCR Focus section; in this Overview we concentrate on improvements of process, definition and measurements that might strengthen the use of PFS as an acceptable endpoint in clinical cancer trials.
Some Statistical Issues and Approaches
A thorough review of the statistical problems and approaches with PFS is given in the paper by Sridhara and colleagues in this CCR Focus (1). The most obvious statistical issue with the use of PFS, as opposed to overall survival (OS), is that the measurement of progression occurs at intervals corresponding to assessment times, and not continuously. At the time of tumor assessment, a decision that disease has progressed according to established criteria means only that progression has occurred sometime between the last assessment and the present one. This gives rise to what is termed interval censored data, for which there is a large body of statistical theory (2). The practical consequence in this context is that estimation of PFS depends on the tumor assessment schedule, so that comparisons between treatment arms will be biased unless assessment schedules are the same. The most common statistical solution to the interval censoring problem in cancer clinical trials is to assume that progression occurs at the assessment time that the criteria are met, which clearly results in an overly optimistic estimate of PFS (too large), though this is not a practical problem unless assessment intervals are long relative to time to progression. While exact solutions are possible they are model dependent (3); there is some evidence that using the midpoint of the interval instead of the endpoint is a sensible alternative with good statistical properties across a range of assumptions (4). Some practical suggestions for estimation and testing with PFS as the endpoint are given by Carroll (5).
A more serious statistical issue, one that can arise in cancer trials in at least two ways, is termed informative censoring. In one scenario, a patient may be taken off protocol treatment due to toxicity or symptomatic deterioration, and then no longer assessed for progression. In a second scenario, protocol defined progression is judged by a retrospective central review, which may overturn a progression call made at the local clinic; in the meantime tumor assessments may have ceased. The actual time to progression for such patients may differ from the typical patient, so treating the observations as right censored, as would be done for incomplete observations resulting from no event having happened by the end of the trial, may not be correct. With this type of informative censoring the most practical suggestion is to perform sensitivity analyses, making assumptions which likely bracket the truth (treating the times as progressions, then as right censored observations (5, 6)). However, a change of definition of progression that corresponded more closely to the clinical judgment that treatment is failing might mitigate the issue in the first scenario, and real time central review of both clinical and imaging data might reduce or eliminate the issue altogether, as discussed in more detail below.
Some Imaging Issues with RECIST
The use of imaging is crucial in the establishment of PFS for patients with solid tumors who are enrolled in clinical trials. Radiology-based imaging methods are well suited for this task because of imaging’s capability to provide both qualitative and quantitative assessment of disease burden before, during and after therapy. The digital nature of most modern day radiology methods is one of the distinct advantages over the use of other measures of benefit (such as tumor markers and other clinically-driven assessments of disease response) because the digital composition of the data allows accurate, reliable and reproducible quantitation when performed correctly, permits automation of measurements, and provides a medium for real-time transmission of studies to centralized locations for advanced image analysis. Digital images can then be archived and stored for decades without loss of fidelity, not only for regulatory and compliance audits but also for data exploration. These repositories of data can then be linked up with other important biological and clinical information to generate unique biomarkers that can serve as surrogates for outcomes or primary outcomes in their own right. Despite these advantages, calculating PFS with the most common and basic of all imaging measurements – unidimensional size measurements according to the RECIST system - still requires human decision making support. The identification of target and non-target lesions creates both variability and bias from reader to reader, as does the perception difference between evaluators for the detection of new lesions. This process becomes even more problematic when subjects with evaluable but not measureable disease are allowed entry into the clinical trial. These so-called interpretation issues are well described by Sullivan and colleagues (7) in this CCR Focus section.
Even when these factors are controlled by centralized interpretations, PFS may not always represent the best alternative to OS or improvement in quality of life (QOL) metrics. There are various reasons why PFS based on imaging metrics may not always parallel clinical outcomes of OS and QOL improvements or be relevant measures of therapeutic efficacy in the age of advanced imaging technologies. First, the lack of tumor shrinkage or tumor growth does not take into account the indolent growth of some tumors. Moreover, many new treatment regimens are cytostatic or target-based rather than cytotoxic. As a result, tumors may not shrink in size but instead become stabilized, increase in size and/or change their texture on imaging (Figure 1). Accordingly, a response assessment that requires a predetermined 20% increase in lesion diameters to declare progressive disease (PD) may not be the most suitable tool in these situations. The article by Villaruz and Socinzki (8) in this edition of CCR Focus provides a very good summary of RECIST, placing it in its historical context and pointing out many of its limitations. Indeed, some have argued that RECIST has outlived its usefulness and “…has stifled implementation of innovative approaches to exploit digital imaging and better measurement of solid tumors” (9). Furthermore, as Villaruz and Socinzki (8) note, crossover clinical trial design, salvage therapy and improvements in supportive care can unlink PFS with OS. Finally, there is an inherent measurement gap in the evaluation of tumor that is difficult to measure using RECIST, such as bone marrow involvement or non-measurable disease involving the pleural, pericardial and peritoneal spaces, or in lesions that are only biologically active as measured on PET. Thus, an opportunity exists for improved radiology based imaging methods that might strengthen the use of PFS as an endpoint if the right parameter(s) can be found (10).
Figure 1. Lesion Response: Change In Tumor Density Versus Size.

The figure above shows the response behavior of a solitary hepatic metastatic lesion on a contrast-enhanced CT scan in a subject with pancreatic carcinoma during experimental therapy. Note the tumor nodularity within the lesion (*) at screening surrounded by fluid (dark appearance on CT). The tumor nodular has disappeared on 90-day scan while the lesion has become more fluid containing (average density within the lesion went from 59HU to 42HU or 29% decrease as indicated in the chart), suggesting intervening tumor necrosis. However, the lesion has not changed substantially in size. This lesion would be considered stable by RECIST criteria but would be a responding lesion by CHOI (21). By 180 days, the lesion has remained stable in size but the nodularity is beginning to reappear (double-head arrow) suggesting tumor recurrence. Thus, a PFS of 180 days would not have been captured by RECIST criteria, leading to a positive bias in favor of experimental therapy.
On Selecting the Right Parameters to Assess PFS
Several issues arise when selecting the best parameters for measuring PFS. Obviously, one that closely correlates with primary efficacy endpoints and is clinically telling would be most attractive, but will ultimately depend upon selecting the most reliable, reproducible and accurate imaging endpoint that tracks clinically relevant outcomes. It is unlikely that such an all-inclusive parameter exists today but the need to create one cannot be overstated. Certainly a common parameter for establishing PFS would allow for the comparison of results across different tumor types and treatments. One approach that has been used is based on consensus driven criteria for response and progression, or developing working definitions that can be tested and modified accordingly (11–13). Indeed, RECIST was established for this very reason.
These modified criteria should ultimately be predicated upon certain principles that take into account the mechanism of action of the experimental treatment, the biologic pathways that will likely be affected downstream from the target(s), and the ultimate killing pathways that will be expressed (such as angiogenic, proliferative, metabolic, apoptotic, stromal, immunologic). Other considerations should include the organ system(s) involved, the pharmacokinetic peaks of drug concentration and effect, and the imaging modality best suited to measure the desired experimental treatment’s activity. Once these principles have been considered, then any decision for selecting a modality specific imaging test should be shaped by an understanding of an imaging modality’s accessibility and inherent accuracy, reliability and reproducibility for detecting a true clinical and biological signal above a background of noise. All of this work, of course, needs to be performed in a manner compliant with current regulations and guidelines, so that it will ultimately be acceptable to the FDA and other governmental agencies that are involved in drug approval.
One generalized parameter that has received a lot of attention recently has been the incorporation of volumetric imaging to assess response and PFS, as highlighted in the article by Sullivan and colleagues (7). Underlying the use of RECIST is the assumption that a uni-dimensional measurement of a lesion’s largest diameter is itself a surrogate marker of tumor volume, assuming that tumors grow in spherical shapes (Figure 2). Unfortunately, this assumption is not always true, as tumors can grow in very complicated shapes, influenced in part by the host tissue tumor interface and the surrounding anatomic boundaries. Nevertheless, volumetric assessments of change might arguably be one of the most quintessential physical parameters to assess tumor response and progression, and overcome some of the limitations inherent in RECIST. Other advantages of volumetric imaging include entire lesion analysis, automation for precise volume determination, and textural evaluation of the tumor (i.e. density or intensity), all of which can be calculated simultaneously with newer quantitation tools. Recent studies (14,15) have shown that semi-automated determination of volumetric change can be an early marker of response and progression in nonsmall cell lung cancer. In addition, volumetric measurements may help to detect subtle changes in indolent disease (16). Finally, the use of volumetric data has been incorporated into neuro-oncology trials as a means of determining response using the RANO criteria (13). However, like uni-dimensional measurements, volumetric calculations may be subject to variability due to both imaging interpretation errors and technical factors. Thus standardization of image acquisition will be required in the future to minimize such variability.
Figure 2. Non-Pulmonary Volumetric Analysis As A Measure of Treatment Responses.
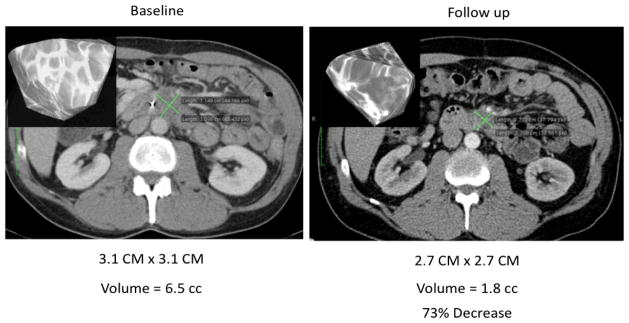
Volumetric analysis of tumor burden was performed (inset top left) at baseline and end of cycle 2 using contrast enhanced CT. Even though the bidimensional measurements of the tumor (green lines) did not change significantly during with therapy, the tumor volume decreased by 73%, suggesting a favorable response to therapy. Quantitative measurements of tumor volume change might be a more sensitive method of assessing tumor response than unidimensional or bidimensional measurements.
Why Don’t We Use Volumetric Assessments of Tumor Response More Often?
With these advantages, why has volumetric imaging not been quickly adopted by the medical and oncology community? First, the lion’s share of data regarding volumetric analysis comes from lung tumors, where evaluating normal tissue and tumor can be quite distinct, allowing for better determination of tumor boundaries than in other organs, where distinguishing tumor from normal tissue is difficult. Secondly, an agreed upon definition of response has not yet been established for volumetric change as it has for RECIST. Simply translating a RECIST response into its volumetric equivalent would reveal that a partial response equates to a 64% reduction in volume while progressive disease would require a 73% change in volume. It is not clear if such large changes in volume are of clinical utility. Certainly, more data for volumetric response would have to be tested before it becomes mainstream. Indeed, many have argued that a continuous measurement scale rather than categorical classification (whether based on diameter or volume) might be the more robust and realistic parameter to use (17,18). Finally, the software tools for volumetric analysis have typically not been available at most local imaging sites (but the distribution and availability of such tools are quickly spreading). Although volumetric analysis can improve response evaluation, it is still a size-based metric. The use of measurements that are not dependent on size, such as apparent diffusion weighted constants and Ktrans for MRI, and Standardized Uptake Values for PET, can be more appealing for PFS determination. However, these parameters have yet to be standardized or validated for determining PFS.
The Use of Blinded Independent Centralized Review (BICR) for PFS Determination
Although the use of new imaging endpoints is growing, the pathway to incorporate these endpoints into clinical trials has yet to be completely clarified. The FDA guidance document regarding centralized image interpretations is a reasonable place to start (available at http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation). In that guidance document, the FDA states that “If the clinical implications are not understood, simply generating an image may not confer benefit to a patient, and an outcome dependent on the interpretation of an imaging test may not be accepted by …[the FDA] as an appropriate endpoint for showing efficacy in a clinical trial”. But the FDA does favor the use of BICR if “…image interpretation results in measurements representing important components of trial eligibility determination or safety or efficacy endpoints, and these measurements are vulnerable to considerable variability among clinical sites…”. Although the support of BICR from the FDA is clear, the use of BICR does have its critics. In particular, there are some studies sponsored by both pharmaceutical industry and academia which estimate a 30–40% discrepancy rate between BICR and local evaluation without any significantly observed clinical impact in outcomes. These conclusions have been based on meta-analysis of a hand full of published phase III oncology trials (19,20) in which BICR were utilized in a retrospective manner predominately using RECIST measurements to determine PFS, as highlighted in the article by Sridhara and colleagues (1) in this CCR Focus series. No analysis has been reported with other measures such as volumetric PFS or tumor density.
An alternative to a blinded review of all cases is to use BICR of a fraction of cases to trigger whether a full review is necessary. Two such auditing methods are evaluated in the paper by Zhang et al in this issue of Focus (21).
Informative Bias from the use of BICR
Perhaps one of the more important criticisms of the use of BICR in phase III clinical trials is the potential introduction of informative censoring, whereby the imaging assessment of subjects may cease due to unconfirmed locally determined progression. As a result, that subject’s data may be compromised, as discussed above. An example of this type of censoring bias has been underlined in a placebo-controlled pivotal trial of everolimus for the treatment of patients with unresectable or metastatic carcinoid tumor (1). The BICR found futility in PFS between the experimental and control arm while the local examiners saw efficacy of the experimental arm. One way to reduce this problem is to conduct simultaneous real time assessments of progression between a centralized resource and the local clinicians. If there is agreement between BICR and local evaluation then the progression assessment is upheld. If discordant conclusions are reached then a rapid adjudication or a short-term follow-up scan could be utilized to decide the ultimate progression assignment. Although there have been barriers in the past that have prevented real time reads due to limited resources, radiologist availability, scan delivery and technical issues, most of those obstacles have been eliminated due to technology solutions that allow for very rapid electronic transmission of imaging data from anywhere in the world with 21 CFR part 11 compliant and validated systems - often within minutes to hours after scan completion. Moreover, competitive pricing for data transmission, and onsite around-the-clock trained clinical trial radiologists’ availability for interpretations of images on a global basis, has made the use of real time reads possible.
Real Time Centralized Review: Can We Get it Right the First Time?
For a variety of reasons, the role of centralized imaging review will likely evolve and grow in the coming years. Advanced quantitative imaging is driving imaging biomarker discovery, which, in turn, will likely be deployed for use in drug development by either helping to select patients most appropriate for targeted therapies based up their personalized context of vulnerability and/or for use in early detection of response. Local site preparation, technology training, credentialing, standardization of acquisition protocols and equipment along with efficient image handling for real time analysis will be more important than ever in the future. Imaging review will become more critical to ensure that the performance of imaging is held to strict standards that will minimize variability in patient preparation, scanner performance and image acquisition, ensuring that even the subtlest of changes in imaging signals will truly reflect real biologic change.
The model of acquiring images locally that are evaluated centrally by a team of experts is not without precedence in medicine. Local pathologists from around the globe are in the habit of sending tissue specimens to other pathologists for expert interpretations including commercial operations that perform genetic analysis (e.g. breast cancer). Thus, we envision that the future desired state of centralized review could parallel this experience by having core labs take a more active role in working with local imaging sites to provide site training and readiness in a form of a “kit” that aids and assists in the education of site personnel, standardization of image acquisition protocols with the use of simultaneously acquired data with pocket phantoms and the assurance that equipment meets certain quality and performance standards. Once acquired, images will be sent to imaging core lab centers of excellence for advanced images analysis and interpretation. Real time analysis will be performed within 2–24 hours after receipt of images. Successful application of this type of activity is beginning to be seen in clinical trials (22). Between better and more tailored criteria for progression, and expert review of images in the context of clinical data, progression-free survival can become both a more clinically relevant and a more reproducible endpoint, making PFS no longer a surrogate but rather an endpoint of choice.
Summary
Progression-free survival (or a variation such as disease-free survival) has long been used as a primary endpoint in situations such as early breast cancer, for which overall survival is so good that the use of OS as a primary endpoint is just not practical. Arguments have been made for the use of PFS instead of OS in other disease settings, on the basis that PFS is a surrogate for OS. A strict definition of surrogacy requires that all the treatment benefit for a new drug or therapy be expressed through an effect on PFS and not some other mechanism (23); most practical definitions require that trials based on PFS would reach the same conclusion as those based on OS, most of the time. There is evidence that this is the case for colorectal cancer, for example (24,25).
The paper in this CCR Focus by Redman and colleagues (26) provides data on the relationship of OS and PFS across several disease categories, and gives a model that explicitly relates PFS to OS. They further provide an intermediate solution to the question of whether to use PFS or OS as a primary endpoint: a phase 2/3 trial, with PFS as the endpoint for a first interim analysis, while OS is retained as the primary overall endpoint for the trial. However, as argued by Villaruz and Socinski in this CCR Focus series (8), effective salvage therapy and cross-over to new targeted agents are increasingly decoupling PFS from OS. It is thus incumbent on the cancer clinical trials community to make PFS a reliable and clinically meaningful endpoint in its own right.
The use of PFS has many merits as well as pitfalls compared to other measures of benefit. Although progression can be defined in several ways, it is a powerful endpoint for evaluating treatment response in tumors. Issues of censoring, criteria for measuring response and the need for real time centralized reads or audits along with the standardization of image acquisition and interpretation will be critical to reduce the variability of detecting progression by imaging. Leading experts in the field will consider these factors in detail in subsequent articles in this special edition of CCR. With the issues highlighted in this Journal, it is our hope is that the scientific and medical community will continue to improve upon those features that will move PFS from a surrogate endpoint to the endpoint of choice.
Acknowledgments
Financial Support: Not applicable
Footnotes
Conflicts of Interest: None
References
- 1.Sridhara R, Mandrekar SJ, Dodd LE. Missing data and measurement variability in assessing progression-free survival endpoint in randomized clinical trials. Clin Cancer Res. 2013;19:xx–xx. doi: 10.1158/1078-0432.CCR-12-2938. [DOI] [PubMed] [Google Scholar]
- 2.Chen D-G, Sun J, Peace KE. Interval-Censored Time-to-Event Data. CRC Press; 2013. [DOI] [PubMed] [Google Scholar]
- 3.Finkelstein DM. A proportional hazards model for interval-censored failure time data. Biometrics. 1986;42:845–54. [PubMed] [Google Scholar]
- 4.Qi Y, Allen Ziegler AL, Hillman SL, Redman MW, Schild SE, Gandara DR, et al. Impact of disease progression date determination on progression-free survival estimates in advanced lung cancer. Cancer. 2012;118:5358–65. doi: 10.1002/cncr.27528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll KJ. Analysis of progression-free survival in oncology trials: Some common statistical issues. Pharmaceutical Statistics. 2007;6:99–113. doi: 10.1002/pst.251. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya S, Fyfe G, Gray RJ, Sargent DJ. Role of sensitivity analyses in assessing progression-free survival in late stage oncology trials. J Clin Oncol. 2009;27:5958–64. doi: 10.1200/JCO.2009.22.4329. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan DC, Schwartz LH, Zhao B. The imaging viewpoint: how imaging affects determination of progression-free survival. Clin Cancer Res. 2013;19:xx–xx. doi: 10.1158/1078-0432.CCR-12-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villaruz LC, Socinzki MA. The clinical viewpoint: definitions, limitations of RECIST, practical considerations of measurement. Clin Cancer Res. 2013;19:xx–xx. doi: 10.1158/1078-0432.CCR-12-2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maitland M. Volumes to learn: Advancing therapeutics with innovative computed tomography image data analysis. Clin Cancer Res. 2010;16:4493–5. doi: 10.1158/1078-0432.CCR-10-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma MR, Maitland ML, Ratain MJ. RECIST: No longer the sharpest tool in the oncology clinical trials toolbox. Clin Cancer Res. 2012;72:5145–9. doi: 10.1158/0008-5472.CAN-12-0058. [DOI] [PubMed] [Google Scholar]
- 11.Byrne MJ, Nowak AK. Modified RECIST criteria for assessment of response in malignant pleural mesothelioma. Annals of Oncology. 2004;15:257–60. doi: 10.1093/annonc/mdh059. [DOI] [PubMed] [Google Scholar]
- 12.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–6. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 13.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: Response assessment in Neuro-Oncology Working Group. J Clin Oncol. 2010;28:1963–72. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 14.Mozley PD, Schwartz LH, Bendtsen C, Zhao B, Petrick N, Buckler AJ. Change in lung tumor volume as a biomarker of treatment response: a critical review of the evidence. Ann Oncol. 2010;21:1751–5. doi: 10.1093/annonc/mdq051. [DOI] [PubMed] [Google Scholar]
- 15.Zhao B, Schwartz LH, Moskowitz CS, Ginsberg MS, Rizvi NA, Kris MG. Lung cancer: computerized quantification of tumor response–initial results. Radiology. 2006;241:892–8. doi: 10.1148/radiol.2413051887. [DOI] [PubMed] [Google Scholar]
- 16.Chang V, Narang J, Schultz L, Issawi A, Jain R, Rock J, et al. Computer-aided volumetric analysis as a sensitive tool for the management of incidental meningiomas. ACTA NeuroChirurgica. 2012;154:589–97. doi: 10.1007/s00701-012-1273-9. [DOI] [PubMed] [Google Scholar]
- 17.Michaelis LC, Ratain MJ. Measuring response in a post-RECIST world: from black and white to shades of grey. Nat Rev Cancer. 2006;6:409–14. doi: 10.1038/nrc1883. [DOI] [PubMed] [Google Scholar]
- 18.Karrison TG, Maitland ML, Stadler WM, Ratain MJ. Design of phase II cancer trials using a continuous endpoint of change in tumor size: application to a study of sorafenib and erlotinib in non small-cell lung cancer. J Natl Cancer Inst. 2007;99:1455–61. doi: 10.1093/jnci/djm158. [DOI] [PubMed] [Google Scholar]
- 19.Amit O, Mannino F, Stone AM, Bushnell W, Denne J, Helterbrand J, et al. Blinded independent central review of progression in cancer clinical trials: Results from a meta-analysis. European J of Cancer. 2011;47:1772–8. doi: 10.1016/j.ejca.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Tang PA, Pond GR, Chen EX. Influence of an independent review committee on assessment of response rate and progression-free survival in phase III clinical trials. Annals of Oncology. 2010;21:19–26. doi: 10.1093/annonc/mdp478. [DOI] [PubMed] [Google Scholar]
- 21.Zhang JJ, Zhang L, Chen H, Murgo AJ, Dodd LE, Pazdur R, et al. Assessment of audit methodologies for bias evaluation of tumor progression in oncology clinical trials. Clin Cancer Res. 2013;19:xx–xx. doi: 10.1158/1078-0432.CCR-12-3364. [DOI] [PubMed] [Google Scholar]
- 22.Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumors treated at a single institution with imatinib mesylate: Proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–9. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 23.Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–440. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 24.Buyse M, Burzykowski T, Carroll K, Michiels S, Sargent DJ, Miller LL, et al. Progression-free survival is a surrogate for survival in advanced colorectal cancer (2007) J Clin Oncol. 2007;25:5218–24. doi: 10.1200/JCO.2007.11.8836. [DOI] [PubMed] [Google Scholar]
- 25.Sidhu R, Rong A, Dahlberg S. Evaluation of progression-free survival as a surrogate endpoint for survival in chemotherapy and targeted agent metastatic colorectal cancer trials. Clin Cancer Res. 2013;19:969–76. doi: 10.1158/1078-0432.CCR-12-2502. [DOI] [PubMed] [Google Scholar]
- 26.Redman MW, Goldman BH, LeBlanc M, Schott A, Baker L. Modeling the relationship between progression-free survival and overall survival: The phase 2/3 trial. Clin Cancer Res. 2013;19:xx–xx. doi: 10.1158/1078-0432.CCR-12-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]