

Abstract
Although previous work identified transcription factors crucial for the specification and migration of parvalbumin (PV)-expressing and somatostatin (SST)-expressing interneurons, the intrinsic factors required for the terminal differentiation, connectivity, and survival of these cell types remain uncharacterized. Here we demonstrate that, within subpopulations of cortical interneurons, Satb1 (special AT-rich binding protein) promotes terminal differentiation, connectivity, and survival in interneurons that express PV and SST. We find that conditional removal of Satb1 in mouse interneurons results in the loss of a majority of SST-expressing cells across all cortical layers, as well as some PV-expressing cells in layers IV and VI, by postnatal day 21. SST-expressing cells initially migrate to the cortex in Satb1 mutant mice, but receive reduced levels of afferent input and begin to die during the first postnatal week. Electrophysiological characterization indicates that loss of Satb1 function in interneurons results in a loss of functional inhibition of excitatory principal cells. These data suggest that Satb1 is required for medial ganglionic eminence-derived interneuron differentiation, connectivity, and survival.
Introduction
Inhibitory interneurons (INs) are strikingly diverse in terms of morphology, distribution, connectivity, physiology, and marker expression (DeFelipe, 1993; Kawaguchi and Kubota, 1996, 1997; Cauli et al., 1997; Gupta et al., 2000; Markram et al., 2004; Batista-Brito and Fishell, 2009). Each subpopulation of INs uniquely shapes cortical processing through GABA-mediated inhibition (McBain and Fisahn, 2001; Pouille and Scanziani, 2001; Wehr and Zador, 2003; Somogyi and Klausberger, 2005; Klausberger and Somogyi, 2008). For instance, somatostatin (SST)-expressing Martinotti cells are distinctly positioned to influence dendritic summation and integration of excitatory inputs onto pyramidal cells (Kapfer et al., 2007; Silberberg and Markram, 2007; Tan et al., 2008; Berger et al., 2009; Murayama et al., 2009; Ma et al., 2010). However, little is known regarding the molecular mechanisms that govern SST cell terminal differentiation.
Cortical INs are primarily derived from the medial ganglionic eminences (MGEs) and caudal ganglionic eminences (CGEs), and migrate tangentially from these transient embryonic structures to their final position in the cortex (Anderson et al., 1999,2001; Parnavelas et al., 2000; Marín and Rubenstein, 2001; Wichterle et al., 2001; Nery et al., 2002; Flames et al., 2007; Welagen and Anderson, 2011). The MGE predominately gives rise to parvalbumin (PV)-expressing and SST-expressing cortical INs, while the CGE gives rise to reelin-positive/SST-negative, calretinin (CR)-expressing, vasoactive intestinal peptide (VIP)-expressing, and neuropeptide Y (NPY)-expressing INs (Nery et al., 2002; Miyoshi et al., 2007; Xu et al., 2008; Lee et al., 2010; Miyoshi et al., 2010). Cortical INs are also derived from the ventrolateral septum, as well as the preoptic areas (Taglialatela et al., 2004; Gelman et al., 2009).
Expression of Nkx2.1, a homeobox protein, is required for IN progenitors to differentiate as PV-expressing and SST-expressing cells (Sussel et al., 1999; Butt et al., 2008). Lim/homeobox protein 6 (Lhx6), a transcription factor that functions downstream of Nkx2.1, is required for marker expression and the proper laminar positioning of PV and SST INs (Liodis et al., 2007; Du et al., 2008). As these populations enter the cortex, Lhx6 activates the expression of the sry-box 6 (Sox6) transcription factor, the loss of which results in a failure of PV cells to develop their characteristic fast-spiking capabilities (Azim et al., 2009; Batista-Brito et al., 2009). However, as most SST cells are spared in Sox6 mutants, it seems probable that Nkx2.1 and/or Lhx6 activate a parallel transcription factor cascade required for SST cell specification.
We recently discovered that the mRNA encoding Satb1 (special AT-rich binding protein), a homeobox protein that coordinates T-cell differentiation, is enriched in MGE-derived IN precursors (Dickinson et al., 1997; Alvarez et al., 2000; Yasui et al., 2002; Cai et al., 2006; Batista-Brito et al., 2008b). In this study, we removed Satb1 in cortical IN precursors and find that the loss of Satb1 results in a profound loss of SST-expressing cortical INs and affects the maturation of some PV-expressing cortical INs. Given that the loss of SST cells coincides with the period when they establish their synaptic connectivity, their loss could be a result of reduced afferent input during the first postnatal week. This hypothesis is supported by the fact that blockade of neuronal activity in MGE-derived IN precursors diminishes Satb1 expression and leads to the death of SST INs in the somatosensory cortex. These data indicate that Satb1 is activity-dependent and is required for late-stage MGE-derived IN differentiation, integration, and survival.
Materials and Methods
Satb1 conditional mice and genotyping
A conditional Satb1 allele was generated via homologous recombination using a targeting construct in which loxP sites were placed in nonconserved regions just 5′ to coding exon 2 and 3′ to coding exon 3 (Fig. 1A). The exon 2/3 and 5′ and 3′ arm fragments were amplified from a bacterial artificial chromosome containing Satb1 genomic sequences derived from the 129 mouse strain (bMQ 399E18 12957Ab2.2, Source BioScience Lifescience). The Cre-mediated deletion of exons 2 and 3 results in premature termination of Satb1 mRNA translation, due to the creation of several in-frame stop codons. The targeting construct also contained a flrted Neo cassette 3′ to the floxed region, and a negative diphtheria toxin fragment A selection cassette flanking the 5′ arm. This construct was electroporated into W4 mouse embryonic stem cells, and 384 G418-resistant colonies were selected for Southern blot screening. Recombinant colonies were identified by NheI digestion of genomic DNA, followed by hybridization with 5′ and 3′ external probes, which resulted in wild-type and mutant allele fragments of 14 and 8 kb, respectively (Fig. 1F). Six positive colonies were selected and karyotyped, and two of these were used for blastocyst injection. The resulting animals were genotyped using the following primers: F1-GTGGCAGACATGCTTCAAGA and R1-TGATAGCACGCAGGGAAA. A PCR cycle consisting of 95°C for 30 s, 59°C for 30 s, and 72°C for 30 s, for a total of 31 cycles resulted in a 278 bp wild-type band, and a 358 bp mutant band after removal of the Neo cassette with FlpE recombination. Null animals were generated by crossing Satb1 conditional mutants with a Cre-deleter strain of mice, and the null allele was detected using the following primers: F2-TCTGCGTTCACTGATTTTGG and the R1 primer described above. The PCR conditions used to detect the null allele were identical to those used to detect the conditional allele, and the null allele PCR yielded a 567 bp band in animals in which the null allele was present and the Neo cassette was removed. We could not detect Satb1 immunofluorescence in the cortices of null animals (Fig. 1G,H).
Figure 1.

Satb1 conditional allele. A, Targeted region of the wild-type Satb1 genomic allele. B, Targeting construct, including coding exons 2 and 3 flanked by loxP sites (▶), PGK-Neo-positive selection cassette in reverse orientation flanked by Frt sites (■), and TK-diphtheria toxin fragment A-negative selection cassette. C, Homologous recombination in mouse W4 embryonic stem cells with the targeting construct results in the insertion of an extra NheI site (N) just 3′ to coding exon 3. D, FlpE recombination results in removal of the Neo cassette. E, Cre recombination results in deletion of coding exons 2 and 3. In addition, downstream mRNA sequences are not translated following recombination due to the creation of multiple in-frame stop codons in the resulting transcription product. F, Southern blot analysis of NheI digested genomic DNA from G418-resistant colonies reveals an expected 14 kb wild-type (WT) band, and an 8 kb band for the targeted mutant allele (Mut) for both the 5′ and 3′ external probes. I, coding exon 1; II, coding exon 2; III, coding exon 3; IV, coding exon 4; F1, forward genotyping primer 1 for use in detecting the conditional allele; R1, reverse genotyping primer 1; F2, forward genotyping primer 2 for use in detecting the null allele. G, Satb1 immunofluorescence in a cortical section from a wild-type animal. H, Satb1 immunofluorescence is undetectable in TKCre;Satb1c/c animals.
Animals
All animals were treated in accordance with the regulations and guidelines of the Institutional Animal Care and Use Committee of the New York University School of Medicine. The Dlx5/6Cre (Stenman et al., 2003), Dlx6aCre (Yu et al., 2011), SSTCre (a gift from Josh Huang), Dlx1/2CreER (Batista-Brito et al., 2008a), X94 (Ma et al., 2006), G42 (Chattopadhyaya et al., 2004), 5HT3aGFP (Gene Expression Nervous System Atlas), RCE:LoxP reporter animals (Sousa et al., 2009) and Satb1 conditional animals were maintained on a mixed background (Swiss Webster and C57BL/6) and genotyped as described previously. To generate productive crosses, Cre-positive/Satb1c/+ males were mated with Satb1c/c;RCE:LoxP+/+ females and the resulting Cre-positive, Satb1c/+;RCE:LoxP+/− and Satb1c/c littermates were analyzed. Dlx1/2CreER induction was performed by gavaging pregnant or nursing females with tamoxifen (4 mg) at either embryonic day 13.5 (E13.5), E15.5, or postnatal day 1 (P1).
Histology and cell counting
Animals were anesthetized using an intraperitoneal injection of Nembutal (0.25 mg/g body weight) followed by transcardiac perfusion of 4% paraformaldehyde in PBS. Brains were then removed and placed in a solution of 30% sucrose in PBS at 4°C overnight, embedded in Tissue-Tek Optimal Cutting Temperature (OCT) compound (Sakura), and frozen at −80°C. Brain tissue was sectioned at a thickness of 16 μm onto Colorfrost Plus microscope slides (Fisher). Sections were blocked using 2% goat serum and 0.1% Triton X-100 in PBS for 1 h, followed by overnight incubation in 0.1% Triton X-100 in PBS with the following primary antibodies: rabbit anti-GFP (1:2000, Invitrogen); rat anti-GFP (1:1000, Nacalai Tesque); mouse anti-PV (1:1000, Sigma-Aldrich); rat anti-SST (1:500, Millipore Bioscience Research Reagents); rabbit anti-NPY (1:500, Incstar); rabbit anti-VIP (1:500, Incstar); mouse anti-CR (1:1000, Millipore Bioscience Research Reagents), and rabbit anti-Satb1 (1:500, Victor Tarabykin). Following primary antibody incubation, slides were washed three times in PBS and incubated for 1 h at room temperature with the appropriate Alexa fluor 488-conjugated or 594-conjugated secondary antibodies (Invitrogen) raised in goat. Slides were then washed three times in PBS and subjected to a 5 min 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain, followed by application of Fluormount-G and coverslipping.
For cell quantification, fluorescent images were captured using a fluorescence microscope (Axioscope A1, Zeiss) with an attached cooled CCD camera (Princeton Scientific Instruments) and MetaMorph software (Universal Imaging). For each animal analyzed, images from five somatosensory cortex sections of equal area were used to count cell densities and numbers in the various cortical layers. Sections in which both the caudoputamen and lateral ventricle were clearly visible were chosen for analysis, and primary somatosensory cortical layers were binned from layer I to layer VI using a DAPI counterstain to identify layer boundaries.
EEGs
EEGs were performed as described previously (Batista-Brito et al., 2009).
Electrophysiology
Brain slice preparation.
Mice were used between P18 and P21 for electrophysiological experiments, except for the assessment of synaptic inputs onto SST INs at early postnatal ages where P4–P6 pups were used. Mice were anesthetized with intraperitoneal injection of pentobarbital (100 mg/kg body weight) and decapitated. The brain was quickly removed and immersed in ice-cold oxygenated artificial CSF (ACSF) containing the following (in mm): 87 NaCl, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 CaCl2, 2 MgSO4, and 10 glucose. Coronal slices (300 μm) through primary somatosensory cortex were generated using a Vibratome 1000 plus (Vibratome) and incubated in a holding chamber at 32–35°C for ∼30 min followed by continued incubation at room temperature for at least 1 h before physiological recordings. A slice was then transferred to a recording chamber attached to the microscope stage and completely submerged in ACSF containing (in mm) the following: 125 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgSO4, and 10 glucose, pH 7.4 (bubbled with 95% O2/5% CO2). ACSF was perfused in the recording chamber at 3 ml/min at 32°C.
Electrophysiological recordings.
Whole-cell patch-clamp recordings were obtained from visually identified neurons using an infrared differential interference contrast video microscopy system. For current clamp, the internal pipette solution contained (in mm) the following: 130 K-gluconate, 0.5 EGTA, 7 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 5 phosphocreatine, pH 7.2, with KOH. For voltage clamp, the pipette solution contained (in mm) the following: 130 Cs-gluconate, 0.5 EGTA, 7 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 5 phosphocreatine, 5 QX-314, pH 7.2, with CsOH. Patch electrodes (3–6 ΩM) were pulled from borosilicate glass capillaries (outer diameter, 1.5 mm). Series resistances were usually 15–30 MΩ upon break-in and were compensated by ∼70%, and only cells with stable series resistance (<20% change throughout the recording) were used for analysis. Data were collected using an Axopatch 700B amplifier (Molecular Devices), low-pass filtered at 5 kHz and digitally sampled at 20 kHz online, and analyzed offline with pClamp9 software (Molecular Devices).
To characterize the intrinsic membrane properties of neurons, current-clamp recordings were made and hyperpolarizing and depolarizing current steps of 1000 ms duration were injected at 50 pA increments at 0.1 Hz. To record spontaneous EPSCs (sEPSCs), slices were perfused with ACSF containing 1 μm GABAzine to block GABAA receptors (GABAARs), and neurons were held at −70 mV. To record spontaneous IPSCs (sIPSCs), slices were perfused with ACSF containing 50 μm AP-5 and 20 μm CNQX to block NMDA and AMPA receptors, respectively, and neurons were held at 0 mV. For paired recordings, whole-cell current-clamp recordings in INs were paired with voltage-clamp recordings (VHOLD = 0 mV) in excitatory neurons in layer IV of primary somatosensory cortex.
Data analysis.
The following parameters were measured to characterize neuronal membrane properties: resting membrane potential was recorded immediately after the rupture of the neuronal membrane; input resistance was determined by measuring the voltage change in response to a small hyperpolarizing current pulse at resting potential (−50 pA, 1000 ms); membrane time constant (τ) was determined using a monoexponential fit to the voltage produced by a small hyperpolarizing current pulse at resting potential (−50 pA, 1000 ms); action potential voltage threshold was defined as dV/dt = 20 mV/ms; action potential current threshold (ITH) was defined as the first 1000 ms rectangular current injection that elicited a spike; spike height was measured from the spike threshold to the peak; action potential duration was measured as the spike width at its half-amplitude; afterhyperpolarization (AHP) was measured as the peak amplitude of AHP from the spike threshold. sEPSCs or sIPSCs were analyzed using Mini Analysis Program (Version 5.6.28, Synaptosoft) and all events were detected above a threshold of 10 pA. Each event was then manually selected based on its rise and decay properties. Analysis of sEPSCs or sIPSCs was performed with cumulative probability plots and the cumulative histograms were compared using the Kolmogorov–Smirnov test for significant differences. Population data are presented as mean ± SEM. To compare the results between different genotype groups, statistical tests (Student's t test for two-group comparisons and ANOVA for multiple-group comparisons) were performed using statistical software (Origin 7.5; OriginLab). In all cases, statistical significance was defined as p < 0.05, unless otherwise indicated.
Mouse electroporations
Mouse in utero electroporations were performed as described previously (De Marco García et al., 2011). Briefly, Swiss Webster pregnant females were anesthetized and underwent a cesarean-section surgery. Dlx5/6-eGFP and Dlx5/6-Kir2.1 plasmids were injected into the lateral ventricle of mouse embryos at E13.5, E14.5, and E15.5. Upon delivery of a square pulse, embryos were returned to the abdominal cavity, which was subsequently sutured. Females were allowed to recover for 3–4 h in a humidified chamber at 37°C. Pups were killed at P5 and perfused with 4% PFA. Analysis was performed on cryostat sections. More than 28 brains were analyzed for each condition and developmental stage. Statistical analysis was performed using a Student's t test (two-tailed distribution, homoscedastic).
Results
MGE-derived INs express Satb1
Previously, we found that Satb1 mRNA expression was enriched in cortical IN progenitors (Batista-Brito et al., 2008b). To determine the timing and IN-subtype specificity of Satb1 protein expression, we performed immunofluorescence analysis using an antibody against Satb1. To visualize IN progenitors during differentiation, we labeled them using animals double-positive for the tamoxifen-inducible Dlx1/2CreER and RCE:LoxP reporter alleles. Dlx1/2CreER is expressed in IN progenitors, allowing us to label these populations in a temporally specific manner. To mark IN progenitors that remained in the MGE or were in the process of tangential migration, we gavaged pregnant Dlx1/2CreER;RCE:LoxP females at E13.5, sectioned E15.5 brains, and performed double immunostaining (Fig. 2A,A'). We saw little or no overlap between EGFP-expressing and Satb1-expressing cells at this stage, indicating that IN progenitors do not yet express Satb1 when they are still in the MGE or migrating tangentially (Fig. 2A,A'). In contrast, when animals were gavaged at E15.5 and sectioned at E17.5 (Fig. 2B,B'), occasional EGFP/Satb1 double-positive cells could be seen entering the cortical plate, indicating that IN progenitors begin to express Satb1 as they enter their radial migration/terminal differentiation phase.
Figure 2.
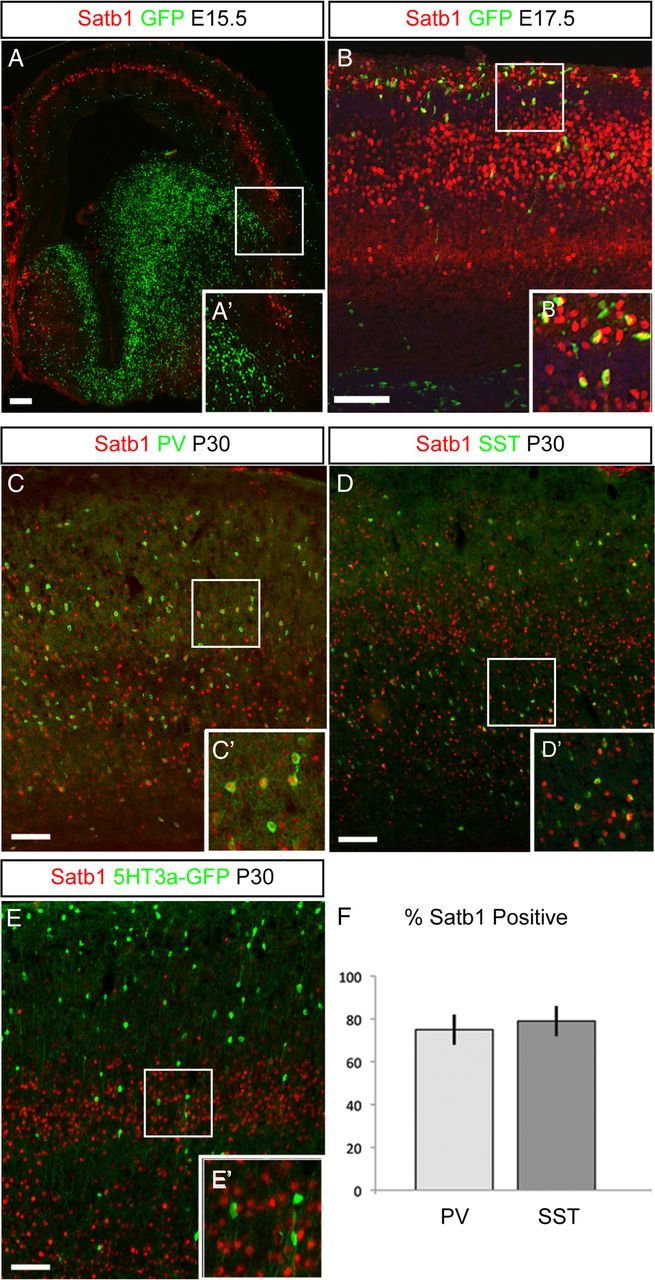
MGE-derived INs express Satb1. A, Dlx1/2CreER;RCE:LoxP pregnant females were gavaged with 4 mg of tamoxifen at E13.5. Embryonic brains were sectioned coronally at E15.5 and immunofluorescence was performed to visualize EGFP-positive IN progenitors (green) and Satb1 (red) expression. No Satb1 expression was seen in IN progenitors in the MGE or in tangentially migrating IN progenitors at this stage. A', Magnification of boxed area in A. B, Dlx1/2CreER;RCE:LoxP pregnant females were gavaged with tamoxifen at E15.5 and embryonic brains were sectioned on E17.5. In this photomicrograph, several EGFP/Satb1 double-positive IN progenitors can be visualized entering the cortical plate at this stage. B', Magnification of boxed area in B. C, Coronal section from a wild-type P30 mouse brain. Immunofluorescence for PV (green) and Satb1 (red) revealed that a majority of PV cells express Satb1 at this stage. C', Magnification of boxed area in C. D, Coronal section from a wild-type P30 mouse brain. Immunofluorescence for SST (green) and Satb1 (red) revealed a majority of SST cells express Satb1 at this stage. D', Magnification of boxed area in D. E, Coronal section from a P30 5HT3a-GFP mouse brain. Few or no CGE-derived 5HT3a-GFP-labeled cells (green) express Satb1 (red) at this stage. E', Magnification of boxed area in E. F, Quantification of PV and SST cell Satb1 expression indicated that a majority of MGE-derived cells express Satb1 at P30 (PV, 75 ± 7%; SST, 79 ± 7%; n = 5). Error bars indicate SEM. Scale bars, 50 μm.
It was recently shown that cortical INs can be divided into three main groups: PV cells derived from the MGE; SST cells derived from the MGE; and ionotropic serotonin receptor-expressing (5HT3aR) neurons, which are derived from the CGE (Lee et al., 2010; Rudy et al., 2011). To determine which subtypes of INs express Satb1, we assessed its expression in PV-, SST-, and 5HT3a-GFP-positive cells. A majority of PV-positive cells expressed Satb1 at P30 (Fig. 2C,C'), as did SST-expressing INs (Fig. 2D,D'). This suggests that MGE-derived IN subtypes express Satb1 at this stage. However, we did not observe Satb1 expression in 5HT3a-GFP-positive INs at P30 (Fig. 2E,E') or in earlier ages (data not shown), indicating that CGE-derived IN subtypes are Satb1-negative. When we quantified Satb1 expression in MGE-derived IN subtypes, we found that 75 ± 7% (n = 5 brains) of PV cells and 79 ± 7% (n = 5 brains) of SST cells were Satb1-positive (Fig. 2F). These data suggest that Satb1 is expressed by a majority of MGE-derived INs as they enter the terminal differentiation phase and into adulthood.
MGE-derived INs require Satb1 for migration and differentiation
To determine whether Satb1 plays a role in IN differentiation, we removed Satb1 conditionally using either the Dlx5/6Cre-IRES-EGFP (Dlx5/6Cre) or the Dlx6aCre driver lines, both of which drive Cre expression in all cortical INs embryonically shortly after these cells become postmitotic (Yu et al., 2011). The Dlx6aCre line was used in all experiments in which transgenic lines expressing EGFP were used to visualize specific cell populations, as the Dlx5/6Cre line contains an IRES-EGFP element that renders all cortical INs EGFP-positive. Use of either the Dlx6aCre or Dlx5/6Cre lines to remove Satb1 resulted in indistinguishable results with regards to their effects on the numbers and distribution of IN subtypes (data not shown).
In P21 animals, both the distribution and numbers of cortical INs in Dlx5/6Cre;Satb1c/+ heterozygote animals (Fig. 3A) was equivalent to that observed in Dlx5/6Cre;Satb1+/+ animals (data not shown). Therefore we used heterozygote littermates as controls. However, Dlx5/6Cre;Satb1c/c animals displayed a marked reduction in the numbers of INs in layer 4 somatosensory cortex (Fig. 3B). Furthermore, when we analyzed MGE-derived IN subtypes, we observed a dramatic reduction in the number of SST-expressing INs in all layers in mutants compared with heterozygotes (Fig. 3C,D). This is most likely the result of a loss of SST cells, rather than a loss of SST expression, as the overall reduction in IN numbers is consistent with what would be expected from the net loss of the SST cell population. To further distinguish between a loss of SST cells and SST expression, the Dlx6aCre line was used to remove Satb1 in a transgenic mouse line (X94 line) in which EGFP is expressed in a subpopulation of SST cells (Ma et al., 2006). In these mutants, we observed a profound loss of EGFP-positive cells, consistent with the notion that SST cells are actually missing in mutant animals (data not shown). While the number of PV cells appeared normal in heterozygote animals (Fig. 3E), they were reduced in layers IV and VI of the somatosensory cortex of mutant animals when compared with heterozygotes (Fig. 3F).
Figure 3.

MGE-derived INs require Satb1 for their migration and differentiation. Dlx5/6Cre was used to remove Satb1 expression in IN precursors and animals were analyzed at P21. All animals also had the RCE:LoxP reporter allele in the background for Cre-dependent EGFP expression in the IN population. A, B, In Satb1c/+ animals (A), Dlx5/6Cre-mediated removal of Satb1 does not affect either the numbers or distribution of EGFP-expressing INs, whereas Dlx5/6Cre;Satb1c/c animals (B) displayed a significant loss of INs in layer IV. C, D, Dlx5/6Cre;Satb1c/+ animals have both normal numbers and distribution of SST-expressing INs (C). However a dramatic reduction in the number of SST-positive cells was observed in Dlx5/6Cre;Satb1c/c animals at P21 (D). E, F, PV cell distribution and numbers appeared normal in Dlx5/6Cre;Satb1c/+ animals (E), but slightly reduced in numbers in layers IV and VI in Dlx5/6Cre;Satb1c/c animals (F). G, Dlx5/6Cre;Satb1c/c animals (gray bars) had significantly fewer INs (EGFP, green) per area (square millimeter) on average in layers IV (41.5 ± 1.8 for mutants vs 86.1 ± 5.23 for controls) and VI (75.8 ± 7.1 for mutants vs 106.44 ± 7.7 for controls), and a greater number of INs in layer V (144.4 ± 6.5 vs 112.1 ± 4.7), compared with heterozygote controls (white bars). H, Compared with controls, SST-expressing IN numbers were significantly reduced in layers II–VI in Dlx5/6Cre;Satb1c/c animals (26.8 ± 3.0 for mutants vs 114.0 ± 8.3 for controls). I, PV cell numbers were significantly reduced in layers IV (21.0 ± 0.8 for mutants vs 45.8 ± 2.7 for controls) and VI (17.2 ± 2.1 for mutants vs 37.5 ± 1.6 for controls), and increased in layer V (60.6 ± 1.8 for mutants vs 46.7 ± 2.9 for controls) after Satb1 removal. (n ≥ 5 animals, with ≥ 3 sections analyzed per animal for all experiments; *p < 0.05; **p < 0.01; ***p < 0.001. Error bars represent SEM.
When the results of Satb1 removal were quantified, we found significant reductions in the number of INs present per square millimeter of somatosensory cortex in layers IV and VI in mutant animals compared with heterozygote controls (41.5 ± 1.8 for mutants vs 86.1 ± 5.23 for controls; Fig. 3G). In addition, we noticed a significant increase in the number of INs per square millimeter within layer V of mutants (144.4 ± 6.5 for mutants vs 112.1 ± 4.7 for controls). When we quantified IN subtype numbers per square millimeter across cortical layers, we observed a significant reduction in the number of SST cells in somatosensory layers II–VI (26.8 ± 3 for mutants vs 114 ± 8.3 for controls; Fig. 3H). To determine whether any subtype of SST cells was spared after Satb1 removal, we colabeled mutant brains with SST and CR, NPY, or neuronal nitric oxide synthase and found that these populations were also reduced in Satb1 mutants (data not shown). Notably, although Dlx5/6Cre-mediated removal of Satb1 resulted in reduced numbers of PV-expressing cells per square millimeter in layers IV (21.0 ± 0.8 for mutants vs 45.8 ± 2.7 for controls) and VI (17.2 ± 2.1 for mutants vs 37.5 ± 1.6 for controls), we observed a concomitant increase in this population within layer V (60.6 ± 1.8 per square millimeter cortex for mutants vs 46.7 ± 2.9 for controls) (Fig. 3H). These results suggest that Satb1 is needed for the terminal differentiation and/or survival of the majority of SST-expressing cells, as well as for the proper radial migration of a subset of PV-expressing cells.
SST cells require postnatal expression of Satb1
SST-expressing and PV-expressing cells continue to express Satb1 into adulthood. To determine whether Satb1 is required postnatally for the differentiation or survival of these cell types, we administered tamoxifen to Dlx1/2CreER;Satb1 conditional animals on P1, and the location and numbers of SST and PV cells were analyzed in the somatosensory cortex at P21. As this approach results in the mosaic targeting of the Dlx1/2-expressing populations, we focused our analysis on EGFP+ cortical cells. The overall number of EGFP+ cells was similar in Dlx1/2CreER;Satb1c/c animals compared with Dlx1/2CreER;Satb1c/+ controls (Fig. 4A,B,D). In addition, we did see a reduction in the percentage of cells that expressed Satb1 at P21, with 72% of EGFP+ cells expressing Satb1 in control animals and 13% of EGFP+ cells expressing Satb1 in Dlx1/2CreER;Satb1c/c animals after tamoxifen treatment at P1 (data not shown). However, the percentage of EGFP+ cells expressing SST was significantly reduced in Dlx1/2CreER;Satb1c/c animals compared with heterozygote controls (10.5% for mutants vs 27.6% for controls; Fig. 4C). There was no significant change in the percentage of EGFP+ cells that expressed PV. Furthermore, we did not observe a significant change in the percentage of cells destined for each of the cortical layers, suggesting that IN radial migration was not affected by later removal of Satb1 (Fig. 4D). This suggests that Satb1 may be required at two stages of MGE IN development: at an early embryonic stage, where it is required to regulate radial migration (in both SST-expressing and PV-expressing INs); and postnatally to regulate SST cell terminal differentiation.
Figure 4.
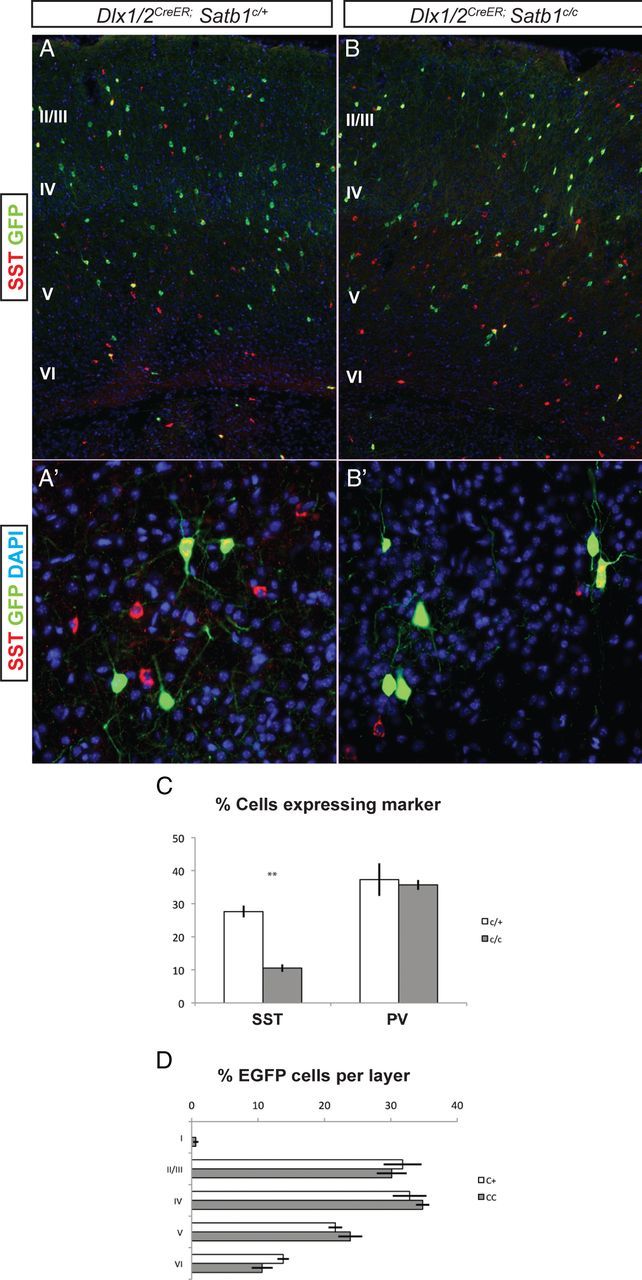
SST INs require postnatal expression of Satb1. Tamoxifen was administered to Dlx1/2CreER;Satb1 conditional animals on P1, and animals were killed at P21 for analysis. All analysis was done on animals positive for the RCE:LoxP reporter allele for Cre-mediated EGFP expression. A, B, The numbers of EGFP-positive INs were similar between Dlx1/2CreER;Satb1c/+ controls (A) and Satb1c/c animals (B). A', B' Higher-powered view of deep-layer cells positive for EGFP after P1 tamoxifen administration. C, Quantification of the percentage of EGFP cells expressing MGE markers. In Dlx1/2CreER;Satb1c/+ animals, 27.6 ± 1.7% of cells were SST-positive, compared with 10.5 ± 1.1% in Satb1c/c animals following P1 administration of tamoxifen. D, The number of cells destined for each cortical layer was similar between Satb1c/+ and Satb1c/c animals. n = 4 for Satb1c/+ animals and n = 5 for Satb1c/c animals. **p < 0.01.
SST cells die postnatally after Satb1 removal
Embryonic removal of Satb1 resulted in a dramatic loss of SST cells. To further dissect the role of Satb1 in the SST subpopulation and to determine the time course of SST cell loss upon early removal, we removed Satb1 using the SSTCre line in combination with the RCE:LoxP reporter allele. SSTCre drives expression of Cre in nearly all cortical SST INs from early embryonic ages (Taniguchi et al., 2011). Analysis of SSTCre;Satb1c/c mice at P5 revealed that the SST cell population is largely intact in these animals when compared with controls (Fig. 5A,B,D), although the number of cleaved caspase-3-positive, apoptotic cells per square millimeter was significantly elevated in layers IV (2.3 ± 0.25 for mutants vs 0.8 ± 0.3 for controls) and VI (5.24 ± 1.1 for mutants vs 2.9 ± 0.5 for controls; Fig. 5C). By P10, the SST cell population is reduced by 40% in SSTCre;Satb1c/c mice (Fig. 5H), and by P21 this population is reduced by 60% in mutants when compared with control animals (Fig. 5E–G). In addition to our findings regarding the loss of SST expression, we found that the number of GFP+ cells per square millimeter was reduced by 40% by P21 in mutant animals when compared with controls, indicating that the loss of SST-expressing cells is due in large part to cell death rather than simply a loss of SST expression (data not shown). Together, these data suggest that the SST IN population, although initially present, begins to die at P5 in the absence of Satb1 gene function. As Satb1 was removed only in the SST population in these experiments, these data also show that Satb1 acts in a cell-autonomous manner to mediate SST cell survival.
Figure 5.
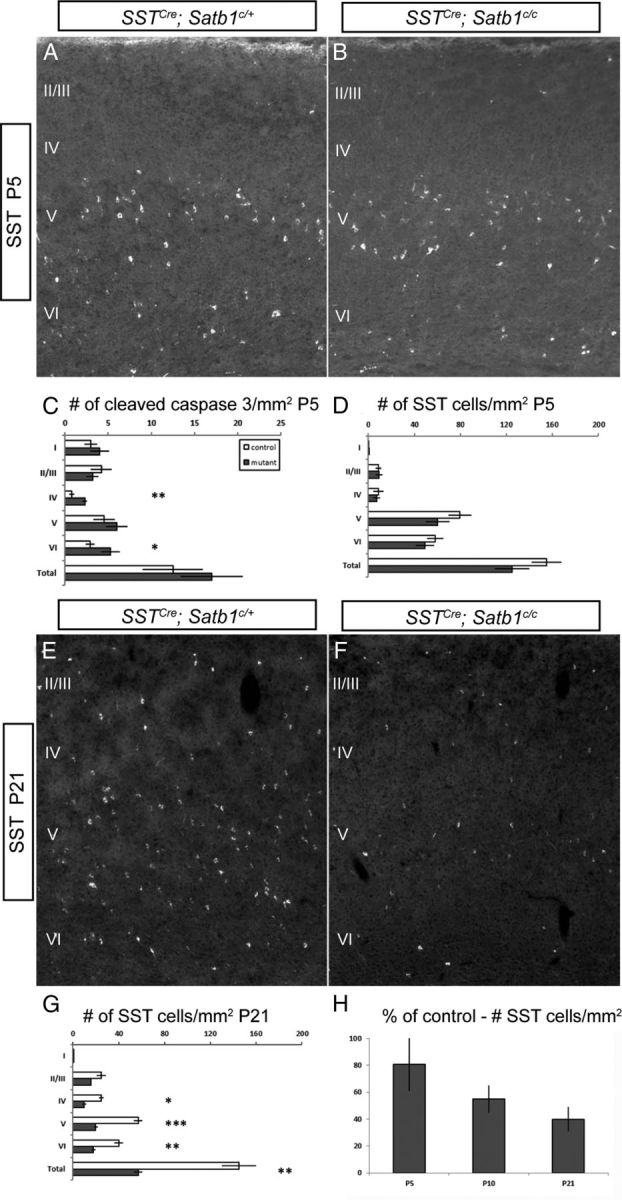
SST cells begin to die at P5 after Satb1 removal. A, SST immunofluorescence in SSTCre;Satb1c/+ animals at P5. B, SST cell numbers are similar in SSTCre;Satb1c/c and control animals at this age. C, Quantification of cleaved caspase 3-positive cells. Significant increases in the number of cleaved caspase 3 cells per square millimeter were observed in layers IV (0.8 ± 0.3 for Satb1c/+ vs 2.3 ± 0.25 for Satb1c/c) and VI (2.9 ± 0.5 for Satb1c/+ vs 5.24 ± 1.1 for Satb1c/c) of mutant animals. D, SST cell numbers were not significantly different in any layer at this age in mutant versus controls. E, F, Immunofluorescence performed on SSTCre;Satb1c/+ animals at P21 indicated normal numbers of SST-expressing cells (E), but the number of SST-expressing cells in SSTCre;Satb1c/c animals was drastically reduced (F). G, Quantification of SST-expressing cell numbers revealed significantly reduced numbers of SST-expressing cells in SSTCre;Satb1c/c animals (gray bars) compared with controls (white bars). H, SST cell numbers per square millimeter of cortex in SSTCre;Satb1c/c are 81 ± 20% of control numbers at P5, and further decline to 55 ± 10% and 40 ± 9% of control numbers by P10 and P21, respectively. n ≥ 5 animals, ≥ 3 sections were analyzed per animal. *p < 0.05, **p < 0.01, ***p < 0.001.
Mutant SST INs receive reduced excitatory inputs at early postnatal ages
Our immunohistochemistry experiments demonstrated that the loss of Satb1 results in a profound loss of SST-expressing INs beginning in the first postnatal week. Because marker expression and position of the SST cells is largely normal in mutant animals at P5 but is lost subsequently, we reasoned that the loss of these cells may occur due to a lack of sufficient connectivity of these cells within the developing cortex. For instance, synaptic activity has been shown to be important for neuronal survival during development (Catsicas et al., 1992; Linden, 1994; Mennerick and Zorumski, 2000). Therefore, we speculated that the inputs to SST INs at early postnatal ages might be compromised after Satb1 removal. To test this hypothesis, Satb1 was conditionally removed using the SSTCre driver line with the RCE:LoxP reporter in the background for visualization, and the sEPSCs of layer IV SST INs were examined at an age (P4–P6) when mutant SST INs begin to die (Fig. 5).
Layer IV SST INs were recorded under voltage-clamp configuration at a holding potential of −70 mV (near the reversal potential for GABA-mediated currents) in the presence of GABAAR antagonist (1 μm GABAzine). For either wild-type animals or Satb1 mutant animals, sEPSCs were readily detected under our recording conditions (Fig. 6A), and were abolished by blockers of NMDA and AMPA receptors (50 μm AP-5 and 20 μm CNQX, respectively), confirming the glutamatergic nature of sEPSCs. Analysis of sEPSCs revealed a highly significant reduction in frequency (mean ± SEM: wild-type, 0.46 ± 0.06 Hz, n = 11; Satb1 mutants, 0.22 ± 0.03 Hz, n = 15; t test, p = 0.001) and a slight, yet significant, reduction in amplitude (mean ± SEM: wild-type, 27 ± 1.9 pA, n = 11; Satb1 mutant, 24 ± 1.6 pA, n = 15; t test, p = 0.04) in P4–P6 mutants (Fig. 6A–C). Cumulative probability plots and Kolmogorov–Smirnov statistical analysis further confirmed an increase in sEPSC interevent interval (Kolmogorov–Smirnov test, p < 0.0001) and a reduction in sEPSC amplitude in Satb1 mutants (Kolmogorov–Smirnov test, p = 0.02) (Fig. 6D,E). Therefore, Satb1 mutant SST INs received significantly reduced excitatory inputs at early postnatal ages, which could be the cellular mechanism underlying the SST IN death that we observed beginning during this period.
Figure 6.
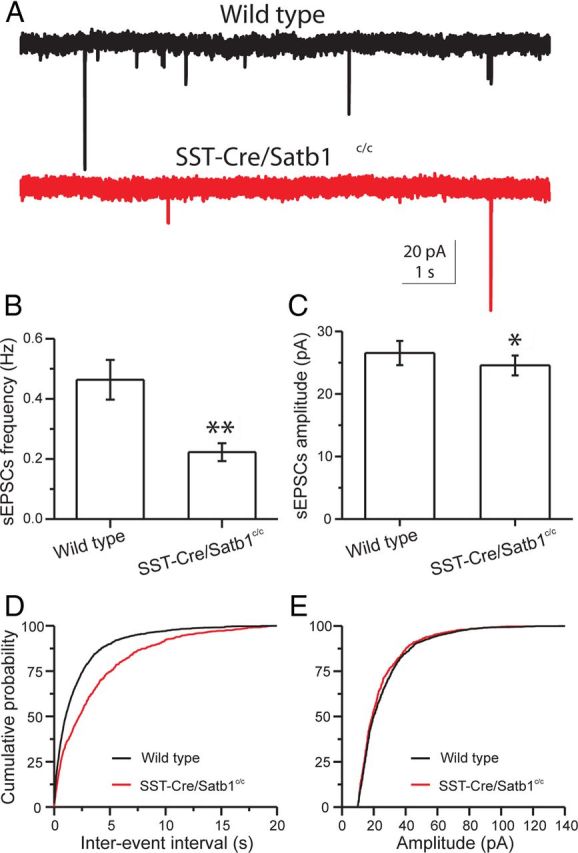
SST INs receive decreased excitatory glutamatergic synaptic inputs at early postnatal ages in SSTCre;Satb1c/c. Recordings obtained in ACSF containing 10 μm GABAzine. A, Representative traces of sEPSCs recorded from somatosensory cortical layer 4 SST INs of P5 wild-type (top, black) and SSTCre;Satb1c/c (bottom, red) mice. The frequency of sEPSCs recorded in SST neurons from SSTCre;Satb1c/c mice was significantly lower. B, C, Bar graphs summarizing the mean frequency and amplitude of sEPSCs recorded from 11 wild-type and 15 Satb1−/− SST INs. The mean frequency of sEPSCs declined significantly in Satb1−/− SST neurons (B). The mean amplitude of sEPSCs was slightly yet significantly reduced in Satb1−/− SST neurons (C). D, Cumulative probability plots of sEPSC interevent interval in SST INs show lower EPSC frequency for SSTCre;Satb1c/c mice (wild-type, black; SSTCre;Satb1c/c, red; Kolmogorov–Smirnov test, p < 0.0001). E, Cumulative probability plots of sEPSC amplitude from SST INs show reduced EPSC amplitudes for SSTCre;Satb1c/c mice (wild-type, black; SSTCre;Satb1c/c, red; Kolmogorov–Smirnov test, p = 0.02).
Neuronal activity regulates the survival of SST INs
Our experiments revealed that Satb1 deletion in SST cells leads to abnormal integration of these INs into nascent cortical circuits, which may subsequently lead to SST cell death. This suggests that Satb1 regulates a genetic program that allows for the formation and/or stabilization of the excitatory inputs necessary for neuronal survival. These results raised the question of whether neuronal activity itself may regulate SST-expressing cell survival. To assess the role of neuronal activity in IN survival, we perturbed electrical activity in developing SST cells by expressing the Kir2.1 channel within cortical IN lineages through in utero electroporation. We electroporated the inward rectifying channel Kir2.1 at different developmental stages and scored for the presence of cortical INs at P5. This strategy allows for the silencing of maturing INs (De Marco García et al., 2011). Since GABAergic subtypes are born at different time points in development, we selectively targeted SST-expressing or CGE-derived interneurons by performing electroporations at different stages. Whereas injections at E13.5 and E14.5 target SST INs, electroporation at E15.5 exclusively targets CGE IN subtypes (Fig. 7A,B) (De Marco García et al., 2011). Our results show that the number of Kir2.1-electroporated cortical INs that survive until P5 is greatly reduced when these electroporations are performed at E13.5 or E14.5, when MGE-derived INs are selectively targeted, compared with electroporation of a control vector (E13.5: 0 surviving neurons/mm2 for Kir2.1 electroporations vs 10 ± 2 for control electroporations; E14.5: 0.6 ± 2 cells for Kir2.1 electroporations vs 8.3 ± 3 for controls; Fig. 7C). By contrast, the number of INs surviving after Kir2.1 electroporation at E15.5, when CGE-derived cells are selectively targeted, is similar to that observed when a control vector is electroporated (9.1 ± 3 for Kir2.1 electroporations vs 9.7 ± 3 for controls). These results suggest that Kir2.1 expression compromises the survival of MGE-derived but not CGE-derived subtypes. To determine whether the expression of Satb1 itself is activity-dependent, we performed genome-wide microarray analysis of control versus Kir2.1-expressing INs in these experiments and found that, while general IN markers, such as Dlx6 and SST, were unchanged, Satb1 expression was strongly downregulated within the Kir2.1-expressing population (data not shown). Together these results suggest that neuronal activity regulates Satb1 expression, which is required for the survival of SST-derived cells.
Figure 7.

Neuronal activity blockade selectively affects the survival of MGE-derived INs. A, B, Representative example of a SST IN electroporated with a Dlx5/6-eGFP plasmid at E13.5. A, Immunohistochemistry for GFP (green) and SST (red). B, Inverted eGFP image depicting neuronal morphology. C, Quantification of the number of cortical INs at P5 after different electroporation stages. Mean values ± SD were obtained from >28 brains for each condition. p values represent the results of a paired t test.
Satb1 removal from IN precursors reduces cortical functional inhibition
We found that Satb1 is essential for the survival and differentiation of PV and SST cells, and characterized the loss of SST cells during the first few postnatal weeks. To examine the functional consequence of Satb1 removal from IN precursors and concomitant IN loss in older animals, we assessed the cortical functional inhibition on excitatory neurons in layer 4, the layer where IN loss was most prominent. Satb1 was conditionally removed using either the Dlx5/6Cre or SSTCre driver line, and sIPSCs were then compared among Dlx5/6Cre;Satb1c/c, SSTCre;Satb1c/c and wild-type P18–P21 animals. In the presence of blockers of NMDA and AMPA receptors (50 μm AP-5 and 20 μm CNQX, respectively), layer 4 excitatory neurons were recorded under voltage-clamp configuration at a holding potential of 0 mV (the reversal potential for glutamate-mediated currents).
For either wild-type animals or Satb1 mutant animals, sIPSCs were readily detected under our recording conditions (Fig. 8A), and were abolished by GABAAR blocker (1 μm GABAzine), confirming the GABAergic nature of sIPSCs. Analysis of sIPSCs revealed a highly significant reduction both in frequency (wild-type: 10.6 ± 0.5 Hz, n = 12; Dlx5/6Cre;Satb1c/c: 5.4 ± 0.5 Hz, n = 10; SSTCre;Satb1c/c: 8.8 ± 0.4 Hz, n = 10; ANOVA, p < 0.0001) and amplitude (wild-type: 109.8 ± 3.2 pA, n = 12; Dlx5/6Cre;Satb1c/c: 79.8 ± 8.4 pA, n = 10; SSTCre;Satb1c/c: 97.7 ± 4.6 pA, n = 10; ANOVA, p = 0.002) in P18–P21 mutants (Fig. 8B,C). Cumulative probability plots and Kolmogorov–Smirnov statistical analysis further confirmed an increase in sIPSC interevent interval in Satb1 mutant animals (wild-type vs Dlx5;6Cre;Satb1c/c, Kolmogorov–Smirnov test, p < 0.0001; wild-type vs SSTCre;Satb1c/c, Kolmogorov–Smirnov test, p < 0.0001) and a reduction in sIPSC amplitude (wild-type vs Dlx5/6Cre;Satb1c/c, Kolmogorov–Smirnov test, p < 0.0001; wild-type vs SSTCre;Satb1c/c, Kolmogorov–Smirnov test, p < 0.0001) (Fig. 8D,E). A separate comparison between Dlx5/6Cre;Satb1c/c and SSTCre;Satb1c/c animals revealed that cortical sIPSCs were more severely compromised in Dlx5/6Cre;Satb1c/c animals (sIPSC frequency: Dlx5/6Cre;Satb1c/c, 5.4 ± 0.5 Hz, n = 10; SSTCre;Satb1c/c, 8.8 ± 0.4 Hz, n = 10; t test, p < 0.001; sIPSC amplitude: Dlx5/6Cre;Satb1c/c, 79.8 ± 8.4 pA, n = 10; SSTCre;Satb1c/c, 97.7 ± 4.6 pA, n = 10; t test, p < 0.05) (Fig. 8B,C), and this observation was further confirmed with cumulative probability plots and Kolmogorov–Smirnov statistical analysis (sIPSC interevent interval: Dlx5/6Cre;Satb1c/c vs SSTCre;Satb1c/c, Kolmogorov–Smirnov test, p < 0.0001; sIPSC amplitude: Dlx5/6Cre;Satb1c/c vs SSTCre;Satb1c/c, Kolmogorov–Smirnov test, p < 0.0001) (Fig. 8D,E). Indeed, the more severe deficit in cortical functional inhibition observed in Dlx5/6Cre;Satb1c/c animals was consistent with the finding that Dlx5/6Cre;Satb1c/c animals displayed more prominent behavioral phenotypes (such as epilepsy and lethality), and clearly relates to the timing of Satb1 removal. When Satb1 was removed from all cortical IN precursors using the Dlx5/6Cre driver line (Fig. 3), IN loss was found in the SST IN population, as well as within the PV IN population, albeit to a lesser extent (Cobos et al., 2005). These results suggest that MGE-derived IN expression of Satb1 is required for the establishment of effective inhibition of cortical pyramidal cells.
Figure 8.
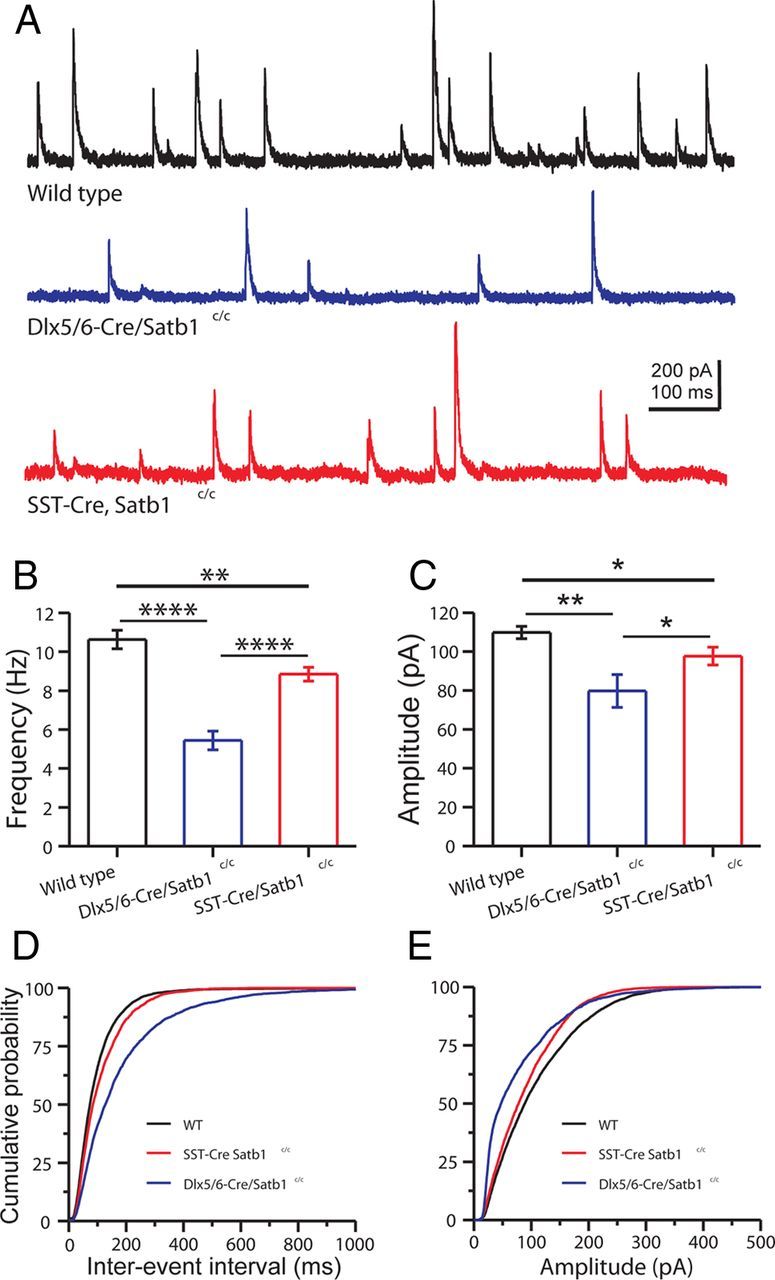
GABAergic synaptic inhibition is decreased in the neocortex of Dlx5/6Cre;Satb1c/c and SSTCre;Satb1c/c mice. A, Representative traces of sIPSCs recorded in somatosensory cortical layer 4 excitatory neurons of P20 wild-type (top, black), Dlx5/6Cre;Satb1c/c (middle, blue), and SSTCre;Satb1c/c (bottom, red) mice. Note that the frequency and amplitude of sIPSCs recorded in the excitatory neurons from Dlx5/6Cre;Satb1c/c or SSTCre;Satb1c/c mice were significantly decreased. B, C, Bar graphs summarizing the mean frequency and amplitude of sIPSCs recorded from 12, 10, and 10 excitatory neurons of wild-type, Dlx5/6Cre;Satb1c/c, and SSTCre;Satb1c/c mice, respectively. B, Statistical comparison of the mean sIPSC frequency in excitatory neurons from wild-type, Dlx5/6Cre;Satb1c/c, and SSTCre;Satb1c/c mice. C, Statistical comparison of the mean sIPSC amplitude in excitatory neurons from wild-type, Dlx5/6Cre;Satb1c/c, and SSTCre;Satb1c/c mice. D, Cumulative probability plots of sIPSC interevent interval show the reduced IPSC frequencies for Dlx5/6Cre;Satb1c/c and SSTCre;Satb1c/c mice, compared with wild types (Kolmogorov–Smirnov test, wild type vs Dlx5/6Cre;Satb1c/c, p < 0.0001; wild type vs SSTCre;Satb1c/c, p < 0.0001; Dlx5/6Cre;Satb1c/c vs SSTCre;Satb1c/c, p < 0.0001). E, Cumulative probability plots of sIPSC amplitudes show reduced IPSC amplitude for Dlx5/6Cre;Satb1c/c and SSTCre;Satb1c/c mice compared with wild types (Kolmogorov–Smirnov test, wild type vs Dlx5/6Cre;Satb1c/c, p < 0.0001; wild type vs SSTCre;Satb1c/c, p < 0.0001; Dlx5/6Cre;Satb1c/c vs SSTCre;Satb1c/c, p < 0.0001). *p < 0.05, **p < 0.01, t test.
Satb1 mutant animals display interictal epileptiform activity during slow-wave sleep
We found that functional inhibition of pyramidal cells was disrupted when Satb1 was removed from inhibitory INs. In addition, we observed massive seizures at ∼P10 in Dlx5/6Cre;Satb1c/c mutant animals (data not shown). To further characterize seizure activity in these animals, EEG recordings were performed between P16 and P18 and analyzed as described previously (Batista-Brito et al., 2009). During slow-wave sleep epochs, we observed interictal epileptiform activity in the cortices of these animals (Fig. 9A,B). No abnormal activity was detected in the hippocampus of these animals. When spectral analysis was performed, we observed a modest reduction in the power of the cortical θ band frequency activity in these animals when compared with controls (Fig. 9C,D). These data further indicate that the balance between cortical excitation and inhibition is disrupted with removal of Satb1 in the IN population, and are thus in agreement with the notion that MGE-derived cells function in the generation of θ oscillations (Le Bon-Jego and Yuste, 2007; Fanselow et al., 2008). Surprisingly, we did not observe any epileptiform activity in animals after removal of Satb1 using the SSTCre driver line, which could reflect a difference in the timing of Satb1 removal, or a requirement for effects on PV cells in this process.
Figure 9.

Satb1−/− mutants display cortical interictal epileptiform activity. Electrodes were implanted in the CA1 region of the right hippocampus, as well as right and left motor cortices (M1R and M1L, respectively) at P14, and EEG recordings were performed between P16 and P18. A, No abnormal activity was observed in cortical or hippocampal traces of Cre-negative littermate control (WT) during slow-wave sleep. B, However, interictal epileptiform spikes were observed in the cortical traces obtained in Dlx5/6Cre;Satb1c/c mutant animals (arrowheads). C, Example slow-wave sleep spectra from a wild-type control animal. D, Example slow-wave sleep spectra from a mutant animal. E, Fast Fourier transformation was performed and the total amplitude per frequency band was quantified (average of 6 10 s epochs, 1 s increments, n = 3 animals). Note the reduction in the θ band power (asterisk) in mutants when compared with controls. Error bars represent SEM.
Satb1 deletion increases neuronal excitability of SST INs
Although a majority of SST INs in Dlx5/6Cre;Satb1c/c mutant animals die before P21, a fraction survive. To determine the consequence of Satb1 deletion on the maturation of SST INs that remain, we next examined the passive and active membrane properties of SST INs at later ages (P18–P21). Resting potential was recorded immediately after the rupture of the neuronal membrane; input resistance was determined by measuring the voltage deflection in response to a hyperpolarizing current pulse (−50 pA, 1000 ms). While the mean resting potential of mutant SST INs was indistinguishable from that of wild-type SST INs (mean ± SEM: wild-type, −65.9 ± 0.8 mV, n = 23 vs Satb1 mutants, −65.3 ± 0.6 mV, n = 18, t test, p = 0.5), Satb1 deletion led to a significant increase in the mean input resistance of surviving layer IV SST INs (mean ± SEM: wild-type, 118.8 ± 7.1 MΩ, n = 23 vs Satb1 mutant, 174.2 ± 22.5 MΩ, n = 18, t test, p < 0.05) (Fig. 10A,B). Moreover, while the action potential voltage threshold of mutant SST INs was not different from that of wild-type SST INs (mean ± SEM: wild-type, −41.8 ± 0.8 mV, n = 23 vs Satb1 mutant, −40.2 ± 0.9 mV, n = 18, t test, p = 0.2), the action potential current threshold of mutant SST INs was significantly lower than that of wild-type SST INs (mean ± SEM: wild-type, 204.4 ± 23.5 pA, n = 23 vs Satb1 mutant, 161.1 ± 20.4 pA, n = 18, t test, p < 0.05) (Fig. 10A,C), consistent with the increase in input resistance. Furthermore, mutant SST INs exhibited a twofold larger membrane time constant than wild-type SST INs (mean ± SEM: wild-type, 8.8 ± 0.4 ms, n = 23 vs Satb1 mutant, 16.2 ± 1.3 ms, n = 18, t test, p < 0.001). In this regard, mutant SST INs fired significantly more action potentials than wild-type SST INs did in response to the same amount of suprathreshold square-current injections (Fig. 10A,D). Together, these results indicate that Satb1 deletion in SST INs not only induced their death at early postnatal ages, but also led to raised excitability of those that survive to later developmental ages. The hyperexcitability of SST INs lacking Satb1 may be a homeostatic compensation that results from the reduced excitatory drive that mutant cells receive. It is also possible that these altered intrinsic properties result from an inability of SST INs to fully mature in the absence of Satb1 expression.
Figure 10.
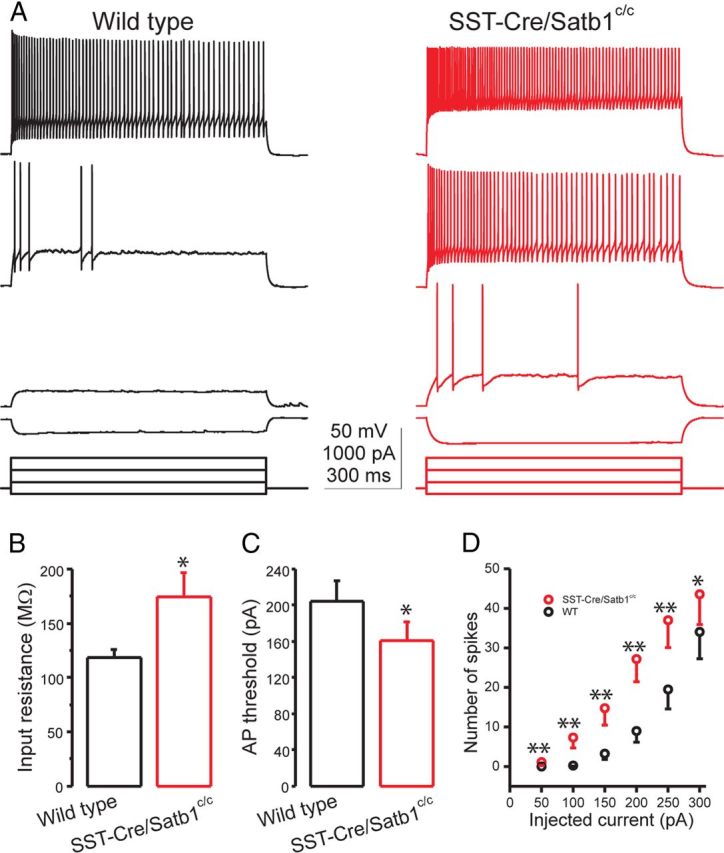
Satb1−/− SST INs exhibit increased membrane excitability at later developmental ages. A, Representative responses to current steps of increasing amplitude recorded from somatosensory cortical layer 4 SST INs in P20 wild-type (left, black) and SSTCre;Satb1c/c (right, red) mice. Note that the SST neuron from the SSTCre;Satb1c/c mouse responds with a larger voltage deflection to the same amplitude of hyperpolarizing current and fires more spikes in response to the same amplitude of depolarizing currents. B, C, Bar graphs summarizing the mean input resistance and action potential current threshold of 23 wild-type and 18 mutant SST neurons. B, The mean input resistance is significantly larger in Satb1−/− SST INs. C, The mean current threshold to elicit a spike is significantly lower in Satb1−/− SST INs. D, Number of spikes plotted against the depolarizing current steps of increasing amplitude. Six 1000 ms depolarizing currents ranging from 50 to 300 pA at 50 pA steps were injected. Note that Satb1−/− SST INs fire more action potentials than wild-type SST INs in response to the same amount of current. *p < 0.5, **p < 0.01, t test.
Satb1 deletion decreases PV IN-mediated synaptic inhibition
We found that cortical inhibition was diminished in Satb1 mutants. In addition, Satb1 deletion had an enduring effect on the membrane properties of surviving SST INs, and deletion of Satb1 had a small but significant effect on the PV cell population. Therefore, we examined the effect of Satb1 removal on PV-expressing IN intrinsic properties and connectivity. As PV expression does not occur until the second postnatal week, Satb1 could not be specifically deleted from PV IN precursors using the PV-Cre driver line. Instead, Dlx6aCre;Satb1c/c mice were crossed with the G42 transgenic line in which EGFP is expressed exclusively by PV-containing INs (Chattopadhyaya et al., 2004). The membrane properties of PV INs were assessed in layer IV of somatosensory cortical slices at P18–P21. In contrast to the alterations in membrane properties of SST INs, no obvious changes were detected in the membrane properties of PV INs compared with those of wild-type PV INs (Table 1).
Table 1.
Comparison of membrane properties between wild-type SST INs or PV INs and surviving Satb1−/− SST INs or PV INs in layer 4 somatosensory cortex
| SST INs |
PV INs |
|||
|---|---|---|---|---|
| Wild type (n = 23) | SSTCre;Satb1c/c (n = 18) | Wild type (n = 12) | Dlx5/6Cre;Satb1c/c (n = 12) | |
| Resting membrane potential (mV) | −65.3 ± 0.6 | −65.9 ± 0.8 | −70.8 ± 1.0 | −69.7 ± 1.2 |
| Membrane resistance (MΩ) | 118.8 ± 7.1 | 174.2 ± 22.5* | 71.3 ± 11.3 | 74.1 ± 11.1 |
| τ (ms) | 8.8 ± 0.4 | 16.2 ± 1.3** | 8.2 ± 0.6 | 7.4 ± 0.6 |
| Action potential threshold (pA) | 204.4 ± 23.5 | 161.1 ± 20.4* | 550 ± 69.9 | 535 ± 40.8 |
| Action potential threshold (mV) | −41.9 ± 0.8 | −40.2 ± 0.9 | −42.4 ± 2.6 | −41.5 ± 1.8 |
| Action potential height (mV) | 64.9 ± 1.7 | 65.1 ± 1.6 | 60.7 ± 3.5 | 60.4 ± 3.1 |
| Action potential width (ms) | 0.41 ± 0.01 | 0.43 ± 0.02 | 0.23 ± 0.01 | 0.22 ± 0.01 |
| Afterhyperpolarization (mV) | 22.4 ± 0.8 | 22.4 ± 1.5 | ||
For an explanation of the parameters, see Materials and Methods. Values shown are expressed as mean ± SEM. Significant differences between mutant and wild-type values are expressed as significant difference from wild type (*p < 0.05; **p < 0.01; t test).
We next examined whether the synaptic inhibition produced by PV INs in Satb1 mutants was affected using paired whole-cell recordings between PV INs and neighboring excitatory neurons in layer 4 somatosensory cortex. Single action potentials were elicited in a PV IN while unitary IPSCs were recorded in a nearby excitatory cell (Fig. 11A, VHOLD = 0 mV). With a soma distance of ∼50 μm (wild-type: 43.8 ± 3.8 μm, n = 14; Dlx6aCre;Satb1c/c: 47.3 ± 3.3 μm, n = 15; t test, p = 0.49), PV INs were found to be connected to excitatory cells with a similarly high connection probability in wild-type and Dlx6aCre;Satb1c/c mice (wild-type, 71.4%; Dlx6aCre;Satb1C/C, 73.3%). However, the amplitude of PV IN-evoked IPSCs was profoundly decreased in Dlx6aCre;Satb1c/c mice (wild-type: 540.5 ± 111.8 pA, n = 10; Dlx6aCre;Satb1c/c: 185.9 ± 45 pA, n = 11; t test, p < 0.01) (Fig. 11A,B). Furthermore, the variation of IPSC amplitude at single connections was significantly greater in Dlx6aCre;Satb1c/c mice (coefficient of variation of IPSC amplitude: wild-type, 0.12 ± 0.02, n = 10; Dlx6aCre;Satb1c/c, 0.21 ± 0.03, n = 11; t test, p < 0.05). The kinetics of PV IN-mediated IPSCs was also slower in Dlx6aCre;Satb1c/c mice. For example, the IPSC rise time (20–80%) was longer (wild-type: 0.41 ± 0.03 ms, n = 10; Dlx6aCre;Satb1c/c: 0.61 ± 0.09 ms, n = 11; t test, p < 0.05) and the IPSC decay time constant was larger (wild-type: 6.52 ± 0.27 ms, n = 10; Dlx6aCre;Satb1c/c: 8.49 ± 0.70 ms, n = 11; t test, p < 0.05). These data suggest that, in Satb1 mutants, the connections from PV-expressing INs onto pyramidal cells are formed, but are less effective than in those of controls, which is in agreement with the reduced levels of functional inhibition observed in such mutants.
Figure 11.

Satb1−/− PV INs produce smaller unitary IPSCs in excitatory neurons. A, Representative paired whole-cell recordings of PV INs and excitatory cells in layer 4 somatosensory cortex of a P19 wild-type mouse (left, black) and a P19 Dlx6aCre;Satb1c/c mouse (right, blue). A single action potential was generated in a PV IN (bottom) and the resulting IPSC was recorded in the excitatory cell (top). Note that the IPSC amplitude was largely reduced in the Dlx6aCre;Satb1c/c mouse. Inset illustrates recording configuration. B, Bar graph summarizing population results showing significantly smaller IPSC amplitudes in principal neurons from Dlx6aCre;Satb1c/c mice. C, Representative IPSCs in an excitatory cell (top) produced by a train of action potentials in PV INs (bottom, 5 action potentials at 40 Hz) of wild-type (left, black) and Dlx6aCre;Satb1c/c mice (right, blue). D, IPSC amplitude plotted as a function of stimulus number in the train. Each IPSC amplitude was normalized to the first response in the train. Data points represent an average of 10 and 11 pairs from wild-type and Dlx6aCre;Satb1c/c mice, respectively. **p < 0.01, t test.
To determine whether the dynamic properties of PV IN synapses are altered by Satb1 deletion, the short-term plasticity of PV IN-mediated IPSCs was assessed with a stimulus train (5 pulses at 40 Hz). IPSC amplitude depressed during the stimulus train for both wild-type and Dlx6aCre;Satb1c/c mice (Fig. 11). To quantify the effect of Satb1 deletion on the kinetics of synaptic depression, the peak amplitude of each IPSC was normalized to the first IPSC in a train and was plotted as a function of stimulus number in the train. The plot revealed a slightly slower short-term depression of IPSCs in Dlx6a-Cre;Satb1c/c mice, yet the difference did not reach significance (Fig. 11). Together these results demonstrate that the early removal of Satb1 has a marked effect on the establishment of PV-expressing IN-mediated inhibition. This suggests that, although the specific role of Satb1 may be different in PV-expressing and SST-expressing IN populations, it is nevertheless required for the proper maturation of both cell types in the developing cortex.
Discussion
In the present study, we describe the role of Satb1 in the differentiation and survival of MGE-derived IN subtypes. We find that Satb1 is expressed in postmitotic INs derived from the MGE, and that it is crucial for the proper differentiation and ultimately the function of these cells. Specifically, Satb1 is required for two phases of IN differentiation: embryonically, for the proper radial migration and synaptic integration of both PV-expressing and SST-expressing cortical INs; and postnatally for the differentiation of SST-expressing INs. Our results thus differentiate between the requirement for Satb1 in migration versus maturation and connectivity. Specifically, while the incorrect positioning of PV-expressing INs may account for the modest reduction in this population, the massive loss of SST-expressing INs upon Satb1 gene removal may reflect their failure to establish proper connectivity within the cortex.
Satb1 deletion disrupts the cortical excitation/inhibition balance
We found that removal of Satb1 in cortical INs had profound effects on the balance between excitation and inhibition in the cortical network. These include reductions in the amplitude and frequency of spontaneous IPSCs in principal cells, as well as interictal epileptiform spikes observed in the EEG of mutant animals. In animals in which Satb1 was removed within all INs, we observed massive seizures beginning in the second postnatal week. We speculate that these seizures contribute to the mortality observed in these animals, as they fail to thrive soon after seizure onset and die by P28. As Satb1-null animals die at a similar time point and in a similar manner (J. Close, unpublished observations), it is probable that the mortality observed in Satb1-null animals is largely explained by the loss of Satb1 within the inhibitory IN population.
Genetic mechanisms underlying Satb1 regulation of SST IN maturation
In work to be published elsewhere, Satb1 appears to function downstream of Lhx6 in the genetic cascade regulating the maturation of SST INs (M. Denaxa and V. Pachnis, personal communication). The special AT-rich binding proteins have been shown to be crucial coordinators of cellular differentiation in multiple cell types (Zhang et al., 2002; Dobreva et al., 2003, 2006; Alcamo et al., 2008; Britanova et al., 2008; Savarese et al., 2009). By regulating multiple genes concurrently via binding to matrix attachment region sequences, chromatin looping, and recruitment of chromatin remodeling factors, Satb1 likely has multiple roles in the normal maturation of SST INs. Further examination of the epigenetic modifications that result from the activity of this gene, as well as the genetic cascades that act downstream of Satb1 will no doubt prove fruitful. Interestingly, although we observe that both PV cells and SST cells express Satb1, its removal has far more profound consequences for the maturation of the SST cell population. This suggests that although PV and SST cells are both derived from the MGE, the progenitor populations that give rise to these respective cell types may be intrinsically divergent at the level of the epigenome. Exploring these differences and the mechanisms by which they are implemented will likely prove crucial in furthering our understanding of how cortical IN diversity is established.
Satb1 differentially regulates PV-IN versus SST-IN development
Recent work from our laboratory suggests that, despite sharing a common origin, PV and SST INs differ in the genetic programs that allow them to achieve their divergent phenotypes. Our characterization of the role of Sox6 in PV-expressing and SST-expressing INs revealed that while Sox6 removal resulted in mispositioning of both PV-expressing and SST-expressing INs, it only significantly affected survival of the PV-expressing IN population (Batista-Brito et al., 2009). As noted above, Satb1 removal also had differential effects depending on the MGE IN subtype in question. Loss of Satb1 resulted in both a general mispositioning of MGE-derived INs and a loss of SST-expressing cells in the somatosensory cortex. Although a portion of the lost SST expression can likely be attributed to downregulation of the SST locus, a large portion of the loss of SST immunoreactivity is due to the death of SST cells. This finding is bolstered by the fact that we see an increase in cleaved Caspase-3 expression at P5 in mutant animals, as well as substantial loss of GFP+ cells in the cortex with Dlx5/6-Cre or SST-Cre removal of Satb1. Because the GFP expression is driven by the constitutively active RCE (R26R CAG-boosted EGFP) reporter after Cre recombination in these experiments, loss of GFP-expressing cells reflects a death of these cells. Notably, even those SST-expressing INs that persist upon Satb1 removal exhibit significant changes in their intrinsic properties, largely resulting in the increased excitability of these cells. This could indicate that, although SST INs survived after Satb1 removal, they remain immature, a result consistent with the higher than normal input resistance observed in these cells. This may also reflect compensation for the loss of the SST IN population. By contrast, PV cell intrinsic properties were largely normal after Satb1 removal. These data indicate that Satb1 acts at multiple levels of MGE IN differentiation, from a general requirement in the radial migration and positioning during early development to a very specific requirement in the maturation and integration of INs into cortical circuitry.
Of note, a previous study showed that removal of Dlx1 also resulted in a loss of SST cells (Cobos et al., 2005). Links connecting Nkx2.1, Lhx6, and the distalless family of transcription factors have not been discovered to date. In fact, previous work has shown that removal of Lhx6 does not alter Dlx1 expression levels in the MGE (Zhao et al., 2008). However, regardless of whether Dlx1 and Satb1 interact molecularly or act in separate, parallel transcription factor cascades, it is clear that both factors affect SST cell migration and differentiation.
SST IN death after Satb1 removal may result from reduced excitatory input
We found that, before their death during the first postnatal week, cortical SST cells within Satb1 mutant animals receive fewer afferent inputs than their wild-type counterparts. Studies of the cochlear nucleus and olfactory bulb have shown that a lack of afferent input to certain neuronal subtypes can result in cell death (Brunjes, 1985; Frazier and Brunjes, 1988; Hashisaki and Rubel, 1989; Corotto et al., 1994; Capurso et al., 1997). In addition, afferent synaptic activity has been shown to be important for the survival of multiple neuronal types during development (Catsicas et al., 1992; Linden, 1994; Mennerick and Zorumski, 2000). This neuroprotective effect is largely mediated by activity-dependent Ca2+ transients during development, which activate neuronal antioxidant defenses to suppress the apoptosis pathway (Papadia et al., 2008; Léveillé et al., 2010). If SST cells are sensitive to afferent input, this lack of synaptic connectivity could provide an explanation of the death of these cells in conditional Satb1 mutants.
Consistent with this hypothesis, we find that suppressing the activity within SST INs results in a reduction in their survival with a time course reminiscent of that seen in Satb1 mutant animals. Furthermore, a number of lines of evidence support the notion that Satb1 expression is regulated by activity. First, a recent study examining Satb1 within cortical pyramidal neurons identifies Satb1 as an immediate early gene whose expression is coordinately regulated in conjunction with other immediate early genes, including Fos and Arc (Balamotis et al., 2012). Second, recent observations by the Pachnis laboratory has demonstrated that KCl-induced activity can strongly upregulate the expression of Satb1 in IN populations in vitro (V. Pachnis, personal communication). Third, complementing the Pachnis findings, we report here that attenuating normal cortical IN activity through Kir2.1 expression results in a reduction in Satb1 expression. Together, these results indicate that Satb1 expression is regulated by activity, likely due to depolarization of terminally differentiating INs by ambient glutamate or GABA levels in the developing cortex. Once established, Satb1 function is required for the maturation and synaptic integration of these cells into cortical circuits, and ultimately this process must be completed to ensure the connectivity and survival of the SST cell population.
Footnotes
We thank Dr. Victor Tarabykin for assistance with Satb1 conditional allele design and his generous gift of the Satb1 antibody, and Lihong Yin for technical assistance.
References
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, McConnell SK. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Alvarez JD, Yasui DH, Niida H, Joh T, Loh DY, Kohwi-Shigematsu T. The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Genes Dev. 2000;14:521–535. [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Marín O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- Anderson S, Mione M, Yun K, Rubenstein JL. Differential origins of neocortical projection and local circuit neurons: role of Dlx genes in neocortical interneuronogenesis. Cereb Cortex. 1999;9:646–654. doi: 10.1093/cercor/9.6.646. [DOI] [PubMed] [Google Scholar]
- Azim E, Jabaudon D, Fame RM, Macklis JD. Sox6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat Neurosci. 2009;12:1238–1247. doi: 10.1038/nn.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balamotis MA, Tamberg N, Woo YJ, Li J, Davy B, Kohwi-Shigematsu T, Kohwi Y. Satb1 ablation alters temporal expression of immediate early genes and reduces dendritic spine density during postnatal brain development. Mol Cell Biol. 2012;32:333–347. doi: 10.1128/MCB.05917-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Brito R, Fishell G. The developmental integration of cortical interneurons into a functional network. Curr Top Dev Biol. 2009;87:81–118. doi: 10.1016/S0070-2153(09)01203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Brito R, Close J, Machold R, Fishell G. The distinct temporal origins of olfactory bulb interneuron subtypes. J Neurosci. 2008a;28:3966–3975. doi: 10.1523/JNEUROSCI.5625-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Brito R, Machold R, Klein C, Fishell G. Gene expression in cortical interneuron precursors is prescient of their mature function. Cereb Cortex. 2008b;18:2306–2317. doi: 10.1093/cercor/bhm258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Brito R, Rossignol E, Hjerling-Leffler J, Denaxa M, Wegner M, Lefebvre V, Pachnis V, Fishell G. The cell-intrinsic requirement of Sox6 for cortical interneuron development. Neuron. 2009;63:466–481. doi: 10.1016/j.neuron.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger TK, Perin R, Silberberg G, Markram H. Frequency-dependent disynaptic inhibition in the pyramidal network: a ubiquitous pathway in the developing rat neocortex. J Physiol. 2009;587:5411–5425. doi: 10.1113/jphysiol.2009.176552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, Sestan N, Molnár Z, Tarabykin V. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 2008;57:378–392. doi: 10.1016/j.neuron.2007.12.028. [DOI] [PubMed] [Google Scholar]
- Brunjes PC. Unilateral odor deprivation: time course of changes in laminar volume. Brain Res Bull. 1985;14:233–237. doi: 10.1016/0361-9230(85)90088-7. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Sousa VH, Fuccillo MV, Hjerling-Leffler J, Miyoshi G, Kimura S, Fishell G. The requirement of Nkx2-1 in the temporal specification of cortical interneuron subtypes. Neuron. 2008;59:722–732. doi: 10.1016/j.neuron.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet. 2006;38:1278–1288. doi: 10.1038/ng1913. [DOI] [PubMed] [Google Scholar]
- Capurso SA, Calhoun ME, Sukhov RR, Mouton PR, Price DL, Koliatsos VE. Deafferentation causes apoptosis in cortical sensory neurons in the adult rat. J Neurosci. 1997;17:7372–7384. doi: 10.1523/JNEUROSCI.17-19-07372.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catsicas M, Péquignot Y, Clarke PG. Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci. 1992;12:4642–4650. doi: 10.1523/JNEUROSCI.12-12-04642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Audinat E, Lambolez B, Angulo MC, Ropert N, Tsuzuki K, Hestrin S, Rossier J. Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci. 1997;17:3894–3906. doi: 10.1523/JNEUROSCI.17-10-03894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. 2005;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- Corotto FS, Henegar JR, Maruniak JA. Odor deprivation leads to reduced neurogenesis and reduced neuronal survival in the olfactory bulb of the adult mouse. Neuroscience. 1994;61:739–744. doi: 10.1016/0306-4522(94)90397-2. [DOI] [PubMed] [Google Scholar]
- DeFelipe J. Neocortical neuronal diversity: chemical heterogeneity revealed by colocalization studies of classic neurotransmitters, neuropeptides, calcium-binding proteins, and cell surface molecules. Cereb Cortex. 1993;3:273–289. doi: 10.1093/cercor/3.4.273. [DOI] [PubMed] [Google Scholar]
- De Marco García NV, Karayannis T, Fishell G. Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature. 2011;472:351–355. doi: 10.1038/nature09865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson LA, Dickinson CD, Kohwi-Shigematsu T. An atypical homeodomain in SATB1 promotes specific recognition of the key structural element in a matrix attachment region. J Biol Chem. 1997;272:11463–11470. doi: 10.1074/jbc.272.17.11463. [DOI] [PubMed] [Google Scholar]
- Dobreva G, Dambacher J, Grosschedl R. SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin mu gene expression. Genes Dev. 2003;17:3048–3061. doi: 10.1101/gad.1153003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Fariñas I, Karsenty G, Grosschedl R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 2006;125:971–986. doi: 10.1016/j.cell.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Du T, Xu Q, Ocbina PJ, Anderson SA. Nkx2.1 specifies cortical interneuron fate by activating Lhx6. Development. 2008;135:1559–1567. doi: 10.1242/dev.015123. [DOI] [PubMed] [Google Scholar]
- Fanselow EE, Richardson KA, Connors BW. Selective, state-dependent activation of somatostatin-expressing inhibitory interneurons in mouse neocortex. J Neurophysiol. 2008;100:2640–2652. doi: 10.1152/jn.90691.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flames N, Pla R, Gelman DM, Rubenstein JL, Puelles L, Marín O. Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci. 2007;27:9682–9695. doi: 10.1523/JNEUROSCI.2750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier LL, Brunjes PC. Unilateral odor deprivation: early postnatal changes in olfactory bulb cell density and number. J Comp Neurol. 1988;269:355–370. doi: 10.1002/cne.902690304. [DOI] [PubMed] [Google Scholar]
- Gelman DM, Martini FJ, Nóbrega-Pereira S, Pierani A, Kessaris N, Marín O. The embryonic preoptic area is a novel source of GABAergic interneurons. J Neurosci. 2009;29:9380–9389. doi: 10.1523/JNEUROSCI.0604-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Wang Y, Markram H. Organizing principles for a diversity of GABAergic interneurons and synapses in the neocortex. Science. 2000;287:273–278. doi: 10.1126/science.287.5451.273. [DOI] [PubMed] [Google Scholar]
- Hashisaki GT, Rubel EW. Effects of unilateral cochlea removal on anteroventral cochlear nucleus neurons in developing gerbils. J Comp Neurol. 1989;283:5–73. doi: 10.1002/cne.902830402. [DOI] [PubMed] [Google Scholar]
- Kapfer C, Glickfeld LL, Atallah BV, Scanziani M. Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat Neurosci. 2007;10:743–753. doi: 10.1038/nn1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci. 1996;16:2701–2715. doi: 10.1523/JNEUROSCI.16-08-02701.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bon-Jego M, Yuste R. Persistently active, pacemaker-like neurons in neocortex. Front Neurosci. 2007;1:123–129. doi: 10.3389/neuro.01.1.1.009.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hjerling-Leffler J, Zagha E, Fishell G, Rudy B. The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci. 2010;30:16796–16808. doi: 10.1523/JNEUROSCI.1869-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé F, Papadia S, Fricker M, Bell KF, Soriano FX, Martel MA, Puddifoot C, Habel M, Wyllie DJ, Ikonomidou C, Tolkovsky AM, Hardingham GE. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci. 2010;30:2623–2635. doi: 10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden R. The survival of developing neurons: a review of afferent control. Neuroscience. 1994;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Liodis P, Denaxa M, Grigoriou M, Akufo-Addo C, Yanagawa Y, Pachnis V. Lhx6 activity is required for the normal migration and specification of cortical interneuron subtypes. J Neurosci. 2007;27:3078–3089. doi: 10.1523/JNEUROSCI.3055-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma WP, Liu BH, Li YT, Huang ZJ, Zhang LI, Tao HW. Visual representations by cortical somatostatin inhibitory neurons—selective but with weak and delayed responses. J Neurosci. 2010;30:14371–14379. doi: 10.1523/JNEUROSCI.3248-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hu H, Berrebi AS, Mathers PH, Agmon A. Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice. J Neurosci. 2006;26:5069–5082. doi: 10.1523/JNEUROSCI.0661-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Fisahn A. Interneurons unbound. Nat Rev Neurosci. 2001;2:11–23. doi: 10.1038/35049047. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Neural activity and survival in the developing nervous system. Mol Neurobiol. 2000;22:41–54. doi: 10.1385/MN:22:1-3:041. [DOI] [PubMed] [Google Scholar]
- Miyoshi G, Butt SJ, Takebayashi H, Fishell G. Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J Neurosci. 2007;27:7786–7798. doi: 10.1523/JNEUROSCI.1807-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Hjerling-Leffler J, Karayannis T, Sousa VH, Butt SJ, Battiste J, Johnson JE, Machold RP, Fishell G. Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J Neurosci. 2010;30:1582–1594. doi: 10.1523/JNEUROSCI.4515-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009;457:1137–1141. doi: 10.1038/nature07663. [DOI] [PubMed] [Google Scholar]
- Nery S, Fishell G, Corbin JG. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci. 2002;5:1279–1287. doi: 10.1038/nn971. [DOI] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnavelas JG, Anderson SA, Lavdas AA, Grigoriou M, Pachnis V, Rubenstein JL. The contribution of the ganglionic eminence to the neuronal cell types of the cerebral cortex. Novartis Found Symp. 2000;228:129–147. doi: 10.1002/0470846631.ch10. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese F, Dávila A, Nechanitzky R, De La Rosa-Velazquez I, Pereira CF, Engelke R, Takahashi K, Jenuwein T, Kohwi-Shigematsu T, Fisher AG, Grosschedl R. Satb1 and Satb2 regulate embryonic stem cell differentiation and Nanog expression. Genes Dev. 2009;23:2625–2638. doi: 10.1101/gad.1815709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–746. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa VH, Miyoshi G, Hjerling-Leffler J, Karayannis T, Fishell G. Characterization of Nkx6–2-derived neocortical interneuron lineages. Cereb Cortex. 2009;19(Suppl 1):1–10. doi: 10.1093/cercor/bhp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23:167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- Taglialatela P, Soria JM, Caironi V, Moiana A, Bertuzzi S. Compromised generation of GABAergic interneurons in the brains of Vax1−/− mice. Development. 2004;131:4239–4249. doi: 10.1242/dev.01299. [DOI] [PubMed] [Google Scholar]
- Tan Z, Hu H, Huang ZJ, Agmon A. Robust but delayed thalamocortical activation of dendritic-targeting inhibitory interneurons. Proc Natl Acad Sci U S A. 2008;105:2187–2192. doi: 10.1073/pnas.0710628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Welagen J, Anderson S. Origins of neocortical interneurons in mice. Dev Neurobiol. 2011;71:10–17. doi: 10.1002/dneu.20857. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- Xu Q, Tam M, Anderson SA. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol. 2008;506:16–29. doi: 10.1002/cne.21529. [DOI] [PubMed] [Google Scholar]
- Yasui D, Miyano M, Cai S, Varga-Weisz P, Kohwi-Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature. 2002;419:641–645. doi: 10.1038/nature01084. [DOI] [PubMed] [Google Scholar]
- Yu M, Xi Y, Pollack J, Debiais-Thibaud M, Macdonald RB, Ekker M. Activity of dlx5a/dlx6a regulatory elements during zebrafish GABAergic neuron development. Int J Dev Neurosci. 2011;29:681–691. doi: 10.1016/j.ijdevneu.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mori M, Burgess DL, Noebels JL. Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J Neurosci. 2002;22:6362–6371. doi: 10.1523/JNEUROSCI.22-15-06362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Flandin P, Long JE, Cuesta MD, Westphal H, Rubenstein JL. Distinct molecular pathways for development of telencephalic interneuron subtypes revealed through analysis of Lhx6 mutants. J Comp Neurol. 2008;510:79–99. doi: 10.1002/cne.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]