

Abstract
Mass spectrometry-based untargeted metabolomics often results in the observation of hundreds to thousands of features that are differentially regulated between sample classes. A major challenge in interpreting the data is distinguishing metabolites that are causally associated with the phenotype of interest from those that are unrelated but altered in downstream pathways as an effect. To facilitate this distinction, here we describe new software called metaXCMS for performing second-order (“meta”) analysis of untargeted metabolomics data from multiple sample groups representing different models of the same phenotype. While the current version of XCMS was designed for the direct comparison of two sample groups, metaXCMS enables meta-analysis of an unlimited number of sample classes to facilitate prioritization of the data and increase the probability of identifying metabolites causally related to the phenotype of interest. metaXCMS is used to import XCMS results that are subsequently filtered, realigned, and ultimately compared to identify shared metabolites that are up- or down-regulated across all sample groups. We demonstrate the software’s utility by identifying histamine as a metabolite that is commonly altered in three different models of pain. metaXCMS is freely available at http://metlin.scripps.edu/metaxcms/.
Metabolites are small molecules within biological systems that serve as the substrates and products of cellular reactions. There is enormous structural diversity among metabolites ranging from polar compounds to lipids and drug derivatives. Untargeted metabolomics describes the process by which these molecules are globally profiled without bias. Using modern mass spectrometers interfaced with either gas or liquid chromatography, tens of thousands of metabolomic features can typically be detected from cells, biofluids, and tissues.1,2 A metabolomics feature represents a peak in the chromatogram and is defined as a molecular entity with a unique mass and retention time. Unbiased metabolomics is performed by first comprehensively identifying every feature within a sample group, and then comparing and statistically assigning the relative intensity of each of them among different sample classes (e.g., healthy versus disease).
Over the course of the past five years, several software tools for differential analysis of mass spectrometry-based metabolomics data have been developed (e.g., XCMS,3,4 MZmine,5,6 and MathDAMP7). These programs identify features whose relative intensity varies between sample groups and are therefore useful in screening for biomarkers of disease. In addition, however, the identification of dysregulated metabolites has been useful in making advances to our understanding of fundamental biochemistry. For example, untargeted metabolomics programs have successfully been applied to reveal new insights related to inborn errors of human metabolism,8 extremophile bacteria,9 viral pathogenesis,10 the gut microbiome,11,12 and stem cell differentiation.13 A major challenge in interrogating complex biological phenomena at the metabolite level, however, is in distinguishing dysregulated pathways that are causally associated with the phenotype of interest from those that are unrelated but altered as a downstream effect. Knockout model organisms provide exciting opportunities to study disease, but metabolomics datasets comparing these organisms to wildtype controls are complicated by the potentially large number of altered features causally unrelated to the pathology. Examining more animal models of the same phenotype increases the likelihood of identifying features associated with underlying disease pathology, but current metabolomics software limits this type of analysis in that only two sample groups can be compared.
It is important to emphasize that metabolomics programs such as XCMS identify dysregulated features, not metabolites. The process of identifying a feature as a metabolite requires searching databases on the basis of accurate mass and comparing the retention time and MS/MS data to that of a model compound for structural confirmation. Growing metabolite databases with advanced functionality have facilitated the procedure of metabolite identification,14–16 but it is still a time consuming and labor-intensive step of the metabolomics workflow. Thus, data reduction is essential to maximizing the physiological relevancy obtained from metabolomics experiments. The challenge is implementing an intelligent methodology to accomplish data reduction at the feature level prior to metabolite identification.
An effective data reduction strategy used in other fields has been performing second-order comparisons to identify shared disturbances among shared phenotypes. Such second-order analyses require the input of multiple sample groups, which previously has not been feasible with existing metabolomics programs. Here we describe new untargeted metabolomics software that can be used in conjunction with XCMS to perform second-order (“meta”) analysis. Pairs of sample groups are first traditionally analyzed with XCMS, and the output files from any number of pair comparisons are then subsequently input into metaXCMS where they are realigned, statistically evaluated, and compared for shared differences. This offers an important metabolomics data reduction tool that has the potential to significantly decrease the number of interesting features selected for subsequent metabolite identification (Figure 1). metaXCMS is freely available as an open-source R-package that includes a graphical user interface. It can be downloaded from http://metlin.scripps.edu/metaxcms/.
Figure 1.

Data reduction with metaXCMS. The workflow is demonstrated using the pain dataset, where pain model A is CFA-treated animals, pain model B is heat-treated animals, and pain model C is KRN-treated animals. The applied fold changes and p-values for the first filtering step were ≥ 1.5 and ≤ 0.05, respectively.
WORKFLOW
The data processing workflow using metaXCMS can be summarized in the following steps. First, metaXCMS is used to import the data from multiple metabolomics experiments as TSV (tab separated) files or Excel sheets (.xlsx), both of which are standard formats exported by XCMS that can be directly imported into metaXCMS without any further preprocessing. After loading of the experimental data, sample class assignments are verified and the control group for each experiment is defined. During the second processing step, feature lists are filtered by fold change (e.g., ≥ 2), p-value (e.g., ≤ 0.01), or predefined patterns of up- and down-regulation (e.g., features that are up-regulated in a first experiment: wildtype 1 vs. knockout 1, but down-regulated in a second experiment: wildtype 2 vs. knockout 2). In addition, feature lists can be designed to be subtracted from the final result so that metabolic changes in a control experiment (such as a reverse knockout) can be disregarded. The next step is the automated alignment of the feature lists from the different experiments on the basis of both m/z and retention time. The alignment method “group.nearest” that is implemented in XCMS is employed to align the data within user-defined m/z and retention time windows. Best results are achieved if the same LC/MS conditions are used for all samples that are to be aligned.
While the number of data sets that can be compared with metaXCMS is generally not limited, a direct visualization of the result as a Venn diagram is only possible for instances in which the number of sample groups is five or less. The Venn diagram shows the number of common features to all sample groups as well as the number of features contained within other possible intersections. It should be noted that in some experiments features that are not shared among sample groups may be the most biologically interesting, depending on the model systems being investigated and the question being asked. All metaXCMS results are displayed in tables that can be exported as Excel sheets.
For a detailed visual verification of the results, the retention time correction for all raw data files is re-calculated using OBI-Warp17 and extracted ion chromatograms (EIC) are generated for all selected features. Furthermore, boxplots are generated to visualize the distribution of feature intensities across the experiments. All graphic results can be exported as PNG or PDF files. The visualization displays are shown in Figure 2 for an example dataset.
Figure 2.

metaXCMS screenshot showing retention time correction curves and parameter settings for extracted ion chromatogram (EIC) generation (left), EIC overlay showing the ion intensity for m/z 112.09±0.01 for all samples from the pain dataset (upper right), and boxplot showing the distribution of integrated feature intensities of the feature m/z 112.09 for all pain samples (lower right). Pain models A, B, and C are defined in the legend of Figure 1.
EXPERIMENTAL SECTION
As a demonstration of the utility of the software, here we briefly describe the experimental application of metaXCMS to the investigation of three mouse models of pain as well as five Halobacterium salinarum knockout organisms. For the pain study, metabolites were extracted from 10 mg pieces of skin isolated from the hind paw of animals. The following animal groups were compared: (A) animals plantar injected with Complete Freund’s Adjuvant (CFA) and control animals, (B) animals to which noxious heat was acutely applied to the hind paw and room temperature-treated control animals; and (C) animals intraperitoneally injected with serum from K/BxN mice (i.e., KRN-treated mice) and vehicle-treated controls. These animals represent an inflammatory model,18 an acute heat model,19 and a spontaneous arthritis model of pain,20 respectively. All experiments were conducted in accordance with the National Institutes of Health and the Scripps Research Institute animal care and use guidelines. Five biological replicates were used for each pain model and control group.
Halobacterium salinarum cultures of four knockout strains (ΔVNG1816G, ΔVNG2094G, ΔVNG1179C, and ΔVNG0314G) were grown to logarithmic-growth phase and compared to their parent control strain (Δura3). Cultures were centrifuged, rinsed with phosphate buffer solution, and lyophilized. Metabolites were extracted from 5 mg of frozen cell pellets from each culture. The genes VNG1816G, VNG2094G, and VNG1179C encode leucine-responsive regulatory protein (Lrp) family transcription factors.21,22 The proteins encoded by VNG1816G and VNG2094G share binding sites that are upstream to genes involved in glutamic acid metabolism. The Cu-responsive transcription factor VNG1179C represses VNG2094G and has been reported to affect glutamic acid metabolism by Cu trafficking in previous studies.22 VNG0314G encodes an enzyme in the shikimate biosynthesis pathway. VNG0314G served as negative control for a metabolic pertubation that does not affect glutamic acid biosynthesis.
From skin tissue and lyophilized cell cultures, metabolites were extracted using cold methanol and acetone as described before.13 Liquid chromatography was performed using a reverse-phase C18 column (Zorbax C18, Agilent, 5 mM, 150×0.5mm diameter column) with a flow rate of 20 μl/min. Samples were analyzed by using electrospray ionization time-of-flight mass spectrometry (Agilent 6510 TOF) with water and acetonitrile for mobile phases A and B, each containing 0.1% formic acid. The chromatography started at 90% mobile phase A with a 45-min linear gradient to 98% mobile phase B.
RESULTS AND DISCUSSION
Although each of the pain models used in this study involves different pathogenic etiologies and mechanisms, we hypothesized that there may be common metabolites involved in triggering the transduction of nociceptive signals. We first compared each of the pain models to its respective control by using XCMS. XCMS performs feature detection as well as nonlinear retention time alignment and calculates statistics (Welch t-test) for each feature. The XCMS result is a table that contains the m/z and retention time coordinates, p-value and fold change for each feature, and the integrated feature intensities from all aligned samples. Each one of the three pain model comparisons resulted in more than 7,000 features, with the total number of summed features from all comparisons being 22,577. The three TSV files that were generated by XCMS were imported into metaXCMS and filtered by fold change (≥ 1.5) and p-value (≤ 0.05). No restrictions were made on up- or down-regulation.
The filter step yielded 380, 837, and 608 differentially regulated features for each one of the pairwise comparisons, resulting in a total number of 1825 dysregulated features (Figure 1, heatmaps). The second-order comparison was applied using a tolerance of 0.01 m/z and 60 seconds retention time. Three features were found to be differentially regulated in all three pain models (Table 1).
Table 1.
Features that were found to be differentially regulated in the pain models A, B, and C as defined in Figure 1. Fold change and p-value are shown as calculated by XCMS for the pairwise comparison.
| m/z | Retention time (s) | Pain model A vs Control A | Pain model B vs Control B | Pain model C vs Control C | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| fold change | p-value | fold change | p-value | fold change | p-value | ||
|
| |||||||
| 112.09 | 107 | 2.2 | 0.04 | 3.0 | 4e-04 | 1.7 | 0.05 |
| 233.10 | 85 | 1.8 | 0.04 | 1.8 | 0.02 | 1.7 | 0.05 |
| 112.18 | 107 | 2.2 | 0.04 | 4.1 | 4e-05 | 2.0 | 0.05 |
Retention time correction was then applied to the raw data for the generation of extracted ion chromatograms for each shared feature. In addition, boxplots were created on the basis of integrated intensity as exported from XCMS. Retention time correction curves for all pain samples is shown in Figure 2, along with overlaid extracted ion chromatograms and boxplots for the feature m/z 112.09. The compound with an observed m/z value of 112.09 was identified as histamine using accurate mass, retention time, and MS/MS fragmentation data as compared with a model compound. As validation of the approach, our result is consistent with the literature in that histamine has been well characterized as a mediator of pain by several mechanisms.23–25 It has been shown that irritation of the skin by mechanical, electrical, or chemical stimuli causes release of histamine by mast cells resulting in sensory nerve ending depolarization.26 In the skin, histamine receptors are found on Aβ fibers, on keratinocytes, on Merkel cells, and on deep dermal Aδ fibers terminating on dermal blood vessels.27 The precise physiological effects of histamine are complex due to its immunomodulatory and neurotransmitter properties,28,29 but our observation that it is commonly dysregulated is consistent with that which has been reported previously.
For the Halobacterium salinarum study, second-order analysis was performed on ΔVNG2094G and ΔVNG1816G with respect to their parent strains. For these mutants, 282 shared differences were detected. Among those, glutamic acid was found to be similarly dysregulated as expected (Figure 3a). The identity of glutamic acid was confirmed using accurate mass and MS/MS data as compared with a model compound. A higher-order analysis was also performed in which the difference profile from ΔVNG1179C with respect to its wildtype was introduced into the analysis. The number of shared differences decreased from 282 to 171 (Figure 3b). Importantly, glutamic acid was similarly dysregulated in all three mutants, supporting the previously reported physiological link between Cu-trafficking and glutamic acid metabolism (ΔVNG2094G: fold change 1.6, p-value 0.01, ΔVNG1816G: fold change 2.2, p-value 0.03, ΔVNG1179C: fold change 1.9, p-value 0.01). Finally, as a negative control, the comparison of ΔVNG0314G to its parent strain was introduced into the analysis. VNG0314G encodes an enzyme involved in shikimate biosynthesis that is unrelated to glutamic acid metabolism and therefore ΔVNG0314G served as a negative control. Glutamic acid was not detected as a differentially regulated metabolite among all four mutants (Figure 3c). The decrease in shared features among all samples with the addition of ΔVNG0314G demonstrates the utility of eliminating features not specifically related to the phenotype of interest with metaXCMS by using a negative control.
Figure 3.
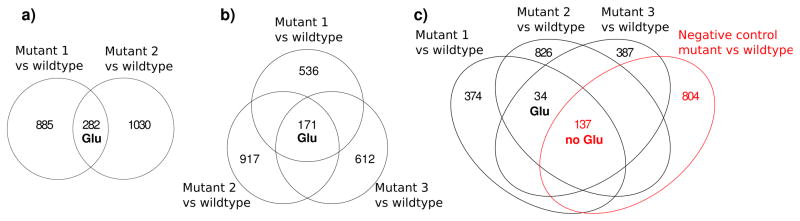
Venn diagrams showing the results of the second-order comparisons of four different Halobacterium salinarum knockout strains using metaXCMS. Mutants 1, 2, and 3 represent the strains ΔVNG2094G, ΔVNG1816G, and ΔVNG1179C, all of which are characterized by pertubations in glutamic acid metabolism. ΔVNG0314G does not affect glutamic acid metabolism, so ΔVNG0314G served as a negative control.
CONCLUSIONS
In summary, metaXCMS provides software for second-order analysis of metabolomics data facilitating meta-comparisons similar to those already used in genomics and transcriptomics.30–33 The introduction of such software in metabolomics is of significant value as it not only provides an analytical tool for distinguishing metabolites fundamentally associated with the underlying origin of a particular phenotype, but it also allows for data reduction at the feature level. Structural characterization of features is a rate-limiting step in the metabolomics workflow, and therefore metaXCMS offers a method to efficiently identify features with a higher likelihood to be biologically relevant prior to the time commitment of compound identification. In addition, metaXCMS provides a tool to analyze large cohorts of clinical samples from different groups or with complex subgroup variability.
Acknowledgments
We thank Matt Petrus at the Genomics Institute of the Novartis Research Foundation for help with the preparation of the animal models used in the study. This work was supported by the California Institute of Regenerative Medicine (TR1-01219), the National Institutes of Health (R24 EY017540-04, P30 MH062261- 10, and P01 DA026146-02), and NIH/NIA L30 AG0 038036 (G. J. P.). Financial support was also received from the Department of Energy (Grants FG02-07ER64325 and DE-AC0205CH11231).
References
- 1.Dunn WB. Phys Biol. 2008;5:011001. doi: 10.1088/1478-3975/5/1/011001. [DOI] [PubMed] [Google Scholar]
- 2.Want EJ, Nordström A, Morita H, Siuzdak G. J Proteome Res. 2007;6:459–68. doi: 10.1021/pr060505+. [DOI] [PubMed] [Google Scholar]
- 3.Smith C, Want E, O’Maille G, Abagyan R, Siuzdak G. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 4.Tautenhahn R, Böttcher C, Neumann S. BMC Bioinformatics. 2008;9:504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katajamaa M, Oresic M. J Chromatogr A. 2007;1158:318–328. doi: 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 6.Pluskal T, Castillo S, Villar-Briones A, Oresic M. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baran R, Kochi H, Saito N, Suematsu M, Soga T, Nishioka T, Robert M, Tomita M. BMC Bioinformatics. 2006;7:530. doi: 10.1186/1471-2105-7-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wikoff WR, Gangoiti JA, Barshop BA, Siuzdak G. Clin Chem. 2007;53:2169–76. doi: 10.1373/clinchem.2007.089011. [DOI] [PubMed] [Google Scholar]
- 9.Kalisiak J, Trauger SA, Kalisiak E, Morita H, Fokin VV, Adams MWW, Sharpless KB, Siuzdak G. J Am Chem Soc. 2009;131:378–86. doi: 10.1021/ja808172n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wikoff WR, Kalisak E, Trauger S, Manchester M, Siuzdak G. J Proteome Res. 2009;8:3578–87. doi: 10.1021/pr900275p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia W, Li H, Zhao L, Nicholson JK. Nat Rev Drug Discov. 2008;7:123–9. doi: 10.1038/nrd2505. [DOI] [PubMed] [Google Scholar]
- 12.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Proc Natl Acad Sci U S A. 2009;106:3698–703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yanes O, Clark J, Wong DM, Patti GJ, Sánchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. Nat Chem Biol. 2010;6:411–7. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horai H, et al. J Mass Spectrom. 2010;45:703–14. doi: 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- 15.Wishart DS, et al. Nucleic Acids Res. 2007;35:D521–526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith CA, Maille GO, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G. METLIN: A Metabolite Mass Spectral Database. Proceedings of the 9th International Congress of Therapeutic Drug Monitoring and Clinical Toxicology; Louisville, Kentucky. 2005. pp. 747–751. [DOI] [PubMed] [Google Scholar]
- 17.Prince JT, Marcotte EM. Analytical Chemistry. 2006;78:6140–6152. doi: 10.1021/ac0605344. [DOI] [PubMed] [Google Scholar]
- 18.Chu YC, Guan Y, Skinner J, Raja SN, Johns RA, Tao YX. Pain. 2005;119:113–23. doi: 10.1016/j.pain.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 19.Bölcskei K, Petho G, Szolcsányi J. Methods Mol Biol. 2010;617:57–66. doi: 10.1007/978-1-60327-323-7_5. [DOI] [PubMed] [Google Scholar]
- 20.Kyburz D, Corr M. Springer Seminars in Immunopathology. 2003;25:79–90. doi: 10.1007/s00281-003-0131-5. [DOI] [PubMed] [Google Scholar]
- 21.Goo YA, Yi EC, Baliga NS, Tao WA, Pan M, Aebersold R, Goodlett DR, Hood L, Ng WV. Mol Cell Proteomics. 2003;2:506–24. doi: 10.1074/mcp.M300044-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Kaur A, Pan M, Meislin M, Facciotti MT, El-Gewely R, Baliga NS. Genome Res. 2006;16:841–54. doi: 10.1101/gr.5189606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajihara Y, Murakami M, Imagawa T, Otsuguro K, Ito S, Ohta T. Neuroscience. 2010;166:292–304. doi: 10.1016/j.neuroscience.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida A, Mobarakeh JI, Sakurai E, Sakurada S, Orito T, Kuramasu A, Kato M, Yanai K. European Journal of Pharmacology. 2005;522:55–62. doi: 10.1016/j.ejphar.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 25.Rosenthal SR, Minard D. J Exp Med. 1939;70:415–25. doi: 10.1084/jem.70.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenthal SR, Sonnenschein RR. Am J Physiol. 1948;155:186–90. doi: 10.1152/ajplegacy.1948.155.2.186. [DOI] [PubMed] [Google Scholar]
- 27.Hough L, Rice FL. J Pharmacol Exp Ther. 2010 doi: 10.1124/jpet.110.171264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aarmstrong D, Dry RML, Keele CA, Markham JW. J Physiol. 1953;120:326–51. doi: 10.1113/jphysiol.1953.sp004898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fjallbrant N, Iggo A. J Physiol. 1961;156:578–90. doi: 10.1113/jphysiol.1961.sp006694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cantor RM, Lange K, Sinsheimer JS. Am J Hum Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borges F, Gomes G, Gardner R, Moreno N, McCormick S, Feijó JA, Becker JD. Plant Physiol. 2008;148:1168–81. doi: 10.1104/pp.108.125229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Güimil S, Chang HS, Zhu T, Sesma A, Osbourn A, Roux C, Ioannidis V, Oakeley EJ, Docquier M, Descombes P, Briggs SP, Paszkowski U. Proc Natl Acad Sci U S A. 2005;102:8066–70. doi: 10.1073/pnas.0502999102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higdon R, Haynes W, Kolker E. OMICS. 2010;14:309–14. doi: 10.1089/omi.2010.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]