

Abstract
The heptapeptide ARHPHPH was identified from biofilms and planktonic cultures of two different strains of Enterococcus faecalis, V583 and ATCC 29212, using matrix assisted laser desorption ionization mass spectrometry (MALDI-MS). ARHPHPH was also imaged at the boundary of cocultured, adjacent E. faecalis and Escherichia coli (ATCC 25922) biofilms, appearing only on the E. faecalis side. ARHPHPH was proteolyzed from κ-casein, a component in the growth media, by E. faecalis microbes. Additionally, top down and bottom up proteomic approaches were combined to identify and spatially locate multiple proteins within intact E. faecalis V583 biofilms by MALDI-MS. The resultant tandem MS data were searched against the NCBInr E. faecalis V583 database to identify thirteen cytosolic and membrane proteins which have functional association with the cell surface. Two of these proteins, enolase and GAPDH, are glycolytic enzymes known to display multiple functions in bacterial virulence in related bacterial strains. This work illustrates a powerful approach for discovering and localizing multiple peptides and proteins within intact biofilms.
I. INTRODUCTION
Peptides and proteins are often used as biomarkers in bacterial identification by mass spectrometry.1, 2 Matrix assisted laser desorption ionization mass spectrometry (MALDI-MS) is well established for the identification of bacterial strains via protein profiling.3–5 Sequencing peptides or proteins using tandem MS and comparing the data with protein databases can reliably identify bacterial species and overcome the limitations of earlier MS-based methods.
The study of protein expression from bacterial biofilms has typically required the separation of proteins from disaggregated biofilms,1 a strategy which loses all information on their spatial distribution within intact biofilms. Imaging proteomics is a MS imaging technique which gives relative abundance and spatial localization of proteins throughout a biological sample.6–8 When imaging proteomics employs trypsin digestion of proteins to peptides, as is frequently the case, identical strategies can be subsequently employed to detect both peptide fragments of proteins and peptides native to a biological sample. Secondary metabolites, endogenous molecules, antibiotics, peptides, and other small molecules have been imaged within intact bacterial colonies by MALDI-MS,9–11 laser desorption postionization MS,12–14 and other MS imaging methods.8, 15
Relatively little work has been reported on in situ imaging proteomics of intact bacterial biofilms, perhaps due to several challenges that must be overcome. Bacterial cells are much smaller than eukaryotic cells.16 Furthermore, the microbes in bacterial biofilms are typically enclosed in an extracellular polysaccharide matrix which hinders easy access for protein digestion and identification. Relatively few bacterial strains have the fully sequenced genomes needed to facilitate protein identification, although new bacterial genomes are reported regularly. However, bacteria have the advantage of much smaller proteomes compared with eukaryotes.
Imaging proteomics can be performed using either a top down or a bottom up approach.17–19 The top down approach identifies low molecular weight proteins by direct MS fragmentation without any preliminary digestion.17, 20 The bottom up approach digests samples prior to MS analysis by depositing trypsin solutions as separate, <200 μm droplets which confines protein digestion and prevents diffusion of digested peptides beyond individual droplets. This strategy aids identification of proteins and permits simultaneous localization of their constituent peptides. Imaging proteomics by the bottom up approach demonstrated previously the feasibility of direct protein identification and imaging in tissues of eukaryotic organisms. For example, on-tissue MALDI-MS analysis of rat brain tissue sections was performed directly from individual trypsin digested spots to identify and localize several proteins.21 Formalin-fixed paraffin embedded rat brain tissues samples were analyzed by MALDI-MS imaging to correlate protein identification and molecular imaging.22 while tissue digestion combined with MALDI-ion mobility MS has also been used for protein identification and imaging directly on rat brain and human cerebellum tissues.23
Traditional proteomics requires proteins be extracted, purified, and concentrated prior to their identification. Imaging proteomics is performed with fewer sample preparation steps and without any protein extraction or concentration steps. However, the imaging technique appears limited to relatively high abundance proteins, at least in mammalian tissues.21–23
Direct analysis and in situ trypsin digestion of intact Enterococcus faecalis biofilms was combined here with MALDI-MS imaging to identify one peptide and over a dozen proteins, establishing the feasibility of imaging proteomics to study biofilms of prokaryotic organisms. E. faecalis is an opportunistic pathogen which is a natural inhabitant of the mammalian gastrointestinal tract and oral cavity. It is known to be a major cause of infections of the urinary tract, respiratory tract, wounds, and root canal.24, 25E. faecalis is known to withstand oxidative stress, desiccation and extreme temperature and pH. It also displays high endogenous resistance to salinity, bile acids, detergents and antimicrobials.25 In particular, the V583 strain of E. faecalis is resistant to the antibiotic vancomycin and was the first vancomycin-resistant clinical isolate reported in the U.S.A.26 The ability of E. faecalis V583 to acquire resistance against most effective antibiotics has attracted global attention.27 Another reason to focus on this strain is that the V583 genome has been completely sequenced, with a total of 3337 predicted protein-encoding open reading frames reported.28 The V583 strain has at least 306 proteins predicted to be covalently anchored to the cell membrane and another 67 proteins non-covalently attached to the membrane.29 These membrane and secreted proteins are known to play a vital role in cell adhesion, apart from their virulence properties.30 Thus, the spatial localization of these proteins within intact biofilms may improve understanding of bacterial virulence mechanisms as a function of culturing conditions. Studies were also performed on biofilms of Escherichia coli (ATCC 25922) and a vancomycin-sensitive, virulent medical isolate strain, E. faecalis ATCC 29212.31 MALDI-MS imaging was demonstrated on cocultured biofilms of E. faecalis V583 and E. coli. E. coli cocultures were included in this study because of the synergistic interaction reported between the two organisms, specifically the higher virulence of E. faecalis observed when in association with E. coli.32
II. MATERIALS AND METHODS
A. Strains and Media
E. faecalis V583 (ATCC 700802), E. faecalis (ATCC 29212), and E. coli (ATCC 25922) were obtained commercially (American Type Culture Collection, Manassus, VA). E. faecalis V583 planktonic culture was grown for 24 h in tryptic soy broth growth medium (Difco, Detroit, MI, USA) containing 10% (w/v) glucose (TSBG) for use in inoculation of drip flow biofilms. Similar cultures were used to inoculate TSBG-amended tryptic soy agar (TSA) for membrane biofilms. Planktonic cultures of E. faecalis (ATCC 29212) and E. coli were grown under similar conditions.
B. Plate and membrane biofilm growth
Single species E. faecalis biofilms were grown either using a drip flow reactor described in detail previously on sterile stainless steel MALDI plates33, 34 or on polycarbonate membranes (Millipore, 0.20 μm pore size, 25 mm diameter, Fisher Scientific). Biosafety protocols were approved by the University of Illinois at Chicago and all biofilm growth was performed inside a certified biosafety cabinet.
Sterile stainless steel MALDI plates were mounted in the flow cell lanes of a sterile drip flow reactor (DFR) and inoculated with 108 colony forming units (CFU) of E. faecalis V583. To allow initial adhesion of the cells to the MALDI plates, the reactor set-up was incubated for 24 h at 37°C and kept flat without any inclination upon addition of 20 mL of TSBG growth media to each of the flow cell lanes. After 24 h of static growth, the drip flow reactor was assembled completely with its stand tilted at an angle of 10°. TSBG growth medium was delivered at a flow rate of 3.6 mL/h for three days using a peristaltic pump to each of the flow cells. At the end of three days, the resultant drip flow biofilms were rinsed with 50 mM ammonium bicarbonate (BICAM) solution (pH 8), followed by submersion in 50 mM BICAM containing 0.5 M sucrose for 2 h, then drying at room temperature prior to further processing.
Membrane biofilms were grown on sterile polycarbonate membranes inoculated with either vancomycin-resistant E. faecalis V583 or with the vancomycin-sensitive ATCC 29212 strain of E. faecalis containing 106 CFU and grown in TSBG-TSA at 37°C for 7 days while replenishing the agar plate daily. For cocultured membrane biofilms, sterile polycarbonate membranes were inoculated with E. faecalis V583 and E. coli cultures containing 106 CFU. The two microbes were spotted at distinct points ~3 mm apart on the same membrane, then grown in TSBG-TSA at 37 °C for 7 days while replenishing the agar plate daily. The two biofilms were observed to grow into one another within 7 days.
C. Biofilm preparation for MALDI analysis
For top down proteomics, 5 mM dithiothreitol in water was twice sprayed on the dried plate biofilms using an airbrush (Testors Corp., Rockford, IL, USA) at 20 psi spray pressure followed by incubation at 37°C for 2 h. Sinapinic acid matrix at 20 g/L concentration in 1:1 (v/v) acetonitrile:trifluoroacetic acid (TFA, 0.1% (v/v) in water) was then sprayed three times with approximately one min of drying time between each spray cycle using the airbrush (20 psi). The plate biofilms were then air dried at room temperature prior to MALDI-MS analysis.
For bottom up proteomics, after treating the plate biofilms with dithiothreitol, 1 g/L trypsin (Sigma-Aldrich) in BICAM was sprayed twice using the airbrush (20 psi) and incubated at 37°C for 24 h prior to spraying with α-cyano-4-hydroxycinnamic acid (CHCA, Sigma-Aldrich) matrix at 20 g/L concentration in 7:3 (v/v) acetonitrile:TFA (0.1% v/v in water) and air drying at room temperature prior to MALDI-MS analysis.
For peptide identification and imaging in single and two species membrane biofilms, membranes were removed from the agar plate and blotted on stainless steel MALDI plate, with care taken to maintain the spatial integrity of the biofilm cells during the transfer. CHCA matrix at 20 g/L concentration in 7:3 (v/v) acetonitrile:TFA (0.1% v/v in water) was then sprayed on the sample and air dried at room temperature prior to MALDI-MS analysis.
D. MALDI-MS
All biofilm samples were analyzed using a MALDI-MS (4700 TOF/TOF, AB SCIEX, Foster City, CA, USA) under MS and MS/MS mode. The MALDI-MS instrument was equipped with a 355 nm Nd: YAG laser operating at 200 Hz with a laser spot size of ~150 μm and laser power set to 2800 arbitrary units, where the maximum of 7000 arbitrary units corresponded to a laser power of ~14 μJ. Helium was used as a collision gas for MS/MS experiments. Data were acquired by commercial software (4000 Series Explorer V3, AB SCIEX). Calibration of the instrument was performed using the standard calibration mixture (mass standards kit for calibration, 4333604, AB SCIEX). MS image acquisition was performed using open source software (4000 Imaging V3, http://maldi-msi.org) with a raster size of 150 μm and 255 laser shots per spot. The MS images acquired were processed further using open source software (BioMap V3803, http://maldi-msi.org).
E. Scanning electron microscopy
CHCA matrix sprayed biofilms were visualized by a scanning electron microscope (S-3000N, Hitachi) with a tungsten electron source operating at 15 keV under high vacuum. Rather than dehydrating by an alcohol/water gradient series, the wet biofilms were simply coated with platinum/palladium alloy.
F. Data analysis for peptide and protein identification
Proteins and peptides were only identified when they were detected in three replicate samples. MS imaging of cocultured biofilms were repeated a minimum of three times with similar mass spectral images observed each time.
Protein database searching was performed using commercial software (MASCOT V 2.2.04 licensed to the University of Illinois at Chicago, National Center for Data Mining, P0127504). All monoisotopic MS/MS data were searched after conversion to MASCOT-compatible format. The entire NCBInr and SwissProt databases were searched using MASCOT without any enzyme, fixed or variable modification selected in the search criteria and the taxonomy selected as all entries (no species selection). The NCBInr database of Enterococcus faecalis V583 (downloaded from NCBI on January 25, 2011, with 6961 records in the database) was also searched. MASCOT searches were performed with a peptide tolerance of ±0.5 Da and a fragment mass tolerance of ±1.0 Da. Searches were performed with one variable modification which included acetylation of protein N-terminal and oxidation of methionines as well as with one missed cleavage and no fixed modifications (since no chemical modification was expected during digestion).
De novo peptide sequencing was performed using commercial software (GPS Explorer TM software-DeNovo Explorer version 3.0, build 291 licensed to The University of Illinois at Chicago). The sequencing was performed with a mass tolerance of ±0.1 Da and without any selection made for enzyme, fixed and variable modifications. Any peptides identified by de novo sequencing were also searched in the EMBL protein database without any species constraints (via MS BLAST similarity search in DeNovo Explorer).
III. RESULTS
A. In situ peptide identification on intact biofilms
Biofilms of two different strains of Gram positive E. faecalis V583 (ATCC 700802) and ATCC 29212 as well as that of Gram negative E. coli (ATCC 25922), were used to test the specificity of MS species observed under various growth conditions. Figure 1 shows representative in situ MALDI-MS spectra for biofilms of E. faecalis V583 grown on a MALDI plate, E. faecalis ATCC 29212 grown on a polycarbonate membrane, and E. coli grown on a polycarbonate membrane. Both strains of E. faecalis biofilms displayed many of the same peaks, despite their growth under different conditions. However, many of those E. faecalis peaks were absent in Gram negative E. coli. Several E. faecalis species-specific peaks with ion counts greater than 500 were observed, including m/z 714.3, 747.4, 780.2 and 851.5. M/z 851.5 and several other peaks were also observed in planktonic cultures of both E. faecalis strains. A peak at m/z 673.3 was observed in both membrane biofilms of E. faecalis and E. coli species. The peak at m/z 655.7 was observed only in E. coli, but not in any E. faecalis biofilms.
Figure 1.

Representative in situ MALDI-MS spectra of E. faecalis plate biofilm (V583, bottom trace), E. faecalis membrane biofilm (ATCC 29212, middle trace) and E. coli membrane biofilm (ATCC 25922, top trace). E. faecalis species specific peaks are labeled and marked with asterisks, Peaks observed in both E. coli and E. faecalis membrane biofilms are marked with Φ (i.e., m/z 673.3). An E. coli specific peak is marked with # (i.e., m/z 655.7).
MALDI-MS/MS (tandem MS) experiments for E. faecalis-specific peaks were performed for de novo sequencing. Figure 2 shows the MALDI-MS/MS spectra of m/z 851.5 with the peptide fragments assigned using the standard notation.35, 36 The peak at m/z 851.5 was identified by de novo sequencing to be the heptapeptide of primary sequence ARHPHPH with a 83.7 MASCOT score. ARHPHPH is part of the primary sequence of κ-casein from Bison bonansus (European bison), specifically the f96 – 102 residues.37 This sequence is likely identical to sequences from other bovine species.
Figure 2.
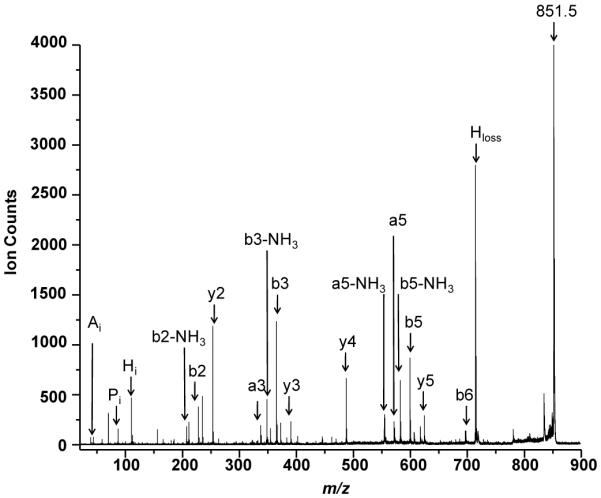
MALDI-MS/MS of m/z 851.5 observed in E. faecalis V583 membrane biofilm with peak assignments indicating the heptapeptide of sequence ARHPHPH.
De novo sequencing also showed that the m/z 780.5 peak corresponded to hexapeptide of sequence RHPHPH (83.6 score, data not shown). The only difference between this peptide and ARHPHPH was the absence of the terminal amino acid residue alanine in the former. It follows that RHPHPH peptide at m/z 780.5 is also derived from κ-casein. However, it is unclear whether RHPHPH exists as a distinct species in cell culture or as only a fragment that subsequently only formed in the gas phase during MS analysis.
Both peptide sequences were further confirmed by trypsin digestion of the E. faecalis biofilms since trypsin cleaves peptides on the c-terminal side of lysine or arginine amino acid residues. Trypsin digestion decreased the intensity of the two peptide peaks (data not shown) which indicated the presence of an arginine residue in the sequences of m/z 780.5 and 851.5.
No peptide sequences or other chemical structures were assigned to any of the other peaks beyond for m/z 780.2 and 851.5. De novo sequencing of the unidentified E. faecalis peaks described above did not yield any unique peptide sequences with a reliable score, indicating that they may not be independent peptides. A regular neutral loss fragment pattern observed in their MS/MS data indicated these peaks might derive from muropeptides of peptidoglycans of E. faecalis. However no conclusive structures were derived for these or any other peaks from their MS/MS data alone.
B. In situ protein identification on intact biofilms
Figure 3 shows representative in situ MALDI-MS data from m/z 3000 to 8500 for proteins identified using the top down proteomics approach. The peaks at m/z 3412.6, 3605.7, 3662.1, 7182.0, 7207.8, 7308.2 and 7325.9 were observed with S/N > 3: all were labeled based on the most intense peak observed in their isotopic distribution. No peaks were observed at higher masses. It was clear that the peak at m/z 3605.7 was a doubly charged peak of m/z 7207.8, based on the isotopic distribution. Two positive hits were observed when the MS/MS data were searched in the protein database using peptide mass fingerprinting (where a protein score greater than 51 was considered significant). This search identified two hypothetical proteins: EF1885 and EF1734 corresponding to m/z 3662.2 and 7325.9, respectively. These m/z values were lower by 975 and 839 Da than the reported masses of these proteins. The MS/MS data from other peaks did not yield a sufficient S/N ratio to generate any successful matches in the protein databases.
Figure 3.
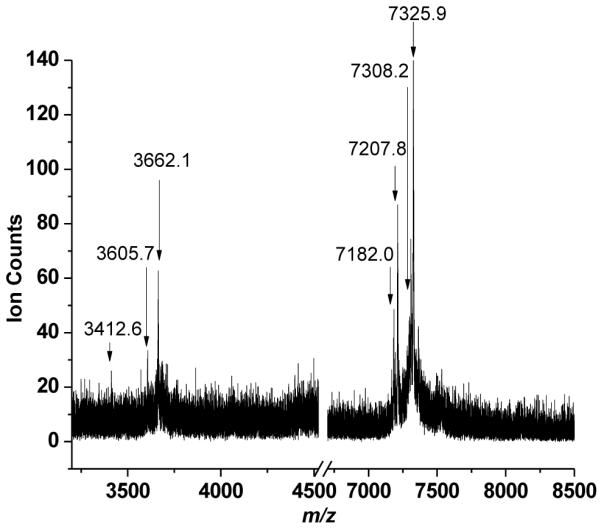
Typical in situ MALDI-MS of intact E. faecalis V583 biofilm.
Figure 4 shows a typical in situ MALDI-MS in the mass range m/z 500 to 2500 of an E. faecalis V583 biofilm for the bottom up proteomic approach with and without trypsin digestion. No peaks were observed at higher masses and matrix interference peaks dominated below m/z 500. The mass spectra of the trypsin digested biofilm (top trace of Figure 4) display several peptide peaks with varying intensities which were not observed in the undigested control biofilm (bottom trace of Figure 4).
Figure 4.

In situ MALDI-MS of E. faecalis V583 biofilm. top trace shows spectrum of biofilm digested with trypsin. Bottom trace shows spectrum of neat untreated control biofilm. Peaks arising after trypsin digestion are marked with asterisks.
MALDI-MS/MS of each trypsin digested peptide (of over 30 different peaks) was performed and the protein database searched. Table 1 summarizes the results, revealing the identity of 11 different proteins found on the intact biofilm using the bottom up proteomic approach. Although most of the trypsin digested peptides peaks were assigned to proteins, a few peptides were not assigned to any protein due to low signal to noise ratios. Peptide sequences were only assigned when they displayed a MASCOT score above 26 which was taken to indicate identity or extensive homology (p<0.05).
Table 1.
List of proteins identified on intact E. faecalis V583 biofilms by top down and bottom up proteomic approaches.
| Top Down | |||||
|---|---|---|---|---|---|
| Database | Sequence Reference | Protein | MASCOT score(a) | Sequence Coverage (%) | Theoretical mol. wt. |
| V583 | gi/29376414 | EF1885 | 65 | 100 | 4637 |
| V583 | gi/29376284 | EF1734 | 104 | 100 | 8165 |
| Bottom Up | ||||||
|---|---|---|---|---|---|---|
| Database | Sequence Reference | Protein | MASCOT score(b) | Peptide m/z used for protein identification | Theoritical mol. wt of the identified protein | Protein subcellular localization |
| V583 | gi|29343947 | enolase | 68 | 717.4, 1188.6, 1851.9 | 46482 | Cytoplasm |
| V583 | gi|29343950 | glyceraldehyde 3-phosphate dehydrogenases | 47 | 749.4, 819.3 | 35749 | Cytoplasm |
| V583 | gi|29342693 | tyrosyl-tRNA synthetase | 37 | 1389.4, 1487.4 | 47231 | Cytoplasm |
| V583 | gi|29344777 | hypothetical protein EF_2843 | 36 | 828.5 | 8359 | Cytoplasm |
| V583 | gi|29344997 | BioY family protein | 34 | 730.1 | 19446 | Membrane |
| V583 | gi|29342815 | excinuclease ABC, subunit B | 34 | 1701.8 | 76054 | Cytoplasm |
| V583 | gi|29344009 | hypothetical protein EF_2033 | 34 | 788.4 | 4498 | Cytoplasm |
| V583 | gi|29342305 | translation elongation factor Tu | 32 | 1139.6, 1702.9, 1862.9, 2184.0 | 43361 | Cytoplasm |
| V583 | gi|29344612 | helicase, putative, RecD/TraA family | 32 | 1863 | 97416 | Cytoplasm |
| V583 | gi|29344600 | spermidine/putrescine ABC transporter, permease protein | 29 | 629.4 | 31484 | Membrane |
| V583 | gi|29344133 | GTP-binding protein | 28 | 712.1 | 46992 | Cytoplasm |
Protein score greater than 51 are significant (p<0.05)
Protein score greater than 26 indicate identity or extensive homology (p<0.05)
Eight peaks between m/z 500 and 1800 were also observed that might be associated with trypsin digested peptides. However, none of these peaks could be reliably assigned as they displayed low MASCOT scores due to low signal to noise, the primary sequences assigned via their MS/MS data were either questionable, and/or lacked correlation with any protein in the database.
C. In situ MALDI-MS imaging
Figure 5 shows the MALDI-MS images of several different ion peaks in cocultures of E. coli and E. faecalis V583 strains grown together until the two biofilms met at the interface: the E. faecalis ARHPHPH peak at m/z 851.5, an endogenous peak at m/z 673.3 observed in both species, and a peak at m/z 655.7 observed predominantly in the E. coli biofilm region. The MS images show the ARHPHPH peptide was observed only in the E. faecalis biofilm region and not in the E. coli region. Both strains grew at approximately equal rates, but the MS images were cropped to emphasize the E. faecalis region.
Figure 5.

MALDI-MS images of cocultured biofilms of E. faecalis V583 and E. coli showing spatial distributions of three ions: (a) m/z 673.3, an endogenous peak observed in both species, (b) m/z 655.7, an E. coli specific peak, and (c) m/z 851.5 corresponding to the ARHPHPH peptide from E. faecalis. (d) is a cartoon showing the relative positions of each strain in the coculture and the absolute size of the images.
MALDI-MS images of trypsin digested peptides on intact E. faecalis V583 single species plate biofilms were also obtained (see Electronic Supplementary Information) to observe the spatial localization of the associated proteins. These results demonstrated the feasibility of imaging proteins on intact biofilms without involving any complicated sample preparation steps employed by traditional proteomics such as protein extraction, purification, and/or concentration. Among the several proteins identified and imaged by MALDI-MS were BioY family protein (a biotin transporter), GTP binding protein (multifunctional protein family), tyrosyl-tRNA synthase (a protein synthesis associated protein), and glyceraldehyde-3-phosphate dehydrogenase (a glycolysis enzyme and possible cell surface virulence factor). Also the MS images of these proteins indicated that different proteins were expressed differentially within the biofilm as a result of heterogeneous environments.
MALDI-MS requires that a matrix compound be sprayed onto the biofilm surface and the presence of this matrix can affect the spatial resolution of MS imaging. The scanning electron micrograph of E. faecalis V583 biofilm sprayed with CHCA matrix showed the ~0.7 μm bacterial cells were apparent within the <10 μm matrix crystals (see Electronic Supplementary Information). However, the spatial resolution of ~150 μm for MS images acquired in this study was not dependent on the matrix crystal size, but rather on the laser spot size (~150 μm diameter) and the step size of MS image acquisition (also 150 μm).
IV. DISCUSSION
A. Summary of results
The ARHPHPH heptapeptide was identified in both vancomycin-resistant and sensitive E. faecalis strains under a range of conditions including mono- and cocultured biofilms as well as planktonic cultures. ARHPHPH was also imaged at the boundary of cocultured, adjacent E. faecalis and E. coli biofilms, appearing only on the E. faecalis side. ARHPHPH was proteolyzed from K-casein, a component in the growth media, by E. faecalis microbes. RHPHPH peptide was also observed, but it could not be determined if this peptide actually existed in culture or was simply a fragment formed within the MS source region from the protonated ARHPHPH parent during the desorption/ionization process. Several peaks specific to E. faecalis other than these two peptides were also observed, but their identities could not be confirmed by de novo sequencing. In addition, eleven different proteins were identified by the bottom up proteomics approach and two proteins were identified by the top down approach. Finally, the feasibility of imaging proteomics on intact E. faecalis V583 bacterial biofilms by MALDI-MS was also demonstrated.
B. Peptide and protein expression
ARHPHPH was found to be associated with E. faecalis biofilms and planktonic culture, but it does not represent any sequence in this organism's proteome. Rather, ARHPHPH corresponds to the f96 – 102 residues of K-casein.37. Casein is one of the components of the trytic soy growth media used here (Difco) and prior work showed that many E. faecalis strains can proteolyze casein.38 Thus, E. faecalis appears to be proteolyzing casein from the growth media to produce ARHPHPH, an observation that has not been previously reported. Other enterococci strains, E. faecium and E. durans, were also found to proteolyze casein, but the E. faecalis strains were report to be more active.38
The imaging of ARHPHPH on intact biofilms demonstrates the potential for MALDI-MS to study the spatial localization of peptides and proteins in their native form. As the E. coli strain examined here did not proteolyze casein, the ARHPHPH peak represents a direct, in situ measurement of species-specific metabolic activity within intact biofilms and could be used to study the spatial interactions of E. faecalis with other microbial species. This is analogous to prior work where surfactin from B. subtilis cocultured with S. aureus was used to map the chemistry associated with the phenotypes by MALDI-MS imaging, with higher concentration of the surfactin observed at the interface suggesting an inhibitory role against S. aureus.11
The eleven proteins identified in vancomycin-resistant E. faecalis V583 by the bottom up strategy include both cytosolic and membrane proteins.29 Several of the cytosolic proteins have `moonlighting' or multiple functional associations with the cell surface as reported for enolase and glyceraldehyde 3-phosphate dehydrogenase (GAPDH).39, 40 These cell surface-associated, multiple function bacterial proteins can be virulence determinants playing important roles in interactions with the host, including adaptive responses to environmental changes, adherence, internalization, toxin synthesis, and escaping the host immune system.29 For instance in Streptococcus agalactiae, the cell surface localized GAPDH protein functions as a virulence factor with B lymphocyte-modulatory activity, while in Streptococcus oralis is important in colonization. GAPDH plays a role in adhesion as a cell binding and binding protein in Streptococcus suis serotype 2. Overall, GAPDH exhibits moonlightling behavior by contributing to bacterial virulence in most gram positive bacteria. Like GAPDH, enolase also plays a role in the virulence behavior of several gram positive bacteria. Enolase is present on the surface of most streptococci and has a strong plasminogen-binding property. Apart from plasminogen-binding, enolase also binds to the salivary mucin (Muc7) in Streptococcus gordonii, contributing to bacterial virulence. Thus, MALDI-MS imaging of these multifunctional proteins in their native form on intact biofilms could permit more detailed studies of their virulence roles in biofilms.30
The top down approach identified only two proteins, EF1885 and EF1734, both predicted to be membrane proteins. This is not surprising since no protein digestion steps were involved in the top down approach. This is consistent with prior results in which only highly abundant membrane proteins were directly detected by MALDI-MS when no cell lysis or protein concentration steps were involved.17, 20, 29 Furthermore, the m/z 12,000 mass limit of the MALDI-MS instrument utilized here severely limits the top down approach to relatively small proteins. While EF1885 and EF1734 were detected intact without any trypsin digestion, the observed masses for their peptides were nonetheless lower than the predicted masses by 839 and 975 Da, respectively. For the protein EF 1734 a predicted cleavage site with the peptide sequence AGGFFLAR of m/z 837.4 was reported.41 The observance of a lower m/z for the protein EF1734 is likely due to the cleavage of this peptide from the protein, although no such cleavage site has yet been reported for the protein EF1885. Fragmentation of intact proteins in the MALDI plume after desorption/ionization may have occurred here, as in-source decay is a common phenomenon in MALDI-MS.17
A further detailed study on imaging these identified proteins in response to various stress factors could give a greater insight and add to the future applications of imaging proteomics to the study of bacterial biofilms. For example, the method can be employed to study the effect of antimicrobial or other culturing perturbations. MALDI-MS and/or laser desorption postionization MS can be used to examine the spatial distribution of antibiotics and/or metabolites in the biofilms14 and can colocalize them with cellular proteins, providing a more complete picture of an antimicrobial challenge and biofilm response.
C. Limits of peptide and protein imaging methodology
Only highly abundant membrane proteins or proteins that have a functional association with the cell surface could be identified and imaged, limiting the number of proteins detected by this technique.21–23 Furthermore, in the bottom-up approach different trypsin digested peptide peaks originating from the same protein could show varying intensity within the biofilm, which could be attributed to differences in digestion efficiency, and/or desorption/ionization efficiency of different trypsin digested peptides.21 The lower sensitivity of the instrument to detect protein peaks of much higher masses due to poor detection efficiencies was also a major reason why more proteins were not detected by the top down approach.21
Optimization and/or incorporation of additional steps in cell lysis and protein denaturation that do not compromise the spatial integrity of the biofilm could aid in detection of additional proteins. The use of different enzymes or a combination of digestion enzymes might also allow identification of a larger number of proteins and their spatial localization on intact bacterial biofilms.
A major challenge with the technique is spot-to-spot variability observed within a single analysis that arises from differences in desorption/ionization efficiency rather than analyte concentration in heterogeneous biofilms. This adverse effect can arise from ion suppression, heterogeneous matrix application, detector noise, and/or sample charging and their net effect is to hinder quantification of analytes. These factors have slowed the progress of MALDI-MS imaging for absolute quantification of analytes in many biological samples.7, 8, 15 Various strategies have been proposed to solve the problem of quantification in MALDI-MS generally.42 For example, stable isotope labeled internal standards have been used for protein quantification in non-imaging proteomic MS.43, 44 Comparison with liquid chromatography MS data45 or use of internal standards46, 47 have also been used to quantify MALDI-MS images of lipids, drugs, and other small molecules. Further, several advanced data processing tools are in the developmental stage to address the noise and surface topography variability in MS imaging.48
Spatial and depth resolution are also an issue in these experiments. The ~150 μm spatial resolution is actually typical for most MALD-MS imaging of biological samples, although resolution below 25 μm is sometimes possible.6–8 The exact depth in the biofilms from which proteins and peptides were detected was not measured, but is also likely to be in the range of tens of microns. For example, MALDI-MS imaging of animal tissue slices found that the matrix solution coated on the sample surface can extract analytes from as deep as 40 μm from the sample surface.49
Nevertheless, the results shown here demonstrate that even relatively low spatial resolution can provide useful information on bacterial biofilms. Spatially resolved chemical identification by MALDI-MS imaging is a useful addition to the toolbox used to study biofilms.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Biomedical Imaging and Bioengineering via grant EB006532. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Biomedical Imaging and Bioengineering or the National Institutes of Health. The authors would like to acknowledge many useful discussions with and assistance by Praneeth D. Edirisinghe, Yang Cui, and Artem Akhmetov as well as Larry Helseth and Alex Schilling of the Research Resources Center.
References
- 1.Jiao Y, D'haeseleer P, Dill BD, Shah M, VerBerkmoes NC, Hettich RL, Banfield JF, Thelen MP. Applied and Environmental Microbiology. 2011;77:5230–5237. doi: 10.1128/AEM.03005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drake RR, Boggs SR, Drake SK. Journal of Mass Spectrometry. 2011;46:1223–1232. doi: 10.1002/jms.2008. [DOI] [PubMed] [Google Scholar]
- 3.Demirev PA, Ho Y-P, Ryzhov V, Fenselau C. Analytical Chemistry. 1999;71:2732–2738. doi: 10.1021/ac990165u. [DOI] [PubMed] [Google Scholar]
- 4.Wilkins CL, Lay JO. Identification of Microorganisms by Mass Spectrometry. Wiley; New York: 2006. [Google Scholar]
- 5.Williamson YM, Moura H, Woolfitt AR, Pirkle JL, Barr JR, Da Gloria Carvalho M, Ades EP, Carlone GM, Sampson JS. Applied and Environmental Microbiology. 2008;74:5891–5897. doi: 10.1128/AEM.00791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seeley EH, Caprioli RM. Proceedings of the National Academy of Sciences, U.S.A. 2008;105:18126–18131. doi: 10.1073/pnas.0801374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chughtai K, Heeren RMA. Chemical Reviews. 2010;110:3237–3277. doi: 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watrous JD, Dorrestein PC. Nature Reviews Microbiology. 2011;9:683–694. doi: 10.1038/nrmicro2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esquenazi E, Coates C, Simmons L, Gonzalez D, Gerwick WH, Dorrestein PC. Molecular bioSystems. 2008;4:562–570. doi: 10.1039/b720018h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu WT, Yang YL, Xu Y, Lamsa A, Haste NM, Yang JY, Ng J, Gonzalez D, Ellermeier CD, Straight PD, Pevzner PA, Pogliano J, Nizet V, Pogliano K, Dorrestein PC. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16286–16290. doi: 10.1073/pnas.1008368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez DJ, Haste NM, Hollands A, Fleming TC, Hamby M, Pogliano K, Nizet V, Dorrestein PC. Microbiology. 2011;157:2485–2492. doi: 10.1099/mic.0.048736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasper GL, Carlson R, Akhmetov A, Moore JF, Hanley L. Proteomics. 2008;8:3816–3821. doi: 10.1002/pmic.200701142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanley L, Zimmermann R. Analytical Chemistry. 2009;81:4174–4182. doi: 10.1021/ac8013675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akhmetov A, Moore JF, Gasper GL, Koin PJ, Hanley L. Journal of Mass Spectrometry. 2010;45:137–145. doi: 10.1002/jms.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vertes A, Hitchins V, Phillips KS. Analytical Chemistry. 2012;84:3858–3866. doi: 10.1021/ac2029997. [DOI] [PubMed] [Google Scholar]
- 16.Anantharaman V, Iyer LM, Aravind L. Annual Review of Microbiology. 2007;61:453–475. doi: 10.1146/annurev.micro.61.080706.093309. [DOI] [PubMed] [Google Scholar]
- 17.Calligaris D, Villard C, Lafitte D. Journal of Proteomics. 2011;74:920–934. doi: 10.1016/j.jprot.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 18.Mallick P, Kuster B. Nature Biotechnology. 2010;28:695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- 19.Speers AE, Wu CC. Chemical Reviews. 2007;107:3687–3714. doi: 10.1021/cr068286z. [DOI] [PubMed] [Google Scholar]
- 20.Dani FR, Francese S, Mastrobuoni G, Felicioli A, Caputo B, Simard F, Pieraccini G, Moneti G, Coluzzi M, della Torre A, Turillazzi S. PLoS ONE. 2008;3:e2822. doi: 10.1371/journal.pone.0002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groseclose MR, Andersson M, Hardesty WM, Caprioli RM. Journal of Mass Spectrometry. 2007;42:254–262. doi: 10.1002/jms.1177. [DOI] [PubMed] [Google Scholar]
- 22.Lemaire R, Desmons A, Tabet JC, Day R, Salzet M, Fournier I. Journal of Proteome Research. 2007;6:1295–1305. doi: 10.1021/pr060549i. [DOI] [PubMed] [Google Scholar]
- 23.Stauber J, MacAleese L, Franck J, Claude E, Snel M, Kaletas BK, Wiel IMVD, Wisztorski M, Fournier I, Heeren RMA. Journal of the American Society for Mass Spectrometry. 2010;21:338–347. doi: 10.1016/j.jasms.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 24.Tailor SA, Bailey EM, Rybak MJ. Annals of Pharmacotherapy. 1993;27:1231–1242. doi: 10.1177/106002809302701014. [DOI] [PubMed] [Google Scholar]
- 25.Klare I, Werner G, W. W. Contributions in Microbiology. 2001;8:108–122. doi: 10.1159/000060406. [DOI] [PubMed] [Google Scholar]
- 26.Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B. Antimicrobial Agents and Chemotherapy. 1989;33:1588–1591. doi: 10.1128/aac.33.9.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, He X, Jiang Z, Wang J, Chen X, Liu D, Wang F, Guo Y, Zhao J, Liu F, Huang L, Yuan J. Journal of Proteome Research. 2010;9:1772–1785. doi: 10.1021/pr901216e. [DOI] [PubMed] [Google Scholar]
- 28.Paulsen IT, Banerjei L, Myers GSA, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. Science. 2003;299:2071–2074. doi: 10.1126/science.1080613. [DOI] [PubMed] [Google Scholar]
- 29.Benachour A, Morin T, Bert LH, Verneuil ALB, Jeune AL, Auffray Y, Pichereau V. Canadian Journal of Microbiology. 2009;55:967–974. doi: 10.1139/w09-052. [DOI] [PubMed] [Google Scholar]
- 30.Coburn PS, Pillar CM, Jett BD, Haas W, Gilmore MS. Science. 2004;306:2270–2272. doi: 10.1126/science.1103996. [DOI] [PubMed] [Google Scholar]
- 31.Swenson JM, Clark NC, Sahm DF, Ferraro MJ, Doern G, Hindler J, Jorgensen JH, Pfaller MA, Reller LB, Weinstein MP, Zabransky RJ, Tenover FC. Journal of Clinical Microbiology. 1995;33:3019–3021. doi: 10.1128/jcm.33.11.3019-3021.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavigne J-P, Nicolas-Chanoine M-H, Bourg G, Moreau J, Sotto A. PLoS ONE. 2008;3:e3370. doi: 10.1371/journal.pone.0003370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu KD, Stewart PS, Xia F, Huang CT, McFeters GA. Applied and Environmental Microbiology. 1998;64:4035–4039. doi: 10.1128/aem.64.10.4035-4039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goeres DM, Hamilton MA, Beck NA, Buckingham-Meyer K, Hilyard JD, Loetterle LR, Lorenz LA, Walker DK, Stewart PS. Nature Protocols. 2009;4:783–788. doi: 10.1038/nprot.2009.59. [DOI] [PubMed] [Google Scholar]
- 35.Roepstorff P, Fohlman J. Biomedical Mass Spectrometry. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 36.Biemann K. Biomedical and Environmental Mass Spectrometry. 1988;16:99–111. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]
- 37.Ong L, Shah NP. Food Science and Technology. 2008;41:1555–1566. [Google Scholar]
- 38.Sarantinopoulos P, Andrighetto C, Georgalaki MD, Rea MC, Lombardi A, Cogan TM, Kalantzopoulos G, Tsakalidou E. International Dairy Journal. 2001;11:621–647. [Google Scholar]
- 39.Pancholi V, Fischetti VA. Journal of Experimental Medicine. 1992;176:415–426. doi: 10.1084/jem.176.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pancholi V, Fischetti VA. Journal of Biological Chemistry. 1998;273:14503–14515. doi: 10.1074/jbc.273.23.14503. [DOI] [PubMed] [Google Scholar]
- 41.Wecker P, Klockow C, Ellrott A, Quast C, Langhammer P, Harder J, Glöckner FO. BMC Genomics. 2009;10:1–16. doi: 10.1186/1471-2164-10-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duncan MW, Roder H, Hunsucker SW. Briefings in Functional Genomics and Proteomics. 2008;7:355–370. doi: 10.1093/bfgp/eln041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monica HE, Derek SS, Carol EP, Christoph B. Journal of Mass Spectrometry. 2009;44:1637–1660. [Google Scholar]
- 44.Anderson DS, Kirchner M, Kellogg M, Kalish LA, Jeong J-Y, Vanasse G, Berliner N, Fleming MD, Steen H. Analytical Chemistry. 2011;83:8357–8362. doi: 10.1021/ac2020905. [DOI] [PubMed] [Google Scholar]
- 45.Koeniger SL, Talaty N, Luo Y, Ready D, Voorbach M, Seifert T, Cepa S, Fagerland JA, Bouska J, Buck W, Johnston RW, Spanton S. Rapid Communications in Mass Spectrometry. 2011;25:503–510. doi: 10.1002/rcm.4891. [DOI] [PubMed] [Google Scholar]
- 46.Goodwin RJA, Mackay CL, Nilsson A, Harrison DJ, Farde L, Andren PE, Iverson SL. Analytical Chemistry. 2011;83:9694–9701. doi: 10.1021/ac202630t. [DOI] [PubMed] [Google Scholar]
- 47.Landgraf RR, Garrett TJ, Prieto Conaway MC, Calcutt NA, Stacpoole PW, Yost RA. Rapid Communications in Mass Spectrometry. 2011;25:3178–3184. doi: 10.1002/rcm.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watrous JD, Alexandrov T, Dorrestein PC. Journal of Mass Spectrometry. 2011;46:209–222. doi: 10.1002/jms.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crossman L, McHugh NA, Hseih Y, Korfmacher WA, Chen J. Rapid Communications in Mass Spectrometry. 2006;20:284–290. doi: 10.1002/rcm.2259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.