

Abstract
Active immunotherapy may prevent the relapse of acute myeloid leukemia (AML) by inducing leukemia-specific T cells. Here, we investigated whether Wilms’ tumor 1 (WT1) and preferentially expressed antigen in melanoma (PRAME)-specific T cells could be induced upon the priming of healthy donor- and AML patient-derived T cells with HLA-A2-matched, peptide-loaded allogeneic dendritic cells. AML-reactive, tetramer (Tm)-binding and interferon-producing, cytotoxic T lymphocytes specific for PRAME could readily be isolated from healthy individuals and maintained in culture. In this setting, priming efficacy was significantly higher for PRAME than for WT1. The priming of T cells from patient-derived material proved to be near-to-impossible: No leukemia-associated antigen (LAA)-specific T cell could be primed in 4 patients that had recently achieved a complete response (CR), and in only 1 out of 3 patients exhibiting a sustained CR we did observe WT1-specific T cells, though with a low frequency. These findings suggest that the functionality and/or repertoire of T cells differ in healthy subjects and AML patients in CR, and may have repercussions for the implementation of active vaccination approaches against AML.
Keywords: acute myeloid leukemia, dendritic cell-based vaccination, immunotherapy, PRAME, T- cell priming, WT1
Introduction
Therapeutic vaccination with dendritic cells (DCs) is regarded as a viable option for the treatment of minimal residual disease (MRD) in acute myeloid leukemia (AML) patients. DC-based vaccination aims at eliciting a leukemia-associated antigen (LAA)-specific immune response against residual leukemic cells, thus preventing relapse.1 In this respect, peptides carrying defined LAA-epitopes are an attractive source of antigens. Among various LAAs, Wilms’ tumor 1 (WT1) and preferentially expressed antigen of melanoma (PRAME) appear to be overexpressed in various hematological malignancies and share the potential for the induction of specific T-cell responses.2,3 These LAAs are often expressed by AML cells, with expression rates ranging from 60–90%.4 Of note, both PRAME and WT1 are involved in leukemogenesis. PRAME influences disease progression by interfering with retinoic acid (RA) receptor signaling,5 while WT1 inhibits cell differentiation through hitherto unknown mechanisms.6,7 The involvement of PRAME and WT1 in leukemogenesis and their expression by malignant stem cells make them attractive candidates for the detection of MRD and valuable targets for immunotherapy.8-12 Cytotoxic T-lymphocyte (CTL) epitopes of WT1 and PRAME have been characterized for both HLA-A2 and HLA-A24 molecules.13-16 Moreover, T cells recognizing endogenous PRAME- and WT1-derived peptides that are presented by HLA-A2 molecules on the surface of tumor cells have been documented in patients affected by various malignancies, including AML.2,17,18
Here, we investigated the relative priming efficiencies of CTLs obtained from the peripheral blood (PB) of either healthy donors (HD), as leukapheresis (LF) material, or AML patients in first complete remission (CR).
Results
WT1 and PRAME Tm-specific T cells can be primed in HDs but not in AML patients
Primed Tm+ T cells were detectable in samples from HDs for both LAA tested, i.e., PRAME and WT1 (Fig. 1). PRAME and WT1 Tm+ T cells were induced in 3 out of 4 and in 2 out of 5 HDs, respectively (Table 1). Multiple parallel priming cultures of CD8+ T cells were set up in separate wells with a fixed number of CD8+ T cells per well (1 × 106 cells). WT1 Tm+ CD8+ T cells were detected in 4 out of a total of 104 cultures (Table 1; 3/28 for donor 1; 1/20 for donor 2). The priming efficiency of PRAME CD8+ T cells in HDs was significantly higher, with 20 Tm+ cultures out of 51 in total (Table 1; 8/12 for donor 1; 6/15 for donor 2; 6/9 for donor 3). Of note, the same HD-derived T cells were used for PRAME and WT1 priming experiments, and WT1- and PRAME-specific T cells were both detected in donor 1 and 2. The highest frequency of PRAME or WT1 Tm+ CD8+ T cells was detected after 1 to 5 rounds of (re-)stimulation (Table 1). Tm+ CD8+ T-cell frequencies ranged from 0.02 to 1.07% for the PRAME epitope and from 0.01 to 0.09% for the WT1 epitope.
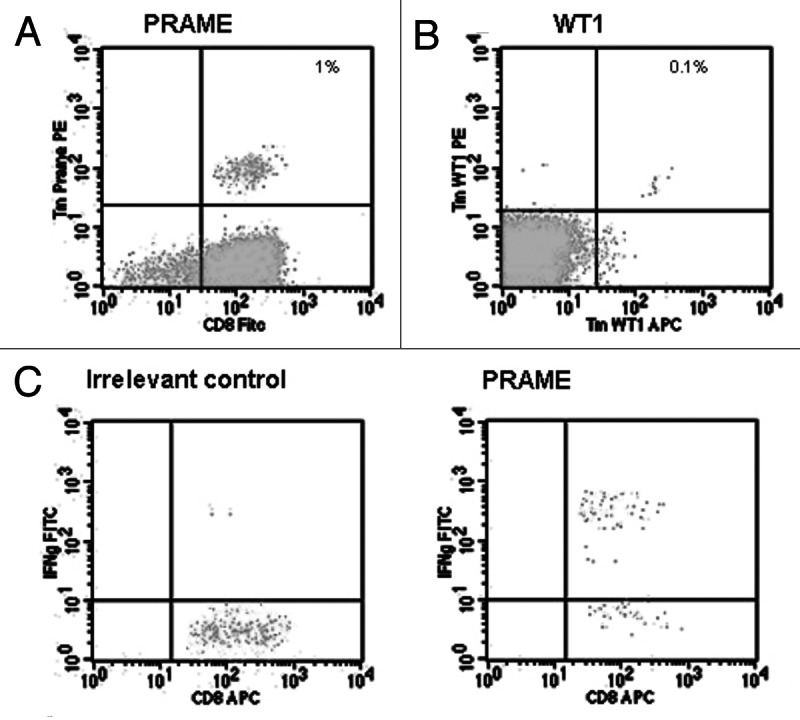
Figure 1. Detection of tetramer+ T cells and interferon γ production by Tm+ T cells expanded from healthy donor-derived peripheral blood mononuclear cells. (A and B) Examples of PRAME100–108 (A) and WT1126–134 (B) tetramer (Tm)+ T cells. (C) Interferon γ (IFNγ) production by PRAME100–108 Tm+ T cells upon incubation with T2 cells loaded with an irrelevant peptide or PRAME100–108.
Table 1. Priming efficiencies of CD8+ T cells obtained from healthy donors or acute myeloid leukemia patients*.
| Tm+ rates after co-culture (per no. of tested HD/patients) | Total no. of Tm+ co-cultures# | Median % Tm+ cells per positive culture (range) | Number of restimulations at maximum of Tm+ | ||
|---|---|---|---|---|---|
| PRAME(100–108) |
HD |
3/4 |
21/50 |
0.10% (0.02–1.07) |
1–5 |
| WT1(126–134) |
HD |
2/5 |
4/104 |
0.07% (0.03–0.09) |
1–4 |
| PRAME(100–108) |
AML-CR1 |
0/4 |
0/44* |
0.00% |
- |
| WT1(126–134) |
AML-CR1 |
0/3 |
0/38 |
0.00% |
- |
| PRAME(100–108) |
AML-LT-CR1 |
0/3 |
0/14 |
0.00% |
- |
| WT1(126–134) | AML-LT-CR1 | 1/3 | 2/14 | 0.05% | 4 |
Patient material was obtained from individuals undergoing short-term (1–6 mo, AML-CR) or long-term (> 1.5 y, AML-LT-CR) complete remission. Priming efficiency was determined upon stimulation with leukemia-associated antigen (LLA)-derived peptide-loaded, HLA-A2-matched, MUTZ-3 cell-derived dendritic cells.
Next, we analyzed the priming efficiencies of CD8+ T cells obtained from AML patients shortly after the achievement of a CR or patients in sustained CR (Table 2). No PRAME and WT1 Tm+ T cells could be expanded from CD8+ T cells isolated from AML patients that had recently achieved a CR (1–6 mo duration, Fisher’s exact test for PRAME in HD vs. AML patients in CR: p < 0.001) (Table 1) and only one patient undergoing a sustained CR exhibited WT1-specific T cells, though with low frequencies in 2 out of 6 wells (UPN 1) (Table 1).
Table 2. Characteristics of acute myeloid leukemia patients in long-term (LT) or short-term (ST) complete remission (CR).
| AML subtype | Duration CR at start priming | |
|---|---|---|
| AML-LT-CR1 UPN 1 |
MDS-RAEB |
1.5 y |
| AML-LT-CR1 UPN 2 |
M1 |
12 y |
| AML-LT-CR1 UPN 3 |
M4 |
14 y |
| AML-CR1 UPN 1 |
AML, not further classified |
6 mo |
| AML-CR1 UPN 2 |
AML-M2 |
1 mo |
| AML-CR1 UPN 3 |
Therapy related AML |
1 mo |
| AML-CR1 UPN 4 | AML-M5B | 2 mo |
Tm+ T cells produce IFNγ upon peptide recognition
Tm+ CD8+ T cells obtained from the PBMCs of HDs produced IFNγ upon recognition of PRAME or WT1 peptides (Fig. 1). PRAME Tm+ T cells were tested for intracellular IFNγ expression upon stimulation with PRAME100–108, and Tm+ populations were indeed able to produce IFNγ (range = 24–71% of Tm+ T cells) (Fig. 1C). CD8+ T cells from two HDs were found to stained positively for the WT1 Tm after priming (Table 1). IFNγ production upon re-stimulation with WT1126–134 was tested in one HD only, exhibiting 27.7% IFNγ+ cells within the Tm+ cell fraction (data not shown).
PRAME-specific T cells kill AML cell lines and patient-derived leukemic cells in a dose-dependent manner
Tm+ T cells recognizing the PRAME epitope were sorted from donor 1 and then cultured in limiting dilution conditions for the generation of PRAME-specific CTL clones. One of such T-cell clones was immortalized by retroviral hTERT transduction,19 followed by the characterization of its functionality. In peptide titration experiments with intracellular IFNγ as readout, this clone was shown to be of intermediate functional avidity, with a half-maximal IFNγ production at 10 to 1 ng/mL PRAME100–108. PRAME100–108-loaded JY cells were killed efficiently by the PRAME-specific T-cell clone, whereas unloaded JY cells remained viable (Fig. 2A). Furthermore, the transduction of K562 cells (which are known to express PRAME)20 with HLA-A2 stimulated IFNγ production by the PRAME-specific CTL clone (data not shown) and rendered them susceptible to its cytotoxic activity, as determined by a flow cytometry-based cytotoxicity assay (Fig. 2A). This demonstrates that the PRAME-specific T-cell clone efficiently recognized the endogenously expressed and processed PRAME epitope. The MHC class I restriction of K562 cell killing was confirmed by the use of an appropriate neutralizing monoclonal antibody (Fig. 2B).

Figure 2. Cytotoxic activity of a healthy donor-derived PRAME100–108-specific cytotoxic T lymphocyte clone toward leukemic cell lines and patient samples. (A and B) Cytotoxic activity of a healthy donor-derived PRAME100–108-specific cytotoxic T lymphocyte (CTL) clone against chronic myeloid leukemia (CML) PRAME+ K562 cells (either HLA-A2- or upon transduction with HLA-A2), acute myeloid leukemia (AML) PRAME-HLA-A2+ ME1 cells, unloaded JY cells and PRAME100–108-loaded JY cells, as monitored in a flow cytometry-based assay with 10:1 effector-to-target (E:T) cell ratios. (B) Cytotoxic activity of PRAME100–108-specific CTLs against HLA-A2-expressing K562 (K562-A2+) cells, in the presence of an MHC class I-blocking antibody (MHC) or appropriate isotype control antibodies (iso). (A) and (B) depict the percentage of viable cells upon incubation with PRAME100–108-specific CTLs. (C and D) Percentage of viable MUTZ-3 cells (C) and patient-derived AML cells (one representative example out of two HLA-A2 matched patients tested) (D) upon incubation with PRAME100–108-specific CTLs at the indicated E:T cell ratios.
To confirm the ability of the PRAME-specific CTL clone to eliminate AML blasts, PRAME-specific T cells were co-cultured with titrated amounts of HLA-A2+ AML target cells, resulting in the killing of both MUTZ-3 cells (PRAME+ AML precursors) (Fig. 2C) and HLA-A2-matched allogeneic primary AML blasts (Fig. 2D, one representative example out of two patient samples tested) in a dose-dependent manner.
Discussion
T cells recognizing WT1 and proteinase 3 have previously been detected in AML patients at diagnosis based on their reactivity in a very sensitive Elispot assay and IFNγ production upon antigen-specific stimulation.18 However, Tm staining-based sorting offers a means of isolation and more extensive functional characterization of specific CTL clones. Since T cells from HDs have not encountered leukemic cells, the outgrowth of PRAME- and WT1-specific CD8+ T cells should be the result of naive T-cell priming. PRAME-specific Tm+ T cells could be readily detected after priming in HDs, whereas the frequency WT1-specific cells was low. Others have studied WT1126–134-loaded DCs for priming, finding that 10 out of 10 low-frequency Tm+ T cells from HDs could be primed, reaching up to 10% Tm+ T cells upon extensive expansion in vitro.21 Such a difference in efficiency between these results and our data may stem from variations in experimental protocols,22 most likely the use of 2-d “fast” DCs and PBMCs in expansion rounds. However, employing the same protocol that we detail here, we have previously observed higher priming efficiencies for other tumor-associated antigens such as hTERT, ERBB3-binding protein 1 (EBP1) and carcinoembryonic antigen (CEA).23 Moreover, Quintarelli and coworkers observed priming efficiencies for the PRAME100–108 epitope in PB samples from HDs that are comparable to those reported here.24 The same group recently reported on the ability of high-avidity PRAME-specific CTLs to eliminate CML lines and primary colony-forming precursors,25 observations that we now confirm for AML. Taken together, these data validate our methodology and the low frequencies of WT1-specific CTL precursors that we observed.
The success of immunotherapeutic strategies relies on an adequately responding immune system. It is not yet clear at what time point the immune system of AML patients has sufficiently recovered from disease and chemotherapy to respond to immunotherapy. Therefore, we analyzed the priming efficiencies in the PB of AML patients that had recently achieved a CR and patients undergoing a sustained CR. Remarkably, no PRAME- and WT1-specific Tm+ T cells could be expanded starting from the CD8+ T cells of patients recently achieving a CR. Such a priming failure can be explained either by a general immunosuppressed state of the patient or by a profound disturbance of the T-cell repertoire. It can take indeed up to six months before normal numbers of T cells are restored once the depleting effects of abundant AML blasts and chemotherapy are relieved.26,27 Furthermore, granulocyte colony-stimulating factor (G-CSF), which is normally given before leukapheresis, can induce T-cell tolerance and hence might have hampered priming efficacy.28 Similar observations have been made in CML patients.24 Conversely, we were able to induce the outgrowth of WT1-specific, but not PRAME-specific, CD8+ T cells upon the stimulation (with WT1126–134-loaded MUTZ-DCs) of material from one (out of 3) AML patient undergoing sustained CR (Table 1). Remarkably, no PRAME-specific T cells could be primed in any of the patients, suggesting that either no PRAME-specific naive T cells were present or PRAME-specific CTLs were anergic and hence unable to expand. Although not conclusive, since we could not expand T cells to numbers compatible with functional tests, these findings strongly suggest that even in the setting of prolonged CR AML-reactive T cells remain relatively dysfunctional (or their repertoire exhausted) and may never fully recover sufficiently to respond to WT1 or PRAME. It remains to be investigated whether this phenomenon is specific for WT1 and PRAME or whether it applies to a wide range of AML-derived antigens and epitopes. Of note, in the case of allogeneic stem cell transplantation (SCT), the immunosuppressive effects of chemotherapy and AML do not affect donor-derived hematopoietic cells, implying that—under these circumstances—T cells are likely to respond similar to HD-derived unprimed T cells. Moreover, allo-reactive T cells specifically recognizing PRAME+ malignant cells might prove to constitute particularly powerful antitumor effector cells.29
In conclusion, we show here that expanding LAA-specific T cells remains a challenge in AML patients recovering from disease and treatment, suggesting that active immunotherapy may be more effective in early-stage AML, including myelodysplastic syndrome (MDS) patients. In this setting, active immunotherapy may be combined with demethylating agents such as 5-azacytidine, as these might augment PRAME expression by leukemic cells and hence PRAME-specific recognition and lysis by CTL clones.30,31 In addition, the ability of PRAME Tm+ T cells derived from HDs to recognize and kill target cells indicates that PRAME is a promising candidate for adoptive T-cell transfer approaches, for instance strategies based on the transduction of TCR-coding genes, and deserves further investigation for the development of induction and consolidation therapeutic regimens against AML.
Materials and Methods
Patient and donor materials
Peripheral blood cells were drawn from 9 AML patients after obtaining the patients’ informed consent. Peripheral blood mononuclear cells (PBMCs) from 3 patients in first CR for 1.5 y or longer (long-term CR1, LT-CR1) (Table 2) were isolated by density centrifugation using Ficoll-Paque (Amersham Pharmacia Biotech). PBMCs (containing > 80% leukemic cells) from 2 patients at diagnosis were isolated using Ficoll-Paque (Amersham Pharmacia Biotech) and stored for later use. Cells were cryopreserved at a controlled rate in liquid nitrogen using RPMI 1640 medium (Gibco) supplemented with 20% heat-inactivated fetal calf serum (FCS) (Greiner) and 10% dimethylsulphoxide (Merck). Autologous leukapheresis products from 4 patients (AML-CR1) (Table 2) were cryopreserved at a controlled rate according to institutional guidelines. Before use, cryopreserved material was rapidly thawed and washed twice in RPMI-1640 supplemented with 40% FCS. Cells were then resuspended in culture medium as described below.
Cell lines
The CD34+ human AML cell line MUTZ-3 [Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ)] was cultured in MEMα medium containing ribonucleosides and deoxyribonucleosides (Life Technologies) supplemented with 20% FCS (Perbio), 100 IU/mL penicillin sodium (Yamanouchi Pharma), 100 μg/mL streptomycin sulfate (Radiumfarma- Fisiopharma), 2.0 mM l-glutamine (Invitrogen), 0.01 mM 2-mercaptoethanol (Merck) and 10% 5637-conditioned medium (5637-CM). The HLA-A2+ Epstein-Barr virus (EBV)-transformed B cell line JY was cultured in Iscove’s modified Dulbecco’s medium (IMDM) (BioWhittaker) supplemented with 10% FCS, 100 IU/mL penicillin sodium, 100 μg/mL streptomycin sulfate, 2.0 mM, l-glutamine and 0.01 mM 2-mercapoethanol. The chronic myeloid leukemia (CML) cell lines K562 (HLA-A2− and PRAME+, from ATCC), K562-A2+ (HLA-A2+ and PRAME+, a kind gift from Dr Carl June) and ME1 (HLA-A2+ and PRAME−, from ATCC) were cultured in IMDM supplemented as above. Culture medium was replenished twice a week.
Isolation of CD8+ T cells from buffy coats and generation of MUTZ-3-derived DCs
Buffy coats were obtained from healthy volunteers according to institutional guidelines in accordance with the Helsinki Declaration of 1975. PBMCs were isolated by density centrifugation using Ficoll-Paque. Subsequently, CD8+ T cells were obtained by negative depletion using an untouched magnetic Microbead-based kit (Miltenyi Biotec GmbH). MUTZ-3-derived DCs (MUTZ3-DCs) were generated as previously described.23 Briefly, MUTZ-3 progenitors were cultured in 12-well tissue culture plates at a concentration of 1 × 105 cell/mL in MEM-α medium in the presence of 100 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF, from Strathmann Biotec), 1,000 U/ml interleukin-4 (IL-4, from Strathmann Biotec) and 2.5 ng/mL tumor necrosis factor α (TNFα, from Strathmann Biotec) for 7 d. Every 3 d fresh cytokines were added. At day 7, the maturation of MUTZ3-DCs was induced by adding monocyte-conditioned medium (MCM) at 30% over a period of 3 d.
Monocyte-conditioned medium
Buffy coats were obtained from healthy volunteers according to institutional guidelines. PBMCs were isolated by density centrifugation using Ficoll-Paque. Subsequently, monocytes were obtained by plating PBMCs onto plastic culture plates coated with 30 μg/mL immunoglobulin. Non-adherent cells were washed away. Adherent cells were incubated for 24 h in IMDM supplemented with 100 IU/mL penicillin sodium, 2.0 mM, l-glutamine and 8% FCS. Supernatants containing the cytokines produced by monocytes were harvested, aliquoted and stored at −20°C until usage. All cultures were performed at 37°C and 5% CO2 in a humidified incubator.
Tetramers and flow cytometry
PE- and/or APC-labeled HLA-A2 Tms presenting the WT1126–134 or PRAME100–108 epitope (Sanquin) were used for cytofluorometric analysis. As a control, a HIV1 Tm was used (kind gift from Dr. Ton Schumacher, NKI). Tm staining was performed in PBS for 15 min at 37°C. Stained cells were analyzed on a FACScalibur cytofluorometer (BD Biosciences) by means of the Cell QuestPro software. To exclude dead cells from the analysis, 0.5 μg/mL propidium iodide (ICN Biomedicals) was used. Tm-guided flow sorting was performed on a FACSAria cytofluorometer (BD Biosciences).
CTL induction
MUTZ3-DCs represent a standardized source of allogeneic DCs that can be used to generate fully functional tumor-specific T cells23,32 In vitro antigen-specific CTLs were generated as described previously.28 Mature MUTZ3-DCs, prepared as described above, were loaded with 10 μg/mL PRAME100–108 (VLDGLDVLL) or WT1126–134 (RMFPNAPYL) in the presence of 3 μg/mL β2-microglobulin (Sigma-Aldrich) for 2–4 h at room temperature and irradiated (50 Gy). PRAME100–108 was synthesized according to the FMOC technology (IHB-LUMC) while WT1126–134 was obtained from Sanquin. Ten thousand peptide-loaded DCs were cultured for 10 d with 1 × 106 CD8+ cells and 1 × 106 irradiated (50 Gy) CD14−/CD8− PBMCs in Yssel’s medium43 supplemented with 1% human AB serum (ICN Biochemicals), 10 ng/mL IL-6 and 10 ng/mL IL-12 in a 24 well tissue-culture plate. At day 1, 10 ng/mL IL-10 (R&D Systems) was added. From day 10 on, CTL cultures were stimulated every week for 5 weeks with 1 × 105 fresh peptide-loaded antigen-presenting cells (10 ng/mL) in the presence of 5 ng/mL IL-7 (Strathmann Biotec). For the first re-stimulation MUTZ3-DCs were used as antigen-presenting cells, while for the second and subsequents re-stimulations HLA-A2+ JY cells were employed. One day prior to each re-stimulation, a sample was taken and analyzed by flow cytometry using both PE- and APC labeled Tms presenting the relevant epitope. Two days after each re-stimulation, 10 U/mL IL-2 (Strathmann Biotec) was added. PRAME Tm+ CTLs were then isolated by Tm+ flow sorting and subsequently cloned by limiting dilution. For this purpose, CTLs were weekly stimulated with irradiated feeder-mixes consisting of allogeneic PBMCs (1:10 ratio with CTLs) and JY cells (1:100 ratio with CTLs) in Yssel’s medium supplemented with 100 ng/mL phytohemagglutin (PHA, from Murex Biotech) and 20 U/mL IL-2. Human telomerase reverse transcriptase (hTERT) was introduced in a PRAME-specific CTL clone as described previously. Briefly PRAME-specific CTLs, stimulated for 48 h with the feeder-mix as described above, were transduced with a retrovirus encoding LZRS-hTERT-IRES-DNGFR, in fibronectin-coated cell culture plates (Retronectin) in the presence of 100 U/ml IL-2. During transduction, plates were centrifuged at 2,000 g for 90 min at 23°C and subsequently incubated at 37°C for 4.5 h. Then, cells were washed and cultured overnight in Yssel’s medium containing 20 U/mL IL-2. Retroviral transduction was repeated one day later, and after 48 h transduction efficiency was determined by the nerve growth factor receptor (NGFR) expression on the cell surface (monitored by fow cytometry).
Intracellular interferon γ detection
To determine the ability of CTL clones to produce interferon γ (IFNγ) upon recognition of a specific target, intracellular IFNγ staining was performed. HLA-A2+PRAME+, HLA-A2−PRAME+ cancer cell lines and JY cells or T2 cells pulsed with either relevant or irrelevant peptides were used as target cells. CTLs were cultured with target cells at an effector:target (E:T) cell ratio of 2:1 in 96-well round-bottom plates, and 0.5 μL of GolgiPlug (BD Biosciences) was added to each well. After 6 h, cells were harvested, washed and stained with PE-labeled tetremers and APC-labeled anti-CD8 monoclonal antibodies. After fixation with 4% paraformaldehyde (Merck) and permeabilization with the BD Perm/wash solution (BD Biosciences), cells were labeled with FITC-conjugated anti-IFNγ antibodies (BD Biosciences) and analyzed by flow cytometry.
FACS-based cytotoxicity assays
The cytotoxic activity of PRAME-specific CTLs was assessed by a flow cytometry-based cytotoxicity assay, as described previously.33 In brief, effector T cells were labeled with 1 μM CFSE (Molecular Probes) for 10 min and subsequently co-cultured with 1 × 104 target cells (cell lines or patient samples) in E:T ratios of 20:1, 10:1 and 5:1, in sterile round-bottom polystyrene tubes (BD Biosciences). All E:T ratios were tested in duplicate. Cell suspension volume was adjusted to 100 μL with complete IMDM. Separate control cultures of effector and target cells alone were performed to check spontaneous apoptosis and secondary necrosis. Co-cultures were kept for 6 h in a 37°C humidified incubator. At the end of cytotoxic contact, target cells were stained at 37°C with PE-conjugated anti-CD34 antibodies plus SYTO62 and 7-AAD.33 MHC-restriction was tested by the addition of 2.5 μg/mL MHC class I-blocking antibodies (W6.32, a kind gift of Dr. S.M. van Ham, Department of Immunopathology, Sanquin Research) or the same amount of appropriate isotype controls (mouse IgG2a, from Sanquin).
Statistical analysis
Statistical significance was determined by two-tailed, paired Student’s t-tests for the results of cytotoxicity assays and by the Fisher’s exact test for priming efficacies. p values < 0.05 were regarded as statistically significant.
Acknowledgments
This research was supported by a grant from Stichting CCA.
Glossary
Abbreviations:
- CR
complete remission
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- FCS
fetal calf serum
- HD
healthy donor
- LAA
leukemia-associated antigen
- LF
leukapheresis
- MRD
minimal residual disease
- MUTZ3-DC
DCs derived from the MUTZ-3 cell line
- PB
peripheral blood
- PBMC
peripheral blood mononuclear cell
- PRAME
preferentially expressed antigen in melanoma
- RA
retinoic acid
- Tm
tetramer
- WT1
Wilms’ tumor 1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
These authors contributed equally to this work.
These authors share senior authorship.
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/23971
References
- 1.van den Ancker W, van Luijn MM, Westers TM, Bontkes HJ, Ruben JM, de Gruijl TD, et al. Recent advances in antigen-loaded dendritic cell-based strategies for treatment of minimal residual disease in acute myeloid leukemia. Immunotherapy. 2010;2:69–83. doi: 10.2217/imt.09.85. [DOI] [PubMed] [Google Scholar]
- 2.Kessler JH, Beekman NJ, Bres-Vloemans SA, Verdijk P, van Veelen PA, Kloosterman-Joosten AM, et al. Efficient identification of novel HLA-A(*)0201-presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. J Exp Med. 2001;193:73–88. doi: 10.1084/jem.193.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Tendeloo VF, Van de Velde AL, Van Driessche A, Cools N, Anguille S, Ladell K, et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci U S A. 2010;107:13824–9. doi: 10.1073/pnas.1008051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greiner J, Schmitt M, Li L, Giannopoulos K, Bosch K, Schmitt A, et al. Expression of tumor-associated antigens in acute myeloid leukemia: Implications for specific immunotherapeutic approaches. Blood. 2006;108:4109–17. doi: 10.1182/blood-2006-01-023127. [DOI] [PubMed] [Google Scholar]
- 5.Oehler VG, Guthrie KA, Cummings CL, Sabo K, Wood BL, Gooley T, et al. The preferentially expressed antigen in melanoma (PRAME) inhibits myeloid differentiation in normal hematopoietic and leukemic progenitor cells. Blood. 2009;114:3299–308. doi: 10.1182/blood-2008-07-170282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greiner J, Bullinger L, Guinn BA, Döhner H, Schmitt M. Leukemia-associated antigens are critical for the proliferation of acute myeloid leukemia cells. Clin Cancer Res. 2008;14:7161–6. doi: 10.1158/1078-0432.CCR-08-1102. [DOI] [PubMed] [Google Scholar]
- 7.Kerst G, Bergold N, Viebahn S, Gieseke F, Kalinova M, Trka J, et al. WT1 protein expression in slowly proliferating myeloid leukemic cell lines is scarce throughout the cell cycle with a minimum in G0/G1 phase. Leuk Res. 2008;32:1393–9. doi: 10.1016/j.leukres.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Bellantuono I, Gao L, Parry S, Marley S, Dazzi F, Apperley J, et al. Two distinct HLA-A0201-presented epitopes of the Wilms tumor antigen 1 can function as targets for leukemia-reactive CTL. Blood. 2002;100:3835–7. doi: 10.1182/blood.V100.10.3835. [DOI] [PubMed] [Google Scholar]
- 9.Candoni A, Tiribelli M, Toffoletti E, Cilloni D, Chiarvesio A, Michelutti A, et al. Quantitative assessment of WT1 gene expression after allogeneic stem cell transplantation is a useful tool for monitoring minimal residual disease in acute myeloid leukemia. Eur J Haematol. 2009;82:61–8. doi: 10.1111/j.1600-0609.2008.01158.x. [DOI] [PubMed] [Google Scholar]
- 10.Matsushita M, Ikeda H, Kizaki M, Okamoto S, Ogasawara M, Ikeda Y, et al. Quantitative monitoring of the PRAME gene for the detection of minimal residual disease in leukaemia. Br J Haematol. 2001;112:916–26. doi: 10.1046/j.1365-2141.2001.02670.x. [DOI] [PubMed] [Google Scholar]
- 11.Østergaard M, Olesen LH, Hasle H, Kjeldsen E, Hokland P. WT1 gene expression: an excellent tool for monitoring minimal residual disease in 70% of acute myeloid leukaemia patients - results from a single-centre study. Br J Haematol. 2004;125:590–600. doi: 10.1111/j.1365-2141.2004.04952.x. [DOI] [PubMed] [Google Scholar]
- 12.Yong AS, Keyvanfar K, Eniafe R, Savani BN, Rezvani K, Sloand EM, et al. Hematopoietic stem cells and progenitors of chronic myeloid leukemia express leukemia-associated antigens: implications for the graft-versus-leukemia effect and peptide vaccine-based immunotherapy. Leukemia. 2008;22:1721–7. doi: 10.1038/leu.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao L, Bellantuono I, Elsässer A, Marley SB, Gordon MY, Goldman JM, et al. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. 2000;95:2198–203. [PubMed] [Google Scholar]
- 14.Greiner J, Döhner H, Schmitt M. Cancer vaccines for patients with acute myeloid leukemia--definition of leukemia-associated antigens and current clinical protocols targeting these antigens. Haematologica. 2006;91:1653–61. [PubMed] [Google Scholar]
- 15.Griffioen M, Kessler JH, Borghi M, van Soest RA, van der Minne CE, Nouta J, et al. Detection and functional analysis of CD8+ T cells specific for PRAME: a target for T-cell therapy. Clin Cancer Res. 2006;12:3130–6. doi: 10.1158/1078-0432.CCR-05-2578. [DOI] [PubMed] [Google Scholar]
- 16.Rosenfeld C, Cheever MA, Gaiger A. WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: therapeutic potential of WT1 targeted therapies. Leukemia. 2003;17:1301–12. doi: 10.1038/sj.leu.2402988. [DOI] [PubMed] [Google Scholar]
- 17.Rezvani K, Yong AS, Savani BN, Mielke S, Keyvanfar K, Gostick E, et al. Graft-versus-leukemia effects associated with detectable Wilms tumor-1 specific T lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood. 2007;110:1924–32. doi: 10.1182/blood-2007-03-076844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheibenbogen C, Letsch A, Thiel E, Schmittel A, Mailaender V, Baerwolf S, et al. CD8 T-cell responses to Wilms tumor gene product WT1 and proteinase 3 in patients with acute myeloid leukemia. Blood. 2002;100:2132–7. doi: 10.1182/blood-2002-01-0163. [DOI] [PubMed] [Google Scholar]
- 19.Hooijberg E, Ruizendaal JJ, Snijders PJ, Kueter EW, Walboomers JM, Spits H. Immortalization of human CD8+ T cell clones by ectopic expression of telomerase reverse transcriptase. J Immunol. 2000;165:4239–45. doi: 10.4049/jimmunol.165.8.4239. [DOI] [PubMed] [Google Scholar]
- 20.Greiner J, Ringhoffer M, Taniguchi M, Li L, Schmitt A, Shiku H, et al. mRNA expression of leukemia-associated antigens in patients with acute myeloid leukemia for the development of specific immunotherapies. Int J Cancer. 2004;108:704–11. doi: 10.1002/ijc.11623. [DOI] [PubMed] [Google Scholar]
- 21.Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naïve repertoire. J Immunol Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 22.Rasaiyaah J, Noursadeghi M, Kellam P, Chain B. Transcriptional and functional defects of dendritic cells derived from the MUTZ-3 leukaemia line. Immunology. 2009;127:429–41. doi: 10.1111/j.1365-2567.2008.03018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santegoets SJ, Schreurs MW, Masterson AJ, Liu YP, Goletz S, Baumeister H, et al. In vitro priming of tumor-specific cytotoxic T lymphocytes using allogeneic dendritic cells derived from the human MUTZ-3 cell line. Cancer Immunol Immunother. 2006;55:1480–90. doi: 10.1007/s00262-006-0142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quintarelli C, Dotti G, De Angelis B, Hoyos V, Mims M, Luciano L, et al. Cytotoxic T lymphocytes directed to the preferentially expressed antigen of melanoma (PRAME) target chronic myeloid leukemia. Blood. 2008;112:1876–85. doi: 10.1182/blood-2008-04-150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quintarelli C, Dotti G, Hasan ST, De Angelis B, Hoyos V, Errichiello S, et al. High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood. 2011;117:3353–62. doi: 10.1182/blood-2010-08-300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. 2009;15:3325–32. doi: 10.1158/1078-0432.CCR-08-3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohnishi K, Yamanishi H, Naito K, Utsumi M, Yokomaku S, Hirabayashi N, et al. Reconstitution of peripheral blood lymphocyte subsets in the long-term disease-free survivors of patients with acute myeloblastic leukemia. Leukemia. 1998;12:52–8. doi: 10.1038/sj.leu.2400891. [DOI] [PubMed] [Google Scholar]
- 28.Rutella S. Granulocyte colony-stimulating factor for the induction of T-cell tolerance. Transplantation. 2007;84(Suppl):S26–30. doi: 10.1097/01.tp.0000269611.66517.bf. [DOI] [PubMed] [Google Scholar]
- 29.Amir AL, van der Steen DM, van Loenen MM, Hagedoorn RS, de Boer R, Kester MD, et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin Cancer Res. 2011;17:5615–25. doi: 10.1158/1078-0432.CCR-11-1066. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez-Cosío S, de la Rica L, Ballestar E, Santamaría C, Sánchez-Abarca LI, Caballero-Velazquez T, et al. Epigenetic regulation of PRAME in acute myeloid leukemia is different compared to CD34+ cells from healthy donors: effect of 5-AZA treatment. Leuk Res. 2012;36:895–9. doi: 10.1016/j.leukres.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 31.Yan M, Himoudi N, Basu BP, Wallace R, Poon E, Adams S, et al. Increased PRAME antigen-specific killing of malignant cell lines by low avidity CTL clones, following treatment with 5-Aza-2′-Deoxycytidine. Cancer Immunol Immunother. 2011;60:1243–55. doi: 10.1007/s00262-011-1024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santegoets SJ, Masterson AJ, van der Sluis PC, Lougheed SM, Fluitsma DM, van den Eertwegh AJ, et al. A CD34(+) human cell line model of myeloid dendritic cell differentiation: evidence for a CD14(+)CD11b(+) Langerhans cell precursor. J Leukoc Biol. 2006;80:1337–44. doi: 10.1189/jlb.0206111. [DOI] [PubMed] [Google Scholar]
- 33.Westers TM, Houtenbos I, Schuurhuis GJ, Ossenkoppele GJ, van de Loosdrecht AA. Quantification of T-cell-mediated apoptosis in heterogeneous leukemia populations using four-color multiparameter flow cytometry. Cytometry A. 2005;66:71–7. doi: 10.1002/cyto.a.20146. [DOI] [PubMed] [Google Scholar]