

Abstract
Aims
Recessive PDX1 (IPF1) mutations are a rare cause of pancreatic agenesis, with three cases reported worldwide. A recent report described two cousins with a homozygous hypomorphic PDX1 mutation causing permanent neonatal diabetes with subclinical exocrine insufficiency. The aim of our study was to investigate the possibility of hypomorphic PDX1 mutations in a large cohort of patients with permanent neonatal diabetes and no reported pancreatic hypoplasia or exocrine insufficiency.
Methods
PDX1 was sequenced in 103 probands with isolated permanent neonatal diabetes in whom ABCC8, KCNJ11 and INS mutations had been excluded.
Results
Sequencing analysis identified biallelic PDX1 mutations in three of the 103 probands with permanent neonatal diabetes (2.9%). One proband and his affected brother were compound heterozygotes for a frameshift and a novel missense mutation (p.A34fsX191; c.98dupC and p.P87L; c.260C>T). The other two probands were homozygous for novel PDX1 missense mutations (p.A152G; c.455C>G and p.R176Q; c.527G>A). Both mutations affect highly conserved residues located within the homeobox domain. None of the four cases showed any evidence of exocrine pancreatic insufficiency, either clinically, or, where data were available, biochemically. In addition a heterozygous nonsense mutation (p.C18X; c.54C>A) was identified in a fourth case.
Conclusions
This study demonstrates that recessive PDX1 mutations are a rare but important cause of isolated permanent neonatal diabetes in patients without pancreatic hypoplasia/agenesis. Inclusion of the PDX1 gene in mutation screening for permanent neonatal diabetes is recommended as a genetic diagnosis reveals the mode of inheritance, allows accurate estimation of recurrence risks and confirms the requirement for insulin treatment.
Introduction
Permanent neonatal diabetes diagnosed before 6 months is a rare condition with a reported incidence of approximately 1:200 000 births in Caucasian populations 1. A genetic diagnosis is possible for ∼70% of cases 2, with the majority of patients harbouring mutation(s) in the potassium channel genes (KCNJ11 or ABCC8) or in the INS gene 2.
Permanent neonatal diabetes attributable to pancreatic agenesis is very rare and, until recently, the causal gene was identified just in five cases, two of them caused by mutations in the PTF1A gene 3 and three with biallelic PDX1 mutations 4–6. Recently, heterozygous mutations in the transcription factor GATA6 were found to be the major cause of pancreatic agenesis in humans, with 15 cases reported 7.
PDX1 is a member of the homeodomain family of proteins necessary for pancreatic development. Mice with a homozygous null mutation of Pdx1 lack a pancreas. Pdx1+/– mice do not show any relevant phenotype at birth, but haploinsufficiency of Pdx1 in β-cells causes impaired glucose tolerance in older mice (18 months) 8.
Biallelic mutations in PDX1 are a known cause of pancreatic agenesis. The first homozygous PDX1 mutation was reported in an infant with pancreatic agenesis 5, with a homozygous deletion of a single nucleotide, resulting in a premature termination codon (Pro63fsX60). The same mutation was later reported in a second case of pancreatic agenesis 6. A third case 4 was a compound heterozygote for two PDX1 missense mutations (E164D, E178K) that were shown to decrease the protein half-life. Recently, Nicolino et al. 9 reported a family where two cousins with permanent neonatal diabetes and no clinical sign of pancreatic insufficiency harboured a homozygous missense mutation in PDX1 (E178G). Biochemical studies revealed subclinical evidence of exocrine insufficiency in both infants. Functional studies showed that the E178G mutant protein had reduced transactivation activity, but normal localization, expression level and chromatin occupancy. They hypothesized that E178G is a hypomorphic mutation, causing the expression of a protein that still retains some residual activity, leading to a milder phenotype.
The finding that PDX1 mutations can cause permanent neonatal diabetes in the absence of clinical features of exocrine pancreatic insufficiency prompted us to screen a cohort of 103 patients with isolated permanent neonatal diabetes to determine the contribution of biallelic hypomorphic PDX1 mutations to the aetiology of this disease.
Materials and methods
Patient cohort
Mutation testing of PDX1 was performed in 103 probands with isolated permanent neonatal diabetes diagnosed before 6 months (median age at diagnosis 13 weeks; interquartile range 1.5–24). The median birthweight was 2600 g (interquartile range 1800–3250). Seventeen out of 103 were born to consanguineous parents. Mutations in ABCC8, KCNJ11 and INS had been excluded. We also excluded EIF2AK3 mutations in patients born to consanguineous parents. None of the patients was reported to have pancreatic agenesis/hypoplasia or show clinical signs of exocrine insufficiency (defined as need for enzyme supplementation therapy). The relevant clinical information was provided by the referring clinicians. The study was conducted in accordance with the Declaration of Helsinki with informed parental consent given on behalf of children.
Molecular genetic analysis
We screened the coding sequence and intron–exon boundaries of the PDX1 gene (NM_000209.3) using Sanger sequencing at standard conditions and following manufacturers' protocols (primers available on request). For case IV we also sequenced the promoter region (3189 bp), the intron and the 3' untranslated region. Sequencing reactions were run on an ABI3730 capillary machine (Applied Biosystems, Warrington, UK) and analysed using Mutation Surveyor, v3.98 (SoftGenetics, State College, PA, USA). We used the bioinformatic tools SIFT and PolyPhen to predict the effect of novel variants on the PDX1 protein (protein reference NP 000200.1).
Results
Molecular genetic testing
Biallelic mutations in PDX1 were found in three of the 103 probands with isolated permanent neonatal diabetes (2.9%) and a heterozygous nonsense mutation was identified in a fourth case.
A male infant (I-1, Table 1) and his affected brother (I-2, Table 1) were compound heterozygotes for a frameshift and a novel missense mutation (p.A34CfsX191; c.98dupC and p.P87L; c.260C>T). The P87L mutation affects a residue located within the proline-rich region of the PDX1 protein (Fig. 1) and was inherited from the mother (see also Supporting Information, Fig. S1). Both the mother and her sister are homozygous for P87L (their parents are first cousins) and had gestational diabetes. The father, diagnosed with diabetes at 32 years of age, is heterozygous for the frameshift mutation and inherited the mutation from his unaffected mother.
Table 1.
Clinical and molecular characteristics of five cases with neonatal diabetes and PDX1 mutations
| No. | Mutation | Protein | Birthweight | Age at diagnosis (days) | Pancreatic imaging result | Pancreatic imaging modality | Pancreatic exocrine: clinical | Pancreatic exocrine: biochemistry | Current insulin daily dose |
|---|---|---|---|---|---|---|---|---|---|
| I-1 | c. 98dup/259C>T | A34CfsX191/P87L | 2.5 kg/40 weeks | 18 | Normal pancreatic size | Ultrasound sonography | Asymptomatic | Faecal elastase 286 μg/g stool (Reference > 200 μg/g) | 0.52 U/kg |
| I-2 | c. 98dup/259C>T | A34CfsX191/P87L | 2.76 kg/40 weeks | 18 | Normal pancreatic size | Ultrasound sonography | Asymptomatic | Faecal elastase 211 μg/g stool (Reference > 200 μg/g) | 0.39 U/kg |
| II | c. 455C>G/455C>G | A152G/A152G | 1.75 kg/40 weeks | 2 | Normal pancreatic size | Computed tomography abdomen | Asymptomatic | Not known | 0.6 U/kg |
| III | c. 527G>A/527G>A | R176Q/R176Q | 1.70 kg, gestational age not recorded | 20 | Normal pancreatic size | Ultrasound sonography | Asymptomatic | Not known | 0.7 U/kg |
| IV | c. 54C>A/Normal | C18X/Normal | 1.60 kg/37 weeks | 8 | Head of pancreas identified | Ultrasound sonography | Asymptomatic | Faecal chymotrypsin 0.36 mkat/kg (Reference > 0.32 mkat/l) | 0.53 U/kg |
FIGURE 1.
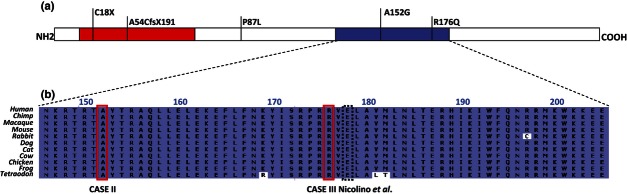
(a) Schematic representation of the PDX1 protein and location of the mutations identified in the four reported cases. The transactivation domain is highlighted in red. The region highlighted in blue is the homeobox domain of the PDX1 protein. (b) Amino acid conservation in the homeodomain. The red rectangles highlight the location of the two homeodomain mutations found in our cohort (cases II and III). The black dashed line indicates the position of the hypomorphic mutation reported by Nicolino et al. 9.
A second proband (II, Table 1) harbours a homozygous mutation in exon 2 of PDX1 (p.A152G; c.455C>G) and both unaffected parents are heterozygous. Another novel homozygous missense mutation (R176Q; c.527G>A) was found in a female infant (III, Table 1). Her unaffected father is heterozygous for the same missense mutation; DNA from the unaffected mother was not available for testing. Both the A152G and R176Q mutations affect highly conserved residues located in the homeodomain of PDX1 (Fig. 1) and were predicted to be deleterious by SIFT and PolyPhen-2.
Heterozygous PDX1 base substitutions were found in four probands. One was a novel nonsense mutation (p.C18X; c.54C>A) (case IV, Table 1). Sequencing of the intron and regulatory regions (promoter and 3' untranslated region) of PDX1 failed to identify a second mutation. Analysis of parental samples showed that the mutation was inherited from the unaffected father.
Two patients were heterozygous for synonymous variants (p.F100F; c.300C>T and p.P244P; c.732C>G) that are unlikely to be pathogenic. We also found a novel missense variant, p.P93R; c.278C>G that affects a residue within a highly variable region of the gene and is likely to be a rare polymorphism.
Clinical characteristics of patients with PDX1 mutations
The first family includes two siblings with permanent neonatal diabetes. Both the proband (I-1, Table 1) and his affected brother were diagnosed with diabetes at 18 days (blood glucose 20.9 mmol/l and 20.6 mmol/l, respectively). Both siblings are treated with insulin pumps. Ultrasound sonography showed a normal pancreas. There were no clinical features of exocrine pancreatic insufficiency and faecal elastase was within the normal range (Table 1).
Proband II and III were born to consanguineous parents (first cousins). Proband II was born at 40 weeks' gestation and developed diabetes at 2 days of life (44.4 mmol/l). The third patient (III, Table 1) was diagnosed with neonatal diabetes mellitus at 20 days (27 mmol/l). There was no clinical evidence of exocrine pancreatic insufficiency in either of these cases, but biochemical testing had not been performed as it was not indicated on clinical grounds.
Finally, we report a female infant in whom a single inactivating PDX1 mutation was identified. The patient (IV, Table 1) was born to unrelated parents without diabetes. She was diagnosed with diabetes at 20 days and insulin treatment commenced. Ultrasound sonography was performed when she was 2 weeks old and showed that at least the head of the pancreas was present. Her faecal chymotrypsin level was normal.
Discussion
Biallelic mutations in PDX1 were identified in four patients from three families with isolated permanent neonatal diabetes. None were reported to have pancreatic agenesis or exocrine pancreatic insufficiency and, in the two cases where faecal elastase was measured, levels were normal.
The first patient and his affected brother are compound heterozygotes for a missense (P87L) and a frameshift mutation (A34CfsX191). Their mother was born to consanguineous parents and is homozygous for the P87L mutation. This mutation is likely to be hypomorphic as she did not develop diabetes until adulthood when she was diagnosed with gestational diabetes during her first pregnancy. The father is heterozygous for the frameshift mutation and any future offspring will be at 50% risk of permanent neonatal diabetes. The two other probands, both from consanguineous families, harbour homozygous missense mutations affecting highly conserved residues within the homeobox domain of the protein (respectively p.R176Q and p.A152G). The R176Q mutation affects a residue just two amino acids upstream of the mutation (E178G) reported by Nicolino et al. 9 (Fig. 1).
A fourth patient was found to be heterozygous for a novel PDX1 nonsense mutation (C18X). The mutation is predicted to introduce a stop codon in the first exon of PDX1 and it is likely that the mutated transcript is subject to nonsense mediated decay. She inherited this mutation from her unaffected father but sequencing analysis of the intronic and regulatory regions of PDX1 failed to identify a second, maternal mutation. This C18X mutation is predicted to cause neonatal diabetes in the homozygous, but not heterozygous state. Our results cannot distinguish between the alternative possibilities of an undetected maternal mutation (e.g. a translocation or inversion) or that the heterozygous mutation is coincidental to her phenotype, consistent with a previous report of heterozygous null PDX1 mutations in population controls 10.
The first cases (three patients from two families) with biallelic PDX1 mutations 4–6 had complete pancreatic agenesis. Subsequently, two cousins with permanent neonatal diabetes and subclinical exocrine insufficiency were reported 9. We report a further four cases with biallelic PDX1 mutations without clinical evidence of exocrine pancreatic insufficiency. In the two cases tested, faecal elastase levels were normal. This suggests that the phenotype caused by biallelic PDX1 mutations is correlated with the mutation type. Null mutations that abolish the protein activity cause pancreatic agenesis, while hypomorphic mutations partially affecting the protein functionality lead to permanent neonatal diabetes with or without exocrine insufficiency.
As expected, mutations in PDX1 were more common in cases of isolated permanent neonatal diabetes born to consanguineous parents compared with outbred cases (2/17 vs. 1/86). Sequencing of PDX1 is recommended in patients with isolated permanent neonatal diabetes, even in the absence of clinical or biochemical features of pancreatic exocrine insufficiency.
Acknowledgments
The authors thank the families for participating in this study. We are grateful to Annet Damhuis, Jess Thompson and Amna Khamis for their technical assistance. SEF was the Sir Graham Wilkins, Peninsula Medical School Research Fellow. ATH was the recipient of a Wellcome Trust Research leave Award. SE and ATH are employed as core members of staff within the NIHR funded Peninsula Clinical Research Facility.
Funding sources
The research leading to these results has received funding from Diabetes UK (grant no. 11/0004193) and the European Community's Seventh Framework Programme (FP7/2007-2013), grant agreement no. FP7-PEOPLE-ITN-2008 (Marie Curie ITN, BOLD).
Competing interests
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Partial pedigree of Family I. The arrow indicates the proband. Colors indicate phenotypic features: black = neonatal diabetes; shading grey = gestational diabetes; grey = adult-onset diabetes. N/N denotes no mutation identified.
{kind=link}
References
- 1.Slingerland AS, Shields BM, Flanagan SE, Bruining GJ, Noordam K, Gach A, et al. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260 000 live births. Diabetologia. 2009;52:1683–1685. doi: 10.1007/s00125-009-1416-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edghill EL, Flanagan SE, Ellard S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev Endocr Metab Disord. 2010;11:193–198. doi: 10.1007/s11154-010-9149-x. [DOI] [PubMed] [Google Scholar]
- 3.Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, J Coleman R, Garrett C, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36:1301–1305. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
- 4.Schwitzgebel VM, Mamin A, Brun T, Ritz-Laser B, Zaiko M, Maret A, et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocrinol Metab. 2003;88:4398–4406. doi: 10.1210/jc.2003-030046. [DOI] [PubMed] [Google Scholar]
- 5.Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 6.Thomas IH, Saini NK, Adhikari A, Lee JM, Kasa-Vubu JZ, Vazquez DM, et al. Neonatal diabetes mellitus with pancreatic agenesis in an infant with homozygous IPF-1 Pro63fsX60 mutation. Pediatr Diabetes. 2009;10:492–496. doi: 10.1111/j.1399-5448.2009.00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lango Allen H, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44:20–22. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas MK, Devon ON, Lee JH, Peter A, Schlosser DA, Tenser MS, et al. Development of diabetes mellitus in aging transgenic mice following suppression of pancreatic homeoprotein IDX-1. J Clin Invest. 2001;108:319–329. doi: 10.1172/JCI12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolino M, Claiborn KC, Senee V, Boland A, Stoffers DA, Julier C. A novel hypomorphic PDX1 mutation responsible for permanent neonatal diabetes with subclinical exocrine deficiency. Diabetes. 2010;59:733–740. doi: 10.2337/db09-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edghill EL, Khamis A, Weedon MN, Walker M, Hitman GA, McCarthy MI, et al. Sequencing PDX1 (insulin promoter factor 1) in 1788 UK individuals found 5% had a low frequency coding variant, but these variants are not associated with Type 2 diabetes. Diabet Med. 2011;28:681–684. doi: 10.1111/j.1464-5491.2011.03269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.