

Abstract
Matrix metalloproteinases (MMPs) are involved in the remodeling of extracellular matrix in various tissues. Their functioning could be related to the formation of complexes, containing MMP9, MMP2, tissue inhibitor of metalloproteinases type 1 (TIMP1), and neutrophil gelatinase-associated lipocalin (NGAL). Such complexes have not been investigated in either myocardial or skeletal muscles. We examined 20 male pigs with heart failure (HF), and 5 sham-operated animals. There were no differences in the mRNA expression of MMP9, MMP2, TIMP1, and NGAL between diseased and healthy animals, in either left ventricle (LV) myocardium or skeletal muscles. In LV from both diseased and healthy animals, in nonreducing and nondenaturing conditions, we demonstrated the presence of high molecular weight (HMW) complexes (130, 170, and 220 kDa) containing MMP9, TIMP1, and NGAL (also MMP2 in 220 kDa complex) without proteolytic activity, and a proteolytically active 115 kDa MMP9 form together with 72 and 68 kDa bands (proMMP2 and MMP2). Proteolytically active bands were also spontaneously released from HMW complexes. In skeletal muscles from both diseased and healthy animals, in nonreducing and nondenaturing conditions, we found no HMW complexes, and proteolytic activity was associated with the presence of 72 and 68 kDa bands (proMMP2 and MMP2).
1. Introduction
Matrix metalloproteinases (MMPs) are the key enzymes orchestrating the turnover of extracellular matrix (ECM) of virtually all tissues [1]. MMP2 and MMP9 are considered to play a pivotal role in myocardial remodeling in a number of cardiovascular diseases, including myocardial infarction (MI) and ischemic and idiopathic dilated cardiomyopathy (DCM) [2–4]. There are premises that they could also be involved in structural and functional changes in skeletal muscles contributing to the development of skeletal myopathy [5], which constitutes an important pathophysiological element aggravating the progression of heart failure (HF) [6].
MMP9 and MMP2 are released as proenzymes (pro-MMPs) into extracellular space, where they are activated through an enzymatic cleavage to mature MMPs [4]. The proteolytic activity of MMPs is critical for the ongoing processes of tissue remodeling and repair [7], and therefore, it is tightly controlled through different mechanisms including interaction with inhibitors (tissue inhibitors of metalloproteinases, TIMP1-4) or stabilizers of active enzymes (neutrophil gelatinase-associated lipocalin, NGAL) [8]. Both pro-MMPs and MMPs can form the noncovalent high molecular weight (HMW) complexes, which may contain different molecules, such as TIMPs [7] or/and NGAL [9, 10]. There are premises that the stability and the proteolytic activity of MMPs forming such complexes vary from the properties of monomeric forms [8]. HMW complex composed of MMP9, MMP2, MMP3, TIMP1, and TIMP2 found in rat chorioallantoid membranes has been shown to be stabile and latent at high physiological calcium concentrations, dissociating at low calcium concentration and releasing active enzymes [11].
There is also available evidence suggesting that the MMP complexes are present and active in biological fluids. Complexes containing proMMP9, NGAL, and TIMP1 have been demonstrated in the medium from cultured human neutrophils [12], MMP9/NGAL complexes—in the synovial fluid of patients with osteoarthritis [13], MMP9/TIMP1 complexes—in the urine of patients with prostate and bladder carcinoma [14], whereas MMP9/NGAL complexes—in the medium from human cholangiocarcinoma cell cultures [15].
Although the presence and proteolytic activity of MMP9 and MMP2 have been confirmed in both normal and failing hearts [4] as well as in skeletal muscles [5], the formation of HMW complexes containing these 2 major proteases and associated enzymatic activity has not been comprehensively studied in these tissues. Due to the obvious difficulties in obtaining serial human tissue samples, we used porcine model of pacing-induced (nonischaemic) dilated cardiomyopathy (DCM) [16]. Our model is based on a relatively slow, long-term right ventricular (RV) pacing, which is able to induce myocardial dysfunction confirmed by both echocardiography and molecular methods, accompanied by an occurrence of clinical features of the HF syndrome [16]. In this paper, we investigated the mRNA and protein expression of MMP9, MMP2, NGAL and TIMP1, along with the levels of complexes and their proteolytic activity in left ventricle (LV) myocardium and skeletal muscles (biceps femoris, BF) from male pigs with and without RV pacing-induced chronic systolic HF.
2. Methods
2.1. Protocol of the Development of Right Ventricle Pacing-Induced Cardiomyopathy
The study was performed in 25 adult pigs of Polish Large White breed (sibling 8-month-old males, initially weighing from 115 to 125 kg). All animals were treated and cared for in compliance with the Guide for the Care and Use of Laboratory Animals as published by the National Institutes of Health (NIH publication No. 85-23, revised in 1996). All experiments were performed in compliance with the Bioethical Committee of the Wroclaw University of Environmental and Life Sciences guidelines for the experimentation on animals.
We developed the experimental porcine model of chronic systolic nonischaemic HF, that is, tachycardia-induced cardiomyopathy (TIC), which was previously established by our group in female animals [16]. Briefly, single-chamber pacemakers (SENSIA SESR01, Medtronic, Poland) were implanted in all 25 pigs, with a bipolar screw-in pacing transvenous lead (CAPSUREFIX NOVUS 58 cm, Medtronic, Poland) positioned at the apex of RV. The pacemakers were programmed for sequential RV pacing at 170 bpm in 20 randomly chosen animals, whereas 5 remaining pigs served as sham-operated controls.
2.2. Clinical Assessment and Classification of Pigs to Subsequent Stages of HF
All animals remained under the same everyday clinical care. There was no difference in the measurement protocols between the paced and nonpaced male pigs. The assessments were performed regularly on a monthly basis and comprised (a) clinical assessments including the recording of HF signs and symptoms (for details see below) and (b) transthoracic echocardiography (for details see below).
The following signs and symptoms of HF occurring in examined male pigs were evaluated semiquantified using a 0–3 scale (0—no sign/symptom, 3—very severe intensity of a particular sign/symptom): appetite, interest in surroundings, willingness to undertake physical activity (after forcing), dyspnea after exertion, lying down after exertion (fatigue), dyspnea at rest, ascites, redness of snouts and ears after exertion, and snout and ears cyanosis at rest. All scores were averaged for each pig for the particular time point. The following ranges of averaged scores were used for the categorization of pigs to subsequent clinical stages of HF: mild HF (≥0 and ≤1), moderate HF (>1 and ≤2), and severe HF (>2 and ≤3). It was prospectively designed that animals developing the consecutive stages of HF (mild, moderate, and severe) during the experiment would be presented for euthanasia. Control animals underwent euthanasia parallel to TIC pigs and were selected for this procedure in a random manner.
All pigs were euthanized with an overdose of pentobarbital, and the postmortem examinations were performed. Tissue sections from left ventricle (LV) myocardial free wall and skeletal muscles (biceps femoris muscle, BF) were taken and immediately frozen in liquid nitrogen before the further molecular analyses were performed.
2.3. Transthoracic Echocardiography
Transthoracic echocardiography was performed using an imaging ultrasound system (Aloka 4000+ with a 3.5 MHz phased array transducer). Right parasternal but not left apical views were readily visible in all animals. Two-dimension and direct M-mode echocardiography was performed at the right parasternal area in left lateral decubitus position. M-mode tracing was carried out from a long four-chamber view, just below the mitral valve. Diastolic measurements were performed at the onset of the QRS complex of ECG. Systolic measurements were performed at the end of T wave. The ratio of left atrium and aorta diameters (LA/Ao) was measured in diastole in a 2D short axis at the level of aortic valve. LV end-diastolic diameter (LVEDd, cm) and LV end-systolic diameter (LVESd, cm) were measured using the leading-edge method from at least 3 consecutive cardiac cycles as recommended by the American Society for Echocardiography [17]. LV fractional shortening (LVFS) was calculated as a ratio of (LVEDd-LVESd) and LVESd and expressed as a percentage. Using the Teicholz formula [18], LV end-diastolic volume (LVEDV, mL) and LV end-systolic volume (LVESV, mL) LV ejection fraction (LVEF, %), and stroke volume (SV, mL) were computed. LVPW (LV posterior wall) thickening was calculated using the equation = [(LVPWs − LVPWd/LVPWd)] × 100 and expressed as a percentage (LVPWs—at systole, LVPWd—at diastole).
Tissue Doppler imaging was performed from the right parasternal short axis view. Doppler cursor was placed at the LV free wall below the mitral annulus at the level of papillary muscle. The recorded velocities (m/s) represented the contraction of circumferential layer of myocardium, that is, negative diastolic waves Em (early diastolic) and Am (late diastolic) waves. Tracings were recorded at a sweep speed of 100 mm/s and measurements were averaged for 3 separate heart beats.
2.4. Neurohormonal Activation
Venous blood samples were drawn from each animal directly before euthanasia and immediately centrifuged, followed by processing and storage as serum and plasma samples at −80°C until further analyses. Plasma renin activity (PRA) was assayed using an indirect radioimmunoassay (REN-CT2, CIS Bio International, POLATOM, Poland) according to the manufacturer's instructions. Values were expressed as ng/mL/h of a generated angiotensin I. Serum B-type natriuretic peptide (BNP, ng/mL) was assessed at 1 : 5 dilution using a Peptide enzyme immunoassay (EIA) Kits (Bachem, St. Helens, UK) according to the manufacturer's instructions. PRA and BNP measurements were performed in duplicates.
2.5. Quantitative RT-PCR
Total RNA was prepared from 30 mg samples of porcine LV myocardium and BF tissues using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Poland) according to the manufacturer's instructions. The protocol included an on-column DNase digestion to remove the genomic DNA. First-strand cDNA was synthesized using a SuperScript III First-Strand Synthesis System with oligo(dT)20 primer (Invitrogen, Poland).
Based on the genomic and cDNA sequences, the primers for MMP9, MMP2, TIMP1, NGAL, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were designed using the Molecular Beacon Software (Bio-Rad) (Table 1). The primers spanned exon junctions to prevent the amplification of genomic DNA. The GAPDH gene was chosen as a reference to normalize the differences in the amount of RNA and in the efficiency of reverse transcription.
Table 1.
Oligonucleotide primers used in RT-PCR experiments.
| Gene | Primers | Sequence 5′-3′ | Genbank accession no. |
|---|---|---|---|
| MMP9 | sMMP9 f | CCACAGGCCCTCCTTCAG | NM001038004 |
| sMMP9 r | TGAACAGCAGCACCTTACC | ||
| MMP2 | sMMP2 f | TACACCTATACCAAGAACTTCCG | NM214192 |
| sMMP2 r | TGTCCGCCAGATGAACCG | ||
| NGAL | sNGAL f | TTAAGAAATACTCTGGATTGC |
AK240091 |
| sNGAL r | TACTCTTGGTTGTTGGAAAC | ||
| TIMP1 | sTIMP1 f | AGCCAGGAGTTTCTCATAGC | NM213857 |
| sTIMP1 r | TCACAGCCAGCAGCATAG | ||
| GAPDH | GAPDHS | TCACTGCCACCCAGAAGA | ABO38240 |
| GAPDHAS | TACCAGGAAATGAGCTTGAC |
MMP9: matrix metalloproteinase type 9; MMP2: matrix metalloproteinase type 2; NGAL: neutrophil gelatinase-associated lipocalin; TIMP1: tissue inhibitor of metalloproteinases type 1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
The relative amounts of porcine MMP9, MMP2, TIMP1, and NGAL in LV myocardium and BF samples were determined using the quantitative real-time PCR using the iQ5 optical system (Bio-Rad) with the kapa mix (Kapa Biosystems, MA, USA) as appropriate. The reactions were performed under the following conditions: an initial denaturation of 94°C for 10 min, 35 cycles of 94°C for 30 s, 65°C for 30 s, followed by 72°C for 1 min. All measurements were performed in triplicates. The specificity of PCR was determined using a melt-curve analysis for each reaction. PCR products for all subsequent investigated genes were sequenced (Genomed, Poland) to confirm their identity.
The amplification efficiency was established by running a template at successive dilutions. Successive dilutions were plotted against the appropriate CT values to generate a standard curve. The slope calculated from the standard curve was used to determine the amplification efficiency (E) according to the formula: E = 101/slope. Since the amplification efficiencies for the target amplicons and GAPDH were not comparable, the Pfaffl method was used to determine the relative expression [19]. mRNA expression was presented in arbitrary units (AU), where the sample from LV myocardium (or BF) from one of the control pigs was chosen as the calibrator, and its mRNA expression was considered as 1.
2.6. Immunoblotting
LV myocardium and BF samples (30 mg of each) were homogenized in 200 μL of an ice-cold extraction buffer (50 mM Tris-HCl, 200 mM NaCl, 10 mM CaCl2, 1% Triton X-100, pH 7.6) [20] containing a 1 : 50 protease inhibitor cocktail (Sigma-Aldrich, Poland). After incubation on ice (30 min) and centrifugation at 9700 ×g, the supernatants were collected and stored on ice. Next, insoluble material was extracted twice after a 10 min incubation with 50 μL of an ice-cold extraction buffer, and supernatants from all extractions were combined. Protein quantification was performed using the Bradford reagent (Sigma-Aldrich), according to the manufacturer's instructions.
Protein samples (0.5–25.0 μg) were mixed with a nonreducing sample buffer (63 mM Tris, 10% glycerol, 2% SDS, 0.1% bromophenol blue, pH 6.8), followed by incubation for 5 min at a room temperature (nonreducing, nondenaturing conditions). Protein samples (25 μg) were also prepared in a reducing sample buffer (Pierce, Poland) (with a final dithiothreitol (DTT) concentration of 0.1 M) followed by incubation for 5 minutes at either a room temperature (reducing and nondenaturing conditions) or 95°C (reducing and denaturing conditions).
All samples were subsequently separated on a 10% gel in a SDS-PAGE and transferred onto the PDVF membrane (Millipore, Poland). The membrane was treated with the Quentix signal enhancer (Pierce), blocked for 1 h with 5% nonfat milk in the PBS containing 0.5% (v/v) Triton X-100 (Sigma-Aldrich), and incubated overnight with polyclonal goat antibodies against MMP9, TIMP1, or NGAL (1 : 500) (R&D System, Poland) or with monoclonal mouse anti-MMP2 antibodies (1 : 1000) (Millipore). Blots were developed using the SuperSignal West Femto ECL substrate (Pierce). Recombinant proteins, NGAL (R&D System, Poland), TIMP1 (R&D System, Poland), proMMP2 (Sigma-Aldrich, Poland) and the culture media from DH82 cell line (MMP9 source) prepared in nonreducing, nondenaturing, as well as in reducing, nondenaturing, and reducing, and denaturing conditions, were used as positive controls.
2.7. Coimmunoprecipitation
For the procedure of coimmunoprecipitation of HMW complexes containing MMP9, homogenates from LV myocardium (suspended in an extraction buffer) were initially incubated with protein G agarose (Sigma-Aldrich). The supernatant was mixed with 25 μL of protein G agarose and anti-MMP9 antibodies (the same antibodies which were used for Western blotting) (5μg). After a 60 min incubation, the protein G agarose beads were washed extensively and proteins were eluted with 50 μL of an elution buffer (pH 2.8, Pierce), neutralized with 5 μL of 1 M Tris (pH 9.0), and electrophoresed on a 10% SDS-PAGE (nonreducing and nondenaturing conditions), followed by immunoblotting. Samples immunoprecipitated with anti-MMP9 antibody were blotted using anti-TIMP1, anti-NGAL, and anti-MMP2 antibodies. In all cases, blotting was performed as described in Section 2.6.
2.8. Gelatin Zymography
For the detection of MMP2 and MMP9 proteolytic activity in porcine LV homogenates, the classic method, that is, gelatin zymography, was used. In this method, enzyme activity is visible as clear zones in the gelatin-containing gel, where the substrate (gelatin) is digested by enzymes having the gelatinase activity [21]. MMP2 and MMP9 have been shown to specifically digest gelatin [21].
LV myocardium and BF samples were prepared as described in Section 2.6. Protein samples (50 μg) were mixed with a nonreducing sample buffer (63 mM Tris, 10% glycerol, 2% SDS, 0.1% bromophenol blue, pH 6.8) and separated at 4°C on a substrate SDS-PAGE (10% acrylamide, 1 mg/mL gelatin) [20, 22]. The gels were washed 3 times in 2.5% Triton X-100 for 30 min and incubated overnight at 37°C in a collagenase buffer (50 mM Tris-HCl, 200 mM NaCl, 5 mM CaCl2, 0.2% Brij-35, and pH 7.6) [23, 24]. Then, the gels were stained using 0.5% Coomassie Blue R-250 (Sigma-Aldrich) in 30% methanol and 10% acetic acid for 60 min and destained in 30% methanol and 10% acetic acid [24]. Gelatinase activity was identified as clear zones against a blue background.
Gelatin zymography of homogenates from porcine LV myocardium was performed twice, that is, first as described above, and second with a modification based on the preincubation of analyzed samples in a collagenase buffer containing 10 mM EDTA (the latter known to inhibit specifically an enzymatic activity of MMPs [25]) in order to prove the specificity of gelatinolytic activity associated with the presence of MMP9 in demonstrated bands. Additionally, the culture media from canine macrophage-like DH82 cell line were used as a positive control of MMP2 and MMP9 activity [26].
2.9. Spontaneous Release of Proteolytically Active MMPs
The preparation of homogenates of porcine LV myocardium was described in Section 2.6. 1 mg of extracts of LV myocardium was mixed with the same volume of Brij buffer (0.5 M NaCl, 5 mM CaCI2, 50 mM Tris, 0.05% Brij, pH 7.6) [27], 40 μL of gelatin-CNBr-Sepharose4B (Sigma-Aldrich) and incubated at room temperature for 60 min. Due to removing gelatin-Sepharose4B (with attached MMP2 and MMP9), the extract of LV myocardium was depleted of any active MMPs. Subsequently, the extracts of LV myocardium were incubated during 60 min with 25 μL protein G agarose (Sigma-Aldrich). The supernatant was mixed with 25 μL of protein G agarose and antibody against MMP9 (5 μg). After an overnight incubation at 4°C (to prevent the enzyme autodegradation) and the extensive washing, immunoprecipitated proteins were eluted with 50 μL of an elution buffer (pH 2.8, Pierce), neutralized, and incubated for 24 hours at 4°C (to prevent the enzyme autodegradation). Subsequently, the samples were analyzed in gelatin zymography (as described in details in the previous paragraph).
2.10. Statistical Analyses
Continuous variables were presented as arithmetic means ± standard errors of a mean (SEM). All molecular and echocardiography assessments were performed in triplicates. Spearman's rank correlatory coefficients were used for all correlatory analyses. P < 0.05 was considered as statistically significant. Statistical analyses were performed using the Polish version of Statistica 9.1 (StatSoft, OK, USA).
3. Results
3.1. Development of Symptomatic Systolic HF in Male Pigs with RV Pacing
In the course of RV pacing, male pigs developed a clinical picture of chronic HF and were presented for euthanasia at subsequent predefined stages of HF: mild (6 ± 2 weeks of pacing, n = 7), moderate (11 ± 3 weeks of pacing, n = 8), and severe HF (18 ± 4 weeks of pacing, n = 5). Sham-operated pigs (n = 5) developed no symptoms of HF.
Echocardiography performed directly before euthanasia revealed that RV pacing-induced progressive development of LV systolic and diastolic dysfunction along with LV dilatation with the most marked echocardiography abnormalities seen in pigs with severe symptoms of HF (Table 2). Echocardiography parameters measured in sham-operated pigs remained normal during the entire study (Table 2). Moreover, the development of symptomatic systolic HF was accompanied by the neurohormonal activation, as evidenced by the higher serum levels of BNP and the increased PRA (Table 2).
Table 2.
Echocardiography parameters reflecting the structure and functioning of left ventricle and the measures of neurohormonal activation in sham-operated male pigs (controls) and right-ventricle-paced male pigs with induced heart failure.
| Variables, units | Controls (n = 5) | Mild HF (n = 7) | Moderate HF (n = 8) | Severe HF (n = 5) | Spearman correlatory rank coefficients R with P | |
|---|---|---|---|---|---|---|
| For all animals | Only for animals with HF | |||||
| Echocardiography parameters | ||||||
| LVEF, % | 52 ± 4 | 42 ± 5 | 28 ± 5 | 20 ± 3 | R = −0.72 | R = −0.61 |
| P < 0.001 | P = 0.004 | |||||
| LVFS, % | 25 ± 3 | 22 ± 7 | 14 ± 7 | 10 ± 4 | R = −0.74 | R = −0.66 |
| P < 0.001 | P = 0.001 | |||||
| LVESV, mL | 93 ± 15 | 134 ± 32 | 213 ± 36 | 223 ± 41 | R = 0.84 | R = 0.72 |
| P < 0.001 | P < 0.001 | |||||
| LVEDV, mL | 192 ± 24 | 241 ± 36 | 284 ± 41 | 277 ± 32 | R = 0.73 | R = 0.49 |
| P < 0.001 | P = 0.03 | |||||
| SV, mL | 94 ± 8 | 107 ± 14 | 88 ± 20 | 55 ± 11 | R = −0.38 | R = −0.46 |
| P = 0.06 | P = 0.04 | |||||
| LVPW thickening, % | 49 ± 3 | 54 ± 4 | 45 ± 10 | 15 ± 7 | R = −0.59 | R = −0.71 |
| P = 0.02 | P < 0.001 | |||||
| LA/Ao | 1.33 ± 0.06 | 1.91 ± 0.10 | 1.9 ± 0.13 | 2.57 ± 0.14 | R = 0.73 | R = 0.56 |
| P < 0.001 | P = 0.01 | |||||
| Em, m/s | 0.19 ± 0.02 | 0.18 ± 0.03 | 0.15 ± 0.04 | 0.15 ± 0.04 | R = −0.42 | R = −0.27 |
| P = 0.04 | P = 0.25 | |||||
| Am, m/s | 0.11 ± 0.03 | 0.08 ± 0.01 | 0.07 ± 0.01 | 0.07 ± 0.01 | R = −0.31 | R = −0.09 |
| P = 0.12 | P = 0.68 | |||||
| Measures of neurohormonal activation | ||||||
| BNP, ng/mL | 0.25 ± 0.25 | 0.29 ± 0.19 | 0.65 ± 0.46 | 0.80 ± 0.30 | R = 0.61 | R = 0.58 |
| P = 0.001 | P = 0.009 | |||||
| PRA, ng/mL/h | 0.3 ± 0.2 | 1.0 ± 0.4 | 2.0 ± 0.7 | 4.1 ± 1.9 | R = 0.66 | R = 0.48 |
| P = 0.001 | P = 0.07 | |||||
HF: heart failure; LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; LVESV: left ventricular end-systolic volume; LVEDV: left ventricular end-diastolic volume; SV: stroke volume; LVPW: left ventricular posterior wall; LA/Ao: left atrial/aorta ratio; Em: early diastolic velocity ratio; Am: late diastolic velocity ratio; BNP: B-type natriuretic peptide; PRA: plasma renin activity. All echocardiography measures were performed directly before an euthanasia. Data are presented as arithmetical means ± standard errors of a mean.
3.2. mRNA Expression of MMP9, MMP2, NGAL, and TIMP1
There were no differences in the mRNA expression of MMP9, MMP2, and TIMP1 in LV myocardium between diseased and healthy animals (Figure 1(a)). The mRNA expression of NGAL in LV myocardium was approximately 5-fold reduced in pigs with moderate and severe HF as compared to controls (P < 0.001) (Figure 1(a)). Also among all examined animals, the reduced mRNA expression of NGAL in LV myocardium was accompanied by LV dilatation (LVEDD: R = −0.53, P = 0.005), diastolic dysfunction (Em: R = 0.52, P = 0.005), and increased PRA (R = −0.48, P = 0.03). Furthermore, there were no differences in the mRNA expression of MMP9, MMP2, NGAL, and TIMP1 in skeletal muscles between diseased and healthy animals (Figure 1(b)). The relative mRNA expression in skeletal muscle was approximately 30-, 4-, 30-, and 10-fold lower than in LV myocardium for MMP9, MMP2, NGAL, and TIMP1, respectively.
Figure 1.
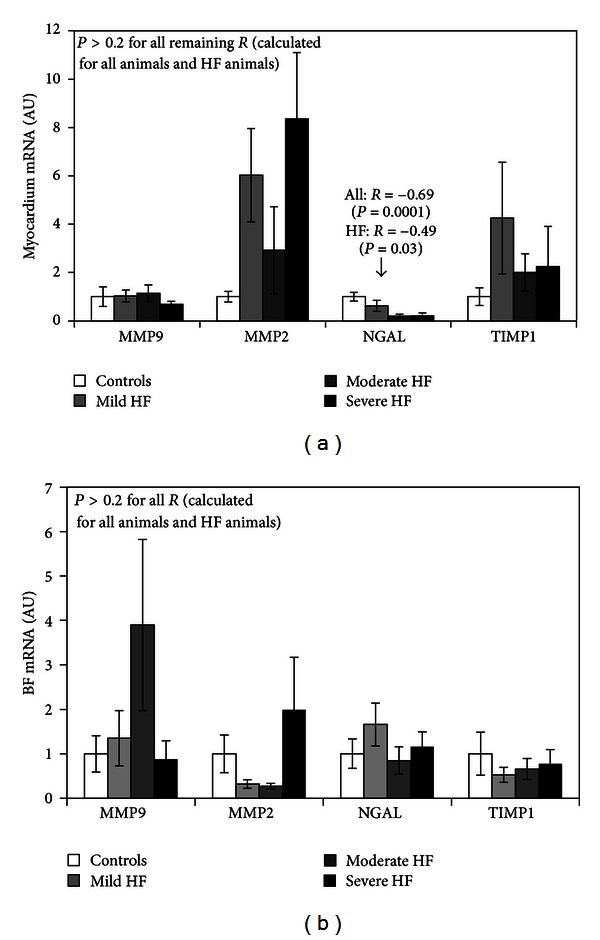
The relative mRNA expression of matrix metalloproteinase type 9 (MMP9), matrix metalloproteinase type 2 (MMP2), neutrophil gelatinase-associated lipocalin (NGAL), and tissue inhibitor of metalloproteinases type 1 (TIMP1) in homogenates from left ventricle (LV) myocardium (a) and biceps femoris (BF) (b) of sham-operated pigs and animals with mild, moderate, and severe HF. Results are expressed in arbitrary units, with a mean value for all control animals considered as 1, separately for each molecule in each tissue. Values are presented as means ± standard error means. R stands for the Spearman' rank correlatory coefficient. The relative mRNA expression of MMP9, MMP2, NGAL, and TIMP1 was, respectively, approximately 30, 4, 30, and 10 times lower in porcine homogenates of BF as compared to porcine homogenates of LV myocardium.
3.3. Protein Expression of MMP9, MMP2, NGAL, and TIMP1 in LV Myocardium
3.3.1. Nonreducing and nondenaturing Conditions
Blots of LV homogenates with MMP antibody from both diseased and healthy pigs in nonreducing and nondenaturing conditions revealed 3 bands of 130, 170, and 220 kDa. The same bands were seen when anti-NGAL or anti-TIMP1 antibodies were used (Figure 2(a)). However, when LV homogenates were blotted using anti-MMP2 antibody, only a 220 kDa band was detected. Coimmunoprecipitation of LV myocardial tissue using both anti-MMP9 and anti-TIMP1 antibodies, or both anti-MMP9 and anti-NGAL antibodies confirmed that all these 3 bands contained MMP9, NGAL, and TIMP1 (Figure 2(d)). Coimmunoprecipitation of LV myocardial tissue using both anti-MMP9 and anti-MMP2 antibodies revealed that a 220 kDa band contained MMP2 in addition to the other 3 proteins (Figure 2(d)).
Figure 2.
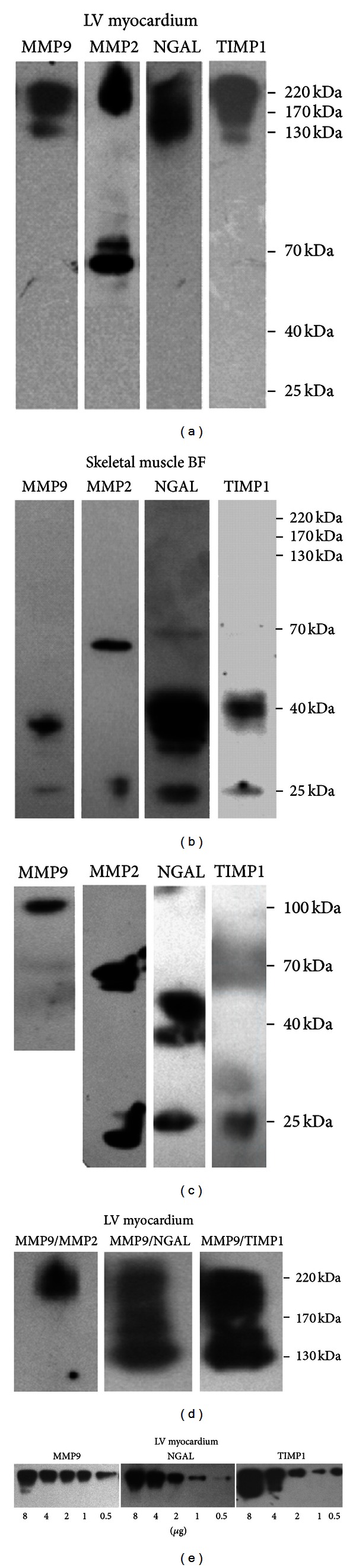
Representative Western blots for MMP9, MMP2, NGAL and TIMP1 detection in porcine homogenates of LV myocardium (a) and BF (b) in nonreducing and nondenaturing conditions. Positive controls in nonreducing and nondenaturing conditions (c). Representative coimmunoprecipitates of porcine lysates of LV myocardium immunoprecipited with anti-MMP9 antibody, and subsequently blotted with anti-MMP2, anti-NGAL, and anti-TIMP1 antibodies (d). Representative semiquantitative Western blots of porcine homogenates of LV myocardium using anti-MMP9, anti-TIMP1, and anti-NGAL antibodies (e).
In order to demonstrate which of the 130, 170, and 220 kDa bands is the most abundant in porcine LV myocardium, Western blotting with either anti-MMP9, anti-TIMP1, or anti-NGAL antibodies with an increasing amount of protein samples (0.5–8 μg) from LV lysates was performed. This experiment demonstrated that the 220-kDa complex was the most abundant in porcine LV myocardium (Figure 2(e)). In LV homogenates, there were no bands corresponding to monomers containing either MMP9, NGAL, or TIMP1, but there were bands of 72 kDa and 68 kDa corresponding to monomeric pro-MMP2 and MMP2, respectively (Figure 2(a)).
3.3.2. Reducing and Nondenaturing Conditions
Western blots on homogenates from LV myocardium performed in reducing conditions (i.e., incubated with DTT) at a room temperature revealed the presence of an intense 220 kDa band containing MMP9, NGAL, and TIMP1 (previously seen in nonreducing conditions), very subtle 130 kDa and 170 kDa bands, along with another intense 115 kDa band containing MMP9, NGAL, and TIMP1. MMP2 was detected in a form of 40-kDa band (Figure 3(a)). The 220 kD complex remained stable in reducing and nondenaturing conditions, suggesting that hydrophobic interactions were involved in the maintenance of its stability. In contrast, the 130 and 170 kDa complexes were destabilized in reducing and nondenaturing conditions, suggesting the involvement of disulphide bonds in their formation. In reducing conditions, there was a 115-kDa band present, which corresponds to active MMP9 band observed in zymography.
Figure 3.
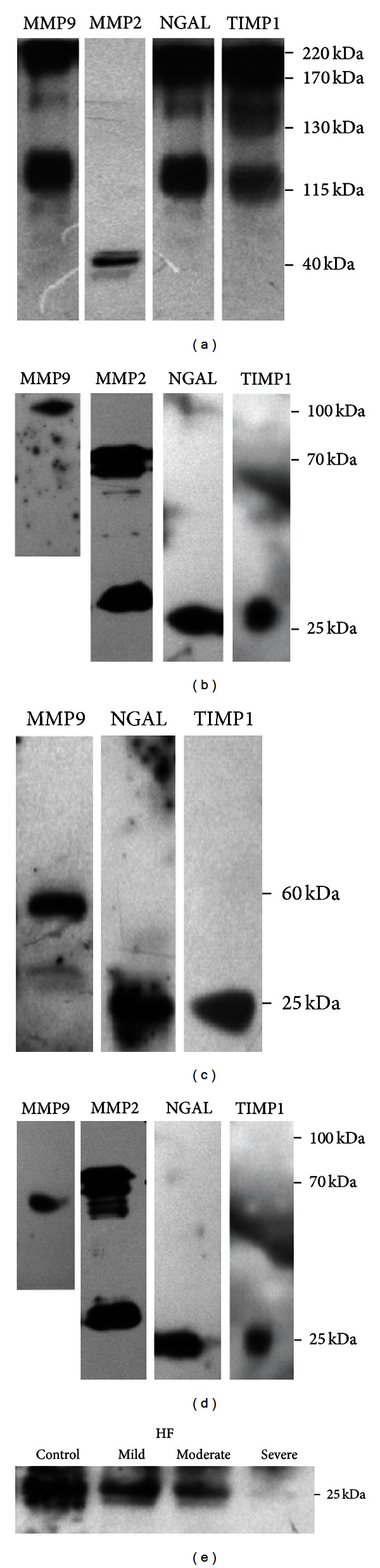
Representative Western blots for MMP9, MMP2, NGAL, and TIMP1 detection in the porcine homogenates of LV myocardium performed in reducing and nondenaturing conditions (a 5-minute incubation in 0.1 M dithiothreitol [DTT] at a room temperature) (a) and in reducing and denaturing conditions (a 5-minute incubation in 0.1 M DTT at 95°C) (c). Positive controls for reducing and nondenaturing conditions (b) and reducing and denaturing conditions (d). Representative Western blots in the porcine homogenates of LV myocardium from a healthy pig and animals with mild, moderate, and severe HF with anti-NGAL antibodies in reducing and denaturing conditions (e).
3.3.3. Reducing and Denaturing Conditions
Western blots performed with the same samples in both reducing (i.e., incubated with DTT for 5 min at a room temperature) and denaturing conditions (i.e., incubated with DTT for 5 min at 95°C) revealed the presence of a 60 kDa-band (blotted with anti-MMP9 antibody) and a 25 kDa-band (blotted with both anti-NGAL and anti-TIMP1 antibodies), which corresponded to monomers of MMP9, NGAL, and TIMP1 (Figure 3(c)). The optic density of a 25 kDa-band blotted with anti-NGAL antibody was lower in pigs with advanced HF as compared to controls (Figure 3(e)). We did not demonstrate any other bands corresponding to HMW complexes and MMP2 was not detected.
3.4. Gelatinase Activity in LV Myocardium
Gelatin zymography of homogenates from LV myocardial tissue from healthy and diseased pigs (performed in nonreducing and nondenaturing conditions) revealed the presence of several active bands ranging from 68 to 115 kDa (Figure 4, line 1). However, the HMW complexes seen in Western blots (130, 170, and 220 kDa) were not detected during gelatin zymography, which confirmed that the proteins of the complex had no enzymatic activity.
Figure 4.
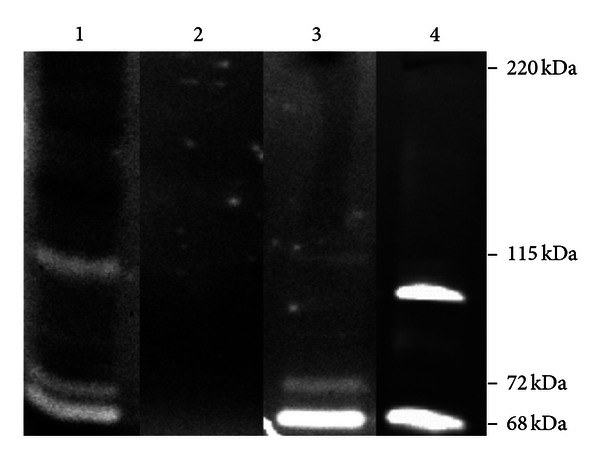
Representative gelatin zymograms of porcine homogenates from LV myocardium with proteolytically active bands of 68, 72, and 115 kDa at baseline (line 1), after the depletion of active gelatinolytic forms using gelatin-Sepharose 4B affinity chromatography (line 2), and subsequently after the 24-hour incubation at 4°C (line 3). Line 4—positive control, the culture media from canine macrophage-like DH82 cell line.
3.5. Release of Active MMP9 and MMP2 from MMP9/MMP2/NGAL/TIMP1 Complexes Derived from LV Myocardium
Homogenates of LV myocardium (Figure 4(a) line 1) were depleted of active MMP2 and MMP9 species using the gelatin Sepharose affinity chromatography (Figure 4(a) line 2). Presumably inactive complexes were immunoprecipitated using anti-MMP9 antibodies. Immunoprecipitates were incubated for 24 h to enable the potential release of molecules from complexes, while they were kept at 4°C to prevent enzymatic autodegradation. The subsequent gelatinase zymography of these immunoprecipitates demonstrated the presence of 68 kDa, 72 kDa, and 115 kDa bands (Figure 4(a) line 3), suggesting that active forms of the 115-kDa MMP9 and the 68 and 72 kDa MMP2 could be spontaneously released from the HMW complexes.
3.6. Protein Expression of MMP9, MMP2, NGAL, and TIMP1 in Skeletal Muscles
Western blots of skeletal muscles from both diseased and healthy pigs (under nonreducing and nondenaturing conditions) and probed with either MMP9, MMP2, TIMP1 or NGAL antibodies revealed the presence of 25 kDa and 40 kDa bands corresponding to monomers and dimers of TIMP1, or/and NGAL. Furthermore, a weak 40 kDa band containing MMP9 and additional 68 and 72 kDa bands containing MMP2 (most likely MMP2 and pro-MMP2, resp.) were also detected. We found no bands of a greater mass corresponding to HMW complexes containing the combination of these 4 molecules (Figure 2(b)).
3.7. Gelatinase Activity in Skeletal Muscles
Gelatin zymography of BF homogenates from healthy and diseased pigs (performed in nonreducing nondenaturing conditions) confirmed the presence of only 2 weak bands with gelatinolytic activity of 68 and 72 kDa, corresponding to MMP2 and proMMP2, respectively (data not shown).
4. Discussion
In the present study we have demonstrated the presence of HMW complexes containing MMP9, MMP2, TIMP1, and NGAL in LV myocardium (but not in skeletal muscles) of male pigs with and without systolic nonischaemic HF. We have shown that HMW complexes release spontaneously the proteolytically active MMP9 and MMP2.
In this study we have demonstrated that RV pacing at 170 bpm applied in male pigs induces the development of symptomatic systolic HF, which has been evidenced by dilated LV cavity accompanied by impaired systolic and diastolic LV function and neurohormonal activation. Previous porcine TIC models have been developed in young, 20–30 kg weight piglets using rapid pacing at 220–240 bpm up to 3 weeks and resulted in the development of acute or subacute rather than chronic HF [28–31]. Low age of animals used in these studies and the standard castration of male animals for experimental purposes might affect the observations [31]. Taking these facts into consideration, we decided to use adult male pigs initially weighting from 115 to 125 kg and low pacing rate (170 bpm) to develop chronic HF. The experiments on male adult pigs have some limitations, for example, the high costs of maintenance and the aggressive behavior of animals. Our TIC model does not require open-chest surgery, associated with potential risk of intraoperative complications, as in coronary ligation model [32]. While coronary ligation and microembolization create an ischemic HF limited to a specific region of the heart [32], our model could be used to study generalized myocardial dysfunction.
We have demonstrated the presence of both mRNA and protein of MMP9, MMP2, TIMP1, and NGAL in LV myocardium and skeletal muscles of male pigs with and without systolic HF. In fact, there are only few studies reporting the expression of these genes in myocardium [33–35] and skeletal muscles [5] and none of these papers compared the expression of these genes between the 2 aforementioned tissues.
We have found significant differences regarding the mRNA and protein expression of MMP9, MMP2, TIMP1, and NGAL between myocardial and skeletal muscles. Firstly, the mRNA expression of these 4 genes was markedly lower in skeletal muscles as compared to LV myocardium (in both healthy and diseased animals). Secondly, the formation of complexes differed between myocardial and skeletal muscle tissues. In nonreducing and nondenaturing conditions, in LV myocardium there were HMW complexes containing MMP9, MMP2, TIMP1, and NGAL, along with pro-MMP2 and MMP2 monomers, whereas in skeletal muscles only dimers and monomers of these 4 aforementioned molecules were present. Thirdly, in LV myocardium, proteolytic activity was associated with the presence of active MMP2 and MMP9 forms and also the presence of HMW complexes being the reservoir of spontaneously released MMPs, whereas in skeletal muscles the gelatinolytic activity was relatively weak and was associated only with the presence of pro-MMP2 and MMP2 monomers (as compared to LV myocardium). The gelatinolytic activity of skeletal muscles was not extensively studied. Schiøtz Thorud et al. [5] demonstrated the presence of gelatinolytically active MMP2 and MMP9 in skeletal muscles of rats after myocardial infarction and sham-operated controls; however, the amount of mRNA and protein of these MMPs did not differ between healthy and diseased animals. The critical role of the MMPs in physiological and pathophysiological tissue remodeling implies that this complex system needs to be precisely controlled on different levels [8]. Only recently, the formation of complexes including MMPs with different components has received an interest as a potential mechanism with the ability to modify the stability of protease structure and their enzymatic activity [8, 11]. The available evidence on the complexes containing MMPs is rather scarce and most of the studies were mainly performed in biological fluids [13–15] and media of cultured cells and tissues [36, 37]. Rouet-Benzineb et al. [38] demonstrated the colocalization of MMP2 and MMP9 in LV myocardium from patients with idiopathic DCM using a confocal microscopic immunoreactive staining. However, only one report demonstrated the presence of HMW complexes containing MMP9, MMP2, MMP3, TIMP1, and TIMP2 in solid rat tissue (chorioallantoid membranes), but none has been reported in myocardium or skeletal muscles [11]. We have revealed that in porcine myocardium MMP9 forms 3 complexes together with MMP2, TIMP1, and NGAL. Whereas estimates of molecular weight of MMP9 monomer ranges from 80 kDa to 92 kDa, according to different authors [11, 36], NGAL monomer weights about 25 kDa (and spontaneously formed dimer ~50 kDa, [15]), TIMP1 monomer ~30 kDa [36], and MMP2 monomer ~72 kDa [38]. Therefore, the following constituents of HMW complexes can be suggested, 220 kDa complex: MMP2, MMP9, NGAL, and TIMP1; 170 kDa complex: MMP9, NGAL (dimer), and TIMP1; 130 kDa complex: MMP9, NGAL, and TIMP1. The 220 kDa complex appears to be stable in reducing and nondenaturing conditions, and labile in strong reducing and denaturing conditions suggesting that hydrophobic interactions may play a role in the maintenance of its stability. In contrast, the other 130 and 170 kDa complexes were destabilized even in reducing and nondenaturing conditions suggesting the involvement of disulphide bonds in their formation. Also the other authors have confirmed that the integrity of heterogeneous MMP9 complexes results from the presence of both disulfide bonds and hydrophobic interactions [39, 40]. To the best of our knowledge, our study is the first report on the presence of HMW heterogeneous complexes containing MMP9, MMP2, NGAL, and TIMP1 in both failing and normal myocardium.
It has been shown in in vitro studies that the formation of MMP9-NGAL complex can attenuate the autodegradation of MMP9 [9]. Also, HMW complexes containing MMP9 have been postulated to serve as a reservoir of active MMP9 protecting it from the rapid degradation when present in biological fluids [13, 15]. The evidence on the analogous role of such complexes in solid tissues is missing. We were able to demonstrate for the first time that HMW complexes containing MMP9 and MMP2 are present in myocardium (but not in skeletal muscles) and are proteolytically inactive but can spontaneously release active MMP9 and MMP2. This observation indicates that the aforementioned HMW complexes may serve as the tissue reservoir of active MMP9, which seems to be unique for myocardial tissue.
Taking into consideration the firmly established role of MMP9 and MMP2 in myocardial remodeling and HF development [4], one might expect differences in the mRNA and protein expression along with proteolytic activity of MMP9 (and also MMP2) between animals with and without systolic HF. In our study, there were no differences in the expression of mRNA and protein of MMP9, MMP2, and TIMP1 in either LV myocardium or skeletal muscles between diseased and healthy animals. These data suggest that the activity of these enzymes may be altered posttranslationally in heart failure and is not dependent on the expression of the mRNA or protein. The only observed difference was the gradual decline in the expression of both mRNA NGAL and the protein amount of NGAL in LV myocardium (but not in skeletal muscles) in male pigs along with the progression of systolic HF. The decline in the mRNA NGAL expression in porcine LV myocardium was accompanied by LV dilatation, the augmented diastolic dysfunction, and the increased PRA activity. So far, the NGAL expression in myocardium has been studied in rodent models of myocardial infarction (MI) and hypoxia [33, 41], where the authors revealed the augmented myocardial expression of rodent NGAL homologue (24p3) in animals with heart dysfunction. Moreover, Aigner et al. [42] showed that an increase in murine myocardial NGAL seen after ischemia and reperfusion is associated with the presence of polymononuclear cells infiltrating myocardial tissue, and NGAL may play a role of an acute phase protein. The evidence on myocardial NGAL expression is scarce, but based on the aforementioned studies and our results, it may be presumed that the pattern on NGAL myocardial expression differs between ischemic and hypoxic myocardium [33, 41] and failing myocardium in animals with nonischaemic HF (our data).
In conclusion, HMW complexes without proteolytic activity containing MMP9, MMP2, TIMP1, and NGAL are present in healthy and failing porcine LV myocardium, but not in skeletal muscles. HMW complexes spontaneously become the source of in vitro proteolytically active MMP9 and MMP2. We should acknowledge that we have not investigated the links between the presence of these complexes in LV myocardium and the magnitude of myocardial remodeling. Further studies are warranted to delineate this process. The presence of MMP in complexes in myocardium should be taken into consideration when designing pharmaceutical interventions inhibiting the MMP system in order to combat the pathological myocardial remodeling.
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
This paper is part of Project “WROVASC-Integrated Cardiovascular Centre” cofinanced by the European Regional Development Fund within Innovative Economy Operational Program 2007–2013 realized in Regional Specialist Hospital, Research and Development Centre in Wroclaw.
References
- 1.Murphy G, Nagase H. Progress in matrix metalloproteinase research. Molecular Aspects of Medicine. 2009;29(5):290–308. doi: 10.1016/j.mam.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ducharme A, Frantz S, Aikawa M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. Journal of Clinical Investigation. 2000;106(1):55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayashidani S, Tsutsui H, Ikeuchi M, et al. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. The American Journal of Physiology. 2003;285(3):H1229–H1235. doi: 10.1152/ajpheart.00207.2003. [DOI] [PubMed] [Google Scholar]
- 4.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiological Reviews. 2007;87(4):1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 5.Thorud HMS, Stranda A, Birkeland JA, et al. Enhanced matrix metalloproteinase activity in skeletal muscles of rats with congestive heart failure. The American Journal of Physiology. 2005;289(2):R389–R394. doi: 10.1152/ajpregu.00067.2005. [DOI] [PubMed] [Google Scholar]
- 6.Strassburg S, Springer J, Anker SD. Muscle wasting in cardiac cachexia. International Journal of Biochemistry and Cell Biology. 2005;37(10):1938–1947. doi: 10.1016/j.biocel.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochimica et Biophysica Acta. 2010;1803(1):55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, Winberg JO. Regulation of matrix metalloproteinase activity in health and disease. The FEBS Journal. 2011;278(1):28–45. doi: 10.1111/j.1742-4658.2010.07920.x. [DOI] [PubMed] [Google Scholar]
- 9.Yan L, Borregaard N, Kjeldsen L, Moses MA. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL): modulation of MMP-9 activity by NGAL. The Journal of Biological Chemistry. 2001;276(40):37258–37265. doi: 10.1074/jbc.M106089200. [DOI] [PubMed] [Google Scholar]
- 10.Tschesche H, Zölzer V, Triebel S, Bartsch S. The human neutrophil lipocalin supports the allosteric activation of matrix metalloproteinases. European Journal of Biochemistry. 2001;268(7):1918–1928. doi: 10.1046/j.1432-1327.2001.02066.x. [DOI] [PubMed] [Google Scholar]
- 11.Meraz-Cruz N, Ortega A, Estrada-Gutierrez G, et al. Identification of a calcium-dependent matrix metalloproteinase complex in rat chorioallantoid membranes during labour. Molecular Human Reproduction. 2006;12(10):633–641. doi: 10.1093/molehr/gal072. [DOI] [PubMed] [Google Scholar]
- 12.Kolkenbrock H, Hecker-Kia A, Orgel D, Kinawi A, Ulbrich N. Progelatinase B forms from human neutrophils. Complex formation of monomer/lipocalin with TIMP-1. Biological Chemistry. 1996;377(7-8):529–533. doi: 10.1515/bchm3.1996.377.7-8.529. [DOI] [PubMed] [Google Scholar]
- 13.Gupta K, Shukla M, Cowland JB, Malemud CJ, Haqqi TM. Neutrophil gelatinase-associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis and Rheumatism. 2007;56(10):3326–3335. doi: 10.1002/art.22879. [DOI] [PubMed] [Google Scholar]
- 14.Roy R, Louis G, Loughlin KR, et al. Tumor-specific urinary matrix metalloproteinase fingerprinting: identification of high molecular weight urinary matrix metalloproteinase species. Clinical Cancer Research. 2008;14(20):6610–6617. doi: 10.1158/1078-0432.CCR-08-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nuntagowat C, Leelawat K, Tohtong R. NGAL knockdown by siRNA in human cholangiocarcinoma cells suppressed invasion by reducing NGAL/MMP-9 complex formation. Clinical and Experimental Metastasis. 2010;27(5):295–305. doi: 10.1007/s10585-010-9327-y. [DOI] [PubMed] [Google Scholar]
- 16.Pasławska U, Gajek J, Kiczak L, et al. Development of porcine model of chronic tachycardia-induced cardiomyopathy. International Journal of Cardiology. 2011;153(1):36–41. doi: 10.1016/j.ijcard.2010.08.033. [DOI] [PubMed] [Google Scholar]
- 17.Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification: a report from the American society of echocardiography’s guidelines and standards committee and the chamber quantification writing group, developed in conjunction with the European association of echocardiography, a branch of the European society of cardiology. Journal of the American Society of Echocardiography. 2005;18(12):1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Teichholz LE, Kreulen T, Herman MV, Gorlin R. Problems in echocardiographic volume determinations: echocardiographic angiographic correlations in the presence or absence of asynergy. The American Journal of Cardiology. 1976;37(1):7–11. doi: 10.1016/0002-9149(76)90491-4. [DOI] [PubMed] [Google Scholar]
- 19.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29(9, article e45) doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mook ORF, van Overbeek C, Ackema EG, van Maldegem F, Frederiks WM. In situ localization of gelatinolytic activity in the extracellular matrix of metastases of colon cancer in rat liver using quenched fluorogenic DQ-gelatin. Journal of Histochemistry and Cytochemistry. 2003;51(6):821–829. doi: 10.1177/002215540305100613. [DOI] [PubMed] [Google Scholar]
- 21.Snoek-van Beurden PAM, von den Hoff JW. Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. BioTechniques. 2005;38(1):73–83. doi: 10.2144/05381RV01. [DOI] [PubMed] [Google Scholar]
- 22.Shapiro SD, Fliszar CJ, Broekelmann TJ, Mecham RP, Senior RM, Welgus HG. Activation of the 92-kDa gelatinase by stromelysin and 4-aminophenylmercuric acetate. Differential processing and stabilization of the carboxyl-terminal domain by tissue inhibitor of metalloproteinases (TIMP) The Journal of Biological Chemistry. 1995;270(11):6351–6356. doi: 10.1074/jbc.270.11.6351. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita C, Hayashi T, Mori T, et al. Angiotensin II receptor blocker reduces oxidative stress and attenuates hypoxia-induced left ventricular remodeling in apolipoprotein E-knockout mice. Hypertension Research. 2007;30(12):1219–1230. doi: 10.1291/hypres.30.1219. [DOI] [PubMed] [Google Scholar]
- 24.Leber TM, Balkwill FR. Zymography: a single-step staining method for quantitation of proteolytic activity on substrate gels. Analytical Biochemistry. 1997;249(1):24–28. doi: 10.1006/abio.1997.2170. [DOI] [PubMed] [Google Scholar]
- 25.Galis ZS, Muszynski M, Sukhova GK, et al. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circulation Research. 1994;75(1):181–189. doi: 10.1161/01.res.75.1.181. [DOI] [PubMed] [Google Scholar]
- 26.Barnes A, Bee A, Bell S, et al. Immunological and inflammatory characterisation of three canine cell lines: K1, K6 and DH82. Veterinary Immunology and Immunopathology. 2000;75(1-2):9–25. doi: 10.1016/s0165-2427(00)00184-7. [DOI] [PubMed] [Google Scholar]
- 27.Moll UM, Youngleib GL, Rosinski KB, Quigley JP. Tumor promoter-stimulated M(r) 92,000 gelatinase secreted by normal and malignant human cells: isolation and characterization of the enzyme from HT1080 tumor cells. Cancer Research. 1990;50(19):6162–6170. [PubMed] [Google Scholar]
- 28.Spinale FG, Fulbright BM, Mukherjee R, et al. Relation between ventricular and myocyte function with tachycardia-induced cardiomyopathy. Circulation Research. 1992;71(1):174–187. doi: 10.1161/01.res.71.1.174. [DOI] [PubMed] [Google Scholar]
- 29.Spinale FG, Zellner JL, Tomita M, Crawford FA, Zile MR. Relation between ventricular and myocyte remodeling with the development and regression of supraventricular tachycardia-induced cardiomyopathy. Circulation Research. 1991;69(4):1058–1067. doi: 10.1161/01.res.69.4.1058. [DOI] [PubMed] [Google Scholar]
- 30.Hendrick DA, Smith AC, Kratz JM, Crawford FA, Spinale FG. The pig as a model of tachycardia and dilated cardiomyopathy. Laboratory Animal Science. 1990;40(5):495–501. [PubMed] [Google Scholar]
- 31.Bauer A, McDonald AD, Donahue JK. Pathophysiological findings in a model of persistent atrial fibrillation and severe congestive heart failure. Cardiovascular Research. 2004;61(4):764–770. doi: 10.1016/j.cardiores.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Schmitto JD, Mokashi SA, Lee LS, et al. Large animal models of chronic heart failure (CHF) Journal of Surgical Research. 2011;166(1):131–137. doi: 10.1016/j.jss.2009.11.737. [DOI] [PubMed] [Google Scholar]
- 33.Yndestad A, Landrø L, Ueland T, et al. Increased systemic and myocardial expression of neutrophil gelatinase-associated lipocalin in clinical and experimental heart failure. European Heart Journal. 2009;30(10):1229–1236. doi: 10.1093/eurheartj/ehp088. [DOI] [PubMed] [Google Scholar]
- 34.Batlle M, Pérez-Villa F, García-Pras E, et al. Down-regulation of matrix metalloproteinase-9 (MMP-9) expression in the myocardium of congestive heart failure patients. Transplantation Proceedings. 2007;39(7):2344–2346. doi: 10.1016/j.transproceed.2007.06.046. [DOI] [PubMed] [Google Scholar]
- 35.Rastogi S, Gupta RC, Mishra S, Morita H, Tanhehco EJ, Sabbah HN. Long-term therapy with the acorn cardiac support device normalizes gene expression of growth factors and gelatinases in dogs with heart failure. Journal of Heart and Lung Transplantation. 2005;24(10):1619–1625. doi: 10.1016/j.healun.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 36.Ogata Y, Itoh Y, Nagase H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by 4-aminophenylmercuric acetate and proteinases. The Journal of Biological Chemistry. 1995;270(31):18506–18511. doi: 10.1074/jbc.270.31.18506. [DOI] [PubMed] [Google Scholar]
- 37.Goldberg GI, Strongin A, Collier IE, Genrich LT, Marmer BL. Interaction of 92-kDa type IV collagenase with the tissue inhibitor of metalloproteinases prevents dimerization, complex formation with interstitial collagenase, and activation of the proenzyme with stromelysin. The Journal of Biological Chemistry. 1992;267(7):4583–4591. [PubMed] [Google Scholar]
- 38.Rouet-Benzineb P, Buhler JM, Dreyfus P, et al. Altered balance between matrix gelatinases (MMP-2 and MMP-9) and their tissue inhibitors in human dilated cardiomyopathy: potential role of MMP-9 in myosin-heavy chain degradation. European Journal of Heart Failure. 1999;1(4):337–352. doi: 10.1016/s1388-9842(99)00048-3. [DOI] [PubMed] [Google Scholar]
- 39.Winberg JO, Kolset SO, Berg E, Uhlin-Hansen L. Macrophages secrete matrix metalloproteinase 9 covalently linked to the core protein of chondroitin sulphate proteoglycans. Journal of Molecular Biology. 2000;304(4):669–680. doi: 10.1006/jmbi.2000.4235. [DOI] [PubMed] [Google Scholar]
- 40.Cha H, Kopetzki E, Huber R, Lanzendórfer M, Brandstetter H. Structural basis of the adaptive molecular recognition by MMP9. Journal of Molecular Biology. 2002;320(5):1065–1079. doi: 10.1016/s0022-2836(02)00558-2. [DOI] [PubMed] [Google Scholar]
- 41.Hemdahl AL, Gabrielsen A, Zhu C, et al. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(1):136–142. doi: 10.1161/01.ATV.0000193567.88685.f4. [DOI] [PubMed] [Google Scholar]
- 42.Aigner F, Maier HT, Schwelberger HG, et al. Lipocalin-2 regulates the inflammatory response during ischemia and reperfusion of the transplanted heart. The American Journal of Transplantation. 2007;7(4):779–788. doi: 10.1111/j.1600-6143.2006.01723.x. [DOI] [PubMed] [Google Scholar]