

Abstract
Redox-inactive metals are found in biological and heterogeneous water oxidation catalysts, but their roles in catalysis are currently not well understood. A series of high oxidation state tetranuclear-dioxido clusters comprised of three manganese centers and a redox-inactive metal (M) of various charge is reported. Crystallographic studies show an unprecedented Mn3M(μ4-O)(μ2-O) core that remains intact upon changing M or the manganese oxidation state. Electrochemical studies reveal that the reduction potentials span a window of 700 mV, dependent upon the Lewis acidity of the second metal. With the pKa of the redox-inactive metal-aqua complex as a measure of Lewis acidity, these compounds display a linear dependence between reduction potential and acidity with a slope of ca. 100 mV per pKa unit. The Sr2+ and Ca2+ compounds show similar potentials, an observation that correlates with the behavior of the OEC, which is active only in the presence of one of these two metals.
Redox-inactive metal ions are critical components in many biological electron transfer reactions.1,2 For example, Ca2+ is essential for activity in the oxygen-evolving complex (OEC) of Photosystem II (PSII), although its exact role in catalysis is still unclear.3–5 There have been numerous studies of electron transfer to synthetic organic substrates,1,6 but studies of electron transfer to metal oxo complexes relevant to the active sites of a number of metalloenzymes have been fewer, possibly due to the challenge of isolating complexes with bound metal ions. Recently, a non-heme FeIVO complex with a bound Sc3+ ion was characterized crystallographically, and it was found that the presence of Sc3+ or Ca2+ in solution allowed the two-electron reduction of the complex using ferrocene.7 The addition of other Lewis acidic metal ions to a different FeIVO compound greatly enhanced electron transfer rates, although the adducts were not structurally characterized.8 With a monomeric MnII complex, faster rates of oxygen reduction were observed in the presence of Ca2+, and a MnIII–(μ-OH)–CaII complex was isolated as the product.9 Substitution of Sr2+ for Ca2+ in this complex showed a similar reduction potential, while substitution with Ba2+ resulted in a more negative reduction potential.10 O-atom transfer from some manganese oxo complexes has also been promoted by the binding of redox-inactive metals such as lithium.11
Redox-inactive metal ions also play a role in non-biological electron transfer reactions such as water oxidation catalyzed by heterogeneous cobalt12–14 and manganese oxides15–17 containing alkali or alkali earth metals. In these examples, there has been speculation that the redox-inactive metal is associated with the transition metal catalyst in cubane-like structures reminiscent of the crystallographically determined structure of the OEC, an oxide-bridged CaMn4 cluster in which the calcium center is associated with three of the manganese centers in a cubane motif (Fig. 1).18,19 A recent study varying the redox-inactive metal (K+, Ca2+, Sr2+, Mg2+) in layered heterogeneous manganese oxides showed that, as in the OEC, the presence of Ca2+ allows for the highest catalytic activity.15 Additionally, heterogeneous mixed oxides of cobalt show different water oxidation behavior dependent on the nature of the redox-inactive metal present in the mixture.20 The role of the redox-inactive metal on electron transfer and catalysis within the material remains unclear in these systems.
Figure 1.
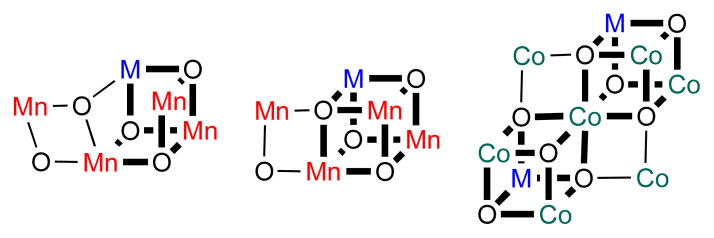
Proposed structures of water oxidation catalysts containing redox-inactive metals (M) in the OEC (left, middle) and in heterogeneous cobalt oxide water oxidation catalysts (right).12 The OEC is known to contain a Mn3M core: one major model is based on EXAFS and electron paramagnetic resonance (EPR) studies (left)41–43 and one on X-ray crystallography (middle).18,19 Bold bonds emphasize the Mn3M and Co3M cluster cores.
Our recent report of a structural model of the CaMn3 subsite of the OEC containing a high oxidation state heterometallic MnIV3CaO4 moiety and comparison to a tetramanganese analog suggested a significant influence of the calcium center on the redox properties of the cluster.21 To study the scope and chemical basis of this phenomenon in multimetallic oxide clusters such as those found in the OEC and in heterogenous systems, access to well-defined and structurally related heterometallic oxido clusters of redox-active and inactive metals is desirable. Based on a multinucleating ligand-based synthetic strategy developed by our group,22,23 we targeted heteronuclear clusters supported by the hexapyridyl trisalkoxido 1,3,5-triarylbenzene ligand (H3L). In the present work, we describe the synthesis of a series of tetranuclear heterometallic trimanganese dioxo clusters [Mn3M(μ4-O)(μ2-O)] containing a redox-inactive cation bridging via oxido moieties to manganese centers. Electrochemical characterization reveals that large changes in the Lewis acidity of the redox-inactive metal have a systematic effect on the redox properties of the cluster.
Results and discussion
Heterometallic clusters have been targeted by several groups as proposed structural models of the OEC as evidence it contained a mixed Mn-Ca-oxido cluster emerged.24–30 Calcium-manganese clusters remain uncommon, though recently there have been a number of such complexes that have been isolated and structurally characterized.9,21,25–30 Because heterometallic clusters are often synthesized by self-assembly, controlling the composition and relative arrangement of metals has been a challenge. Furthermore, these synthetic protocols are not necessarily easily extended to the incorporation of other redox-inactive metals instead of calcium. To develop general syntheses of heterometallic clusters, we employed a multinucleating ligand that affords versatile trimetallic (MII3) precursors.23 These MnII3 species could be elaborated to site-differentiated tetramanganese cubanes, Mn4O4, as well as heteronuclear Mn3CaO4 clusters.21 Strategies for general synthetic protocols to related heteronuclear complexes were then explored.
A dicationic calcium-bridged hexamanganese complex ([LMn2IIIMnIIO(OAc)3]2Ca(OTf)2) in which each trimanganese unit is coordinated by a μ3-oxide was prepared and proposed to be an intermediate in the synthesis of LMnIV3 CaO4(OAc)3(THF).21 In an effort to isolate other manganese clusters of high oxidation state relevant for the preparation of structural mimics of the OEC, [LMn2IIIMnIIO(OAc)3]2Ca(OTf)2 was treated with Ca(OTf)2 and PhIO in 1,2-dimethoxyethane (DME) to form a red-purple compound (Fig. 2). The same compound was also independently synthesized in high yield (84%) in one step from the more reduced LMnII3(OAc)3 precursor (Fig. 2). A single crystal X-ray diffraction (XRD) study of this species confirmed the material to be a calcium trimanganese dioxo complex ([1-Ca(DME)(OTf)]2+ (1 = LMnIVMnIII2O2(OAc)2), Fig. 3). The metal oxidation states were assigned based on crystallographic, XAS, and magnetism data (vide infra). In this complex, as in LMnII3(OAc)3, the three manganese centers are bridged by three alkoxide donors from L, forming a six-membered ring, and the pyridine nitrogens of each dipyridyloxymethyl moiety coordinate to adjacent metal centers. Ca2+ is bridged to the trimanganese cluster by a μ4-oxido, to the MnIV center by a μ2-oxido, and to the remaining MnIII centers by bridging acetate moieties. The Ca2+ is further coordinated by a bidentate DME ligand and a trifluoromethanesulfonate anion. Two trifluoromethanesulfonate ions remain outer-sphere. The isolated compounds reported here display diagnostic 1H NMR spectra, although the paramagnetically broadened and shifted signals have not been assigned. The addition of excess water to a CD2Cl2 solution of [1-Ca(DME)(OTf)]2+ forms a new species by 1H NMR spectroscopy (Supplementary Fig. S2) that was identified by XRD as the tris(aqua) complex [1-Ca(OH2)3]3+ (Supplementary Fig. S26). Addition of DME to the CD2Cl2 solution of [1-Ca(OH2)3]3+ converts the complex back to [1-Ca(DME)(OTf)]2+ (1H NMR spectroscopy, Supplementary Fig. S9). In the solid state, [1-Ca(DME)(OTf)]2+ is stable for weeks under ambient conditions.
Figure 2.

Synthesis of tetrametallic trimanganese dioxido complexes. Reduced precursor LMn3(OAc)3 was oxidized in the presence of metal triflate salts to form [LMn3MO2(OAc)2] compounds (M = Na, Ca, Sr, Y). Complex [1-Ca(DME)(OTf)]2+ (1 = LMnIVMnIII2O2(OAc)2) undergoes reversible chemical reduction, substitution of Zn2+ for Ca2+, or exchange of the DME ligand for coordinated water molecules.
Figure 3.

Solid-state structures of reported complexes (thermal ellipsoids shown at 50% level). Hydrogen atoms and outer-sphere anions not shown for clarity. (a) Full structure of [1-Ca(DME)(OTf)][OTf]2. (b) Truncated view of [1-Ca(DME)(OTf)][OTf]2. (c) Truncated view of [2-Ca(DME)(OTf)][OTf]. (d) Truncated view of [1-Sr(DME)(OTf)][OTf]2. (e) Truncated view of [2-Sr(DME)(OTf)][OTf]. (f) Truncated view of [1-Na]2[OTf]4. (g) Truncated view of [1-Zn(CH3CN)][OTf]3. (h) Truncated view of [2-Y(DME)(OTf)][OTf]2.
Treatment of a DME suspension of [1-Ca(DME)(OTf)]2+ with the one-electron reductant decamethylferrocene yields the singly reduced product [2-Ca(DME)(OTf)]+ (2 = LMnIII3O2(OAc)2). The chemical reversibility of this conversion is evidenced upon treatment of [2-Ca(DME)(OTf)]+ with AgOTf to convert back to [1-Ca(DME)(OTf)]2+. 1H NMR analysis is consistent with clean interconversion of these two clusters. A XRD study of [2-Ca(DME)(OTf)]+ reveals a cluster isostructural to [1-Ca(DME)(OTf)]2+ with only slight bond distance changes (vide infra), indicating little rearrangement of the cluster upon reduction.
The variable temperature magnetic susceptibilites of [1-Ca(DME)(OTf)][OTf]2 and [2-Ca(DME)(OTf)][OTf] were studied (Supplementary Fig. S11). For [1-Ca(DME)(OTf)][OTf]2, dominant ferromagnetic coupling between Mn ions is observed (see Supplementary Table S1 for fitting parameters). At 14 K, the χMT value increases to a maximum of 18.7 cm3 mol−1 K, which is close to the expected spin-only value of a S = 11/2 system (17.9 cm3 mol−1 K, g = 2). Different values would be expected for a more oxidized S = 5 system (15 cm3 mol−1K, g = 2) or a more reduced S = 6 system (21 cm3 mol−1K, g = 2). These results support the oxidation state assignment of [1-Ca(DME)(OTf)]2+ as MnIVMnIII2. The χMT value of [2-Ca(DME)(OTf)][OTf] approaches 10.2 cm3 mol−1 K at 300 K, which is near the expected spin-only value of three uncoupled MnIII ions (S = 2 spins, 3 cm3 mol−1 K, g = 2). The χMT value decreases at low temperatures, reaching 2.6 cm3 mol−1 K at 4 K, which shows a dominant antiferromagnetic interaction (Supplementary Table S1). To further confirm the oxidation state assignment of the isolated species, Mn XANES spectra were collected for [1-Ca(DME)(OTf)](OTf)2 and [2-Ca(DME)(OTf)](OTf) (Fig. 4a). The rising edge energy, taken as a zero-crossing point of the 2nd derivative spectrum (Fig. 4b), is shifted to a higher energy by ca. 1.0 eV step from [2-Ca(DME)(OTf)](OTf) (6548.66 eV) to [1-Ca(DME)(OTf)](OTf)2 (6549.76 eV). Such step-wiseedge shift is consistent with an one-electron oxidation state change in redox-active Mn complexes,31 when the geometry and type of ligands are highly conserved. This result, correlated with charge balance in the solid state structure, magnetism, and chemical reactivity, therefore, supports the formal oxidation state assignments for [1-Ca(DME)(OTf)](OTf)2 and [2-Ca(DME)(OTf)](OTf) as MnIVMnIII2 and MnIII3 respectively.
Figure 4.

(a) Mn XANES spectra and (b) second derivative spectra of [2-Ca(DME)(OTf)][OTf] (dotted) and [1-Ca(DME)(OTf)][OTf]2 (solid). The shift in the rising edge energy of the two complexes (6548.66 and 6549.76 eV respectively) suggests a one-electron oxidation state change.
To study the effects of the redox-inactive center on the properties of the [Mn3O2] core, the analogous Sr2+-, Y3+-, Na+-, and Zn2+-capped trimanganese dioxo complexes were targeted (Figs 2 and 3). Treatment of LMnII3(OAc)3 with PhIO and M(OTf)n (M = Na, Sr, Y), led to new species with 1H NMR spectroscopic characteristics similar to compounds [1-Ca(DME)(OTf)]2+ and [2-Ca(DME)(OTf)]+. Complexes [1-Sr(DME)(OTf)]2+ and [2-Y(DME)(OTf)]2+ are structurally analogous to [1-Ca(DME)(OTf)]2+ with the redox-inactive metal bridged by two oxido moieties to the trimanganese cluster and further coordinated by a DME molecule and a triflate anion (Fig. 3d, h). The yttrium-capped dioxo compound was isolated in the more reduced MnIII3 state rather than the MnIVMnIII2 state observed under the same reaction conditions for the calcium and strontium dioxo compounds. Similar to the reduction of [1-Ca(DME)(OTf)]2+ to [2-Ca(DME)(OTf)]+, the reduced strontium compound [2-Sr(DME)(OTf)]+ was prepared by treating [1-Sr(DME)(OTf)]2+ with decamethylferrocene. The Na-capped dioxo complex [1-Na]24+ was isolated in the solid-state as a dimer of Mn3NaO2 moieties via acetate bridges. Each Mn3NaO2 core is structurally similar to the Ca2+, Sr2+ and Y3+ analogs, with the Na+ cation bridged by two oxidos to the trimanganese moiety (Fig. 3f). Preparation of the Zn-capped compound [1-Zn(CH3CN)]3+was accomplished by addition of Zn(OTf)2 to a CH3CN solution of [1-Ca(DME)(OTf)]2+, leading to substitution of Ca2+ with Zn2+. A XRD study of [1-Zn(CH3CN)]3+ grown from an acetonitrile/diethyl ether mixture shows one acetonitrile coordinated to the Zn2+ center. The Mn3ZnO2 core is structurally similar to the other Mn3MO2 moieties reported here. The magnetic susceptibilities of the MnIVMnIII2 complexes [1-Sr(DME)(OTf)][OTf]2, [1-Na]2[OTf]4, and [1-Zn(CH3CN)][OTf]3 were measured, and all demonstrate ferromagnetic coupling of spins close to that of [1-Ca(DME)(OTf)][OTf]2, supporting the same formal oxidation state assignment (Supplementary Fig. S11).
Analysis of the solid-state structures show that the distances from the redox-inactive metals to the briding oxido moieties vary as expected based on the ionic radius of the ion M, with the M–O distances increasing from [1-Zn(CH3CN)]3+ [2.008(3), 2.090(3) Å] to [1-Sr(DME)(OTf)]2+ [2.510(5), 2.651(4) Å] (Table 1). Upon reduction of the MnIVMnIII2 clusters [1-Ca(DME)(OTf)]2+ and [1-Sr(DME)(OTf)]2+ to form the MnIII3 complexes [2-Ca(DME)(OTf)]+ and [2-Sr(DME)(OTf)]+, the M–(μ4-O) distances contract, indicating a stronger interaction with the redox-inactive metal as the interaction of the μ4-oxido with the trimanganese core weakens due to increased population of σ-antibonding orbitals in d4, MnIII centers. As expected, the Mn–O distances increase upon reduction (compounds 2 vs 1), but only small variations are observed based upon the redox-inactive metal. [2-Y(DME)(OTf)]2+ displays the longest Mn-oxido average distance, consistent with the yttrium center, the most Lewis acidic metal of the series, drawing more electron density from the oxido moieties and weakening the Mn-O interactions. Notably, the bond distances of the Mn3O2 core are essentially the same between [1-Ca(DME)(OTf)]2+ and [1-Sr(DME)(OTf)]2+. These observations are consistent with XAS studies of Sr-substituted PSII indicating no significant structural change in the OEC from Ca-substituted PSII.3
Table 1.
Selected bond lengths (Å) for reported complexes. The Mn–O bond lengths vary depending on manganese oxidation state, but not significantly due to the redox-inactive metal M. M–O bond lengths vary with ionic radius. All e.s.d.s were calculated rigorously from the full covariance matrix.40
| [1-Ca(DME)(OTf)]2+ | [2-Ca(DME)(OTf)]+ | [1-Sr(DME)(OTf)]2+ | [2-Sr(DME)(OTf)]+ | [1-Na]24+ | [1-Zn(CH3CN)]3+ | [2-Y(DME)(OTf)]2+ | |
|---|---|---|---|---|---|---|---|
| M–Mn1 | 3.317(1) | 3.283(1) | 3.476(1) | 3.424(1) | 3.216(3) | 3.0005(8) | 3.3011(6) |
| M–Mn2 | 3.749(1) | 3.802(1) | 4.005(1) | 3.923(1) | 3.739(3) | 3.3495(9) | 3.7533(6) |
| M–Mn3 | 4.042(1) | 4.034(1) | 4.149(1) | 4.166(1) | 3.942(3) | 3.7595(9) | 3.8592(6) |
| Mn1–Mn2 | 3.0480(9) | 3.0111(9) | 3.062(1) | 3.020(2) | 3.076(2) | 3.065(1) | 3.2110(7) |
| Mn1–Mn3 | 3.0486(9) | 3.1537(9) | 3.051(1) | 3.142(2) | 3.000(2) | 3.038(1) | 3.0494(7) |
| Mn2–Mn3 | 3.0179(9) | 3.0541(9) | 3.025(1) | 3.064(2) | 3.004(2) | 3.0739(9) | 3.1223(7) |
| M–O2 | 2.349(3) | 2.368(3) | 2.510(5) | 2.508(5) | 2.300(6) | 2.008(3) | 2.269(2) |
| M–O1 | 2.452(3) | 2.397(3) | 2.651(4) | 2.536(5) | 2.422(6) | 2.090(3) | 2.232(2) |
| Mn1–O2 | 1.842(3) | 1.887(3) | 1.841(4) | 1.869(5) | 1.840(5) | 1.862(3) | 1.889(2) |
| Mn1–O1 | 2.017(3) | 1.939(3) | 2.022(4) | 1.943(5) | 1.995(5) | 1.981(3) | 2.150(2) |
| Mn2–O1 | 1.913(3) | 2.159(3) | 1.932(4) | 1.856(5) | 1.936(5) | 1.946(3) | 2.177(2) |
| Mn3–O1 | 1.958(3) | 1.860(3) | 1.936(4) | 2.172(5) | 1.889(5) | 1.960(3) | 1.895(2) |
| Avg. Mn– O1 | 1.96 | 1.99 | 1.96 | 1.99 | 1.94 | 1.96 | 2.07 |
We and others have reported studies that suggest a significant effect of redox-inactive metals on the electron transfer properties of metal-oxido species, phenomenon of particular relevance in biological systems and heterogeneous mixed metal oxides.7,8,15,21,24,32–34 With a series of well-defined and structurally analogous Mn3MO2 complexes in hand, the effect of metal M on the reduction potential of the clusters was investigated. The cyclic voltammogram (CV) of a 10:1 CH2Cl2/DME solution of [1-Ca(DME)(OTf)]2+ (0.1 M NBu4PF6) shows two quasireversible redox couples at −70 and −530 mV versus the ferrocene/ferrocenium couple (Fc/Fc+) (Supplementary Fig. S15). Since [1-Ca(DME)(OTf)]2+ is chemically reduced to [2-Ca(DME)(OTf)]+ by the addition of one equivalent of decamethylferrocene (EO ~ −0.48 V vs. Fc/Fc+ in CH2Cl2), the couple centered at −70 mV is assigned as the [MnIVMnIII2/MnIII3] couple. A 10:1 CH2Cl2/DME solution of [1-Ca(DME)(OTf)]2+ (0.05 M LiOTf electrolyte) was electrolyzed at a potential of −0.25 V vs. Ag/Ag+ to form a new species by 1H NMR spectroscopy. The amount of current passed at that potential supports the assignment of the wave at −80 mV as the one electron reduction of [1-Ca(DME)(OTf)]2+. Independently prepared [2-Ca(DME)(OTf)]+ reacts with excess LiOTf to cleanly form a product whose 1H NMR spectrum matches that of the controlled potential electrolysis product described above (Supplementary Fig. S13). Under the same conditions, [1-Ca(DME)(OTf)]2+ does not react with LiOTf. The more negative redox couple at −530 mV is presumed to correspond to the [MnIII3/MnIII2MnII] couple, but the more reduced product has not yet been isolated.
Since the electrochemical studies are presumed to be of intact clusters, the solution stability of the [MMn3O2] core was studied. Isotopically labelled LCaMn318O2(OAc)2(DME)(OTf)3 ([1*-Ca(DME)(OTf)][OTf]2) was prepared and mixed with one equivalent of natural abundance [1-Ca(DME)(OTf)]2+ in dichloromethane at room temperature, and the mixture was analyzed over time using electrospray ionization mass spectrometry (ESI-MS). Less than 20% isotopic scrambling occurs on the timescale of the electrochemical experiments (ca. 18% after 1 h at RT). Additionally, methylene chloride solutions of [1-Ca(DME)(OTf)]2+ are stable at room temperature under anhydrous conditions for days (1H NMR spectroscopy). The addition of 10 equivalents of Ca(OTf)2 to the solution of [1-Ca(DME)(OTf)]2+ does not change the reduction potential of the complex (Supplementary Fig. S17) indicating that any equilibrium toward dissociation of the clusters lies toward Ca2+ association. To interrogate the effect of the Ca-coordinated ligands, under the same conditions, the reduction of [1-Ca(OH2)3]3+ occurs within 30 mV of that of [1-Ca(DME)(OTf)]2+ (Supplementary Fig. S16) suggesting that the capping ligands on the Ca2+ center do not significantly affect the reduction potentials of the clusters.
CVs of [1-Sr(DME)(OTf)]2+, [2-Y(DME)(OTf)]2+, [1-Zn(CH3CN)]3+, and [1-Na]24+ all display the [MnIVMnIII2/MnIII3] couple observed for [1-Ca(DME)(OTf)]2+, although at different potentials (Fig. 5a). Although the peak-to-peak separations of the observed couples are large when measured at a glassy carbon electrode (ΔEp ~ 400 mV), indicating slow electron transfer to the complexes, the E1/2 values calculated from the CVs of [1-Ca(DME)(OTf)]2+, [1-Sr(DME)(OTf)]2+, and [1-Na]24+ are in good agreement with CVs collected using a hanging drop mercury electrode, with ΔEp ~ 200 mV (Supplementary Fig. S14). The E1/2 corresponding to the reduction of [1-Ca(DME)(OTf)]2+ to [2-Ca(DME)(OTf)]+ was also measured by monitoring the electronic absorption spectrum upon titration with dimethylferrocene (Supplementary Fig. S18), and the calculated values (−0.1 V vs. Fc/Fc+) are close to the value from the CV (−0.08 V vs. Fc/Fc+).
Figure 5.

The redox potentials of the [MMn3O2] complexes are correlated with the Lewis acidity of the redox-inactive metal. (a) Cyclic voltammograms of reported complexes in 0.1 M NBu4PF6 10:1 CH2Cl2/DME (CV of [1-Na]24+ in CH2Cl2) using a glassy carbon disc electrode at a scan rate of 100 mV/s. (b) Dependence of E1/2 of MnIVMnIII2/MnIII3 couple on pKa of M(aqua)n+ ion.36 Error bars correspond to the standard deviation of the reduction potentials measured from three independent samples.
As structural characterization was obtained only for clusters displaying the MnIVMnIII2 and MnIII3 oxidation states, these are the reduction potentials that are compared below. Moreover, the more highly oxidized species are pertinent to the moieties present in the OEC and proposed for water oxidation catalysts. The E1/2 values of the [MnIVMnIII2/MnIII3] couple become more positive as the charge of the redox-inactive metal increases. This trend suggests that the increased charge of the proximal redox-inactive cation facilitates reduction of the manganese centers. The E1/2 value for the Zn2+ complex appears at potentials ca. 230 mV more positive compared to the Ca2+ and Sr2+ species. Although [1-Zn(CH3CN)]3+ is tricationic while [1-Ca(DME)(OTf)]2+ and [1-Sr(DME)(OTf)]+ are dicationic, the E1/2 of the [MnIVMnIII2/MnIII3] couple of [1-Ca(OH2)]3+, also a tricationic complex, is more positive than that of [1-Ca(DME)(OTf)]2+ by less than 30 mV. The more positive potential of the Zn2+ compound is inconsistent with a purely electrostatic explanation of the change in reduction potentials, as proposed for oxo-bridged manganese dimers with alkali and alkali earth metal ions associated via salen ligands modified with crown ether moieties.33
The effect of redox-inactive metals on the kinetics of electron transfer to a non-heme FeIVO species has been previously linked to the Lewis acidity of the metal.8,35 The E1/2 values of the [MnIVMnIII2/MnIII3] couples measured above in organic solvents were plotted against the pKa of the metal aqua ions, M(aqua)n+ in water,36 used here as a measure of the Lewis acidity of the metal M. A remarkable linear correlation is observed (Fig. 5b) that clearly links the effect of the redox-inactive metal to the cluster reduction potential in terms of the metal’s Lewis acidity. The slope provides a quantitative measure of this correlation, with each pKa unit shifting the potential by ca. 100 mV. This effect is likely a consequence of the interaction between the oxido moieties and redox-inactive metals vs. manganese centers. The stronger Lewis acid is expected to draw more electron density from the oxido ligands and to destabilize the higher oxidation state manganese centers.
The above findings have implications for biological and heterogeneous metal catalysts for water oxidation and other redox processes. Catalysis occurs at discrete multinuclear sites, consisting of five metal centers for the OEC in PSII and less than ten metal centers for heterogeneous manganese and cobalt oxides. The potential of the cluster is expected to vary based on structure and number of oxido ligands. For example, the reduction potential of the[MnIVMnIII2CaO2/MnIII3CaO2] couple reported here is more positive than that of the [MnIV3CaO4/MnIV2MnIIICaO4] couple of the CaMn3 cubane cluster,21 despite the higher manganese oxidation states in the latter complex, likely due to the greater number of oxido ligands. Changing the nature of the redox-inactive metal component of the cluster, without a structural change of the cluster, allows for drastic variation of the reduction potential in both directions, potentially from values that render the chemistry of interest (e.g. water oxidation) thermodynamically unfavorable to values that make it favorable. Tuning of the reduction potentials of mixed metal-oxide cluster by Lewis acids is an appealing strategy for designing practical catalysts for water splitting.
It is notable in the present series that the Ca2+ and Sr2+ variants have essentially the same potentials, which is consistent with the observation that substitution of Sr2+ for Ca2+ in PSII retains reactivity and a similar electronic structure.37 Although a previously proposed role of Ca2+ (or Sr2+) of the OEC in facilitating the attack of a water or hydroxide moiety on an electrophilic manganese oxo is still possible,38 the reported results support a significant role in redox tuning of the cluster.
In summary, a series of tetrametallic dioxo complexes containing redox-inactive metal ions in the +1 to +3 oxidation states was synthesized by employing a trinucleating ligand framework. The structural characteristics of these complexes, with the oxido ligands bridging the redox-inactive metals and the manganese centers, make them particularly relevant to biological and heterogeneous metal-oxido clusters. Electrochemical studies of these compounds show that the reduction potentials are highly dependent upon the Lewis acidity of the redox-inactive metal, identifying the chemical basis for the observed differences in electrochemistry. This correlation provides evidence for the role of the Ca2+ ion in modulating the redox potential of the OEC and of other redox-inactive ions in tuning the redox potentials of other metal oxide electrocatalysts. The observed linear dependence between cluster potential and Lewis acidity provides a rational strategy for tuning the redox properties of heterometallic metal-oxido clusters of interest for catalysis. Current studies are focused on further understanding the relationship between the structures of mixed metal manganese-oxido clusters and their reactivity.
Methods
Unless indicated otherwise, reactions were carried out in oven-dried glassware in a glovebox under a nitrogen atmosphere. Anhydrous tetrahydrofuran (THF) was purchased from Aldrich in 18 L Pure-Pac™ containers. Anhydrous dichloromethane, diethyl ether, and THF were purified by sparging with nitrogen for 15 minutes and then passing under nitrogen pressure through a column of activated A2 alumina. Anhydrous 1,2-dimethoxyethane (DME) was dried over sodium/benzophenone ketyl and vacuum-transferred onto molecular sieves. CD2Cl2 was purchased from Cambridge Isotope Laboratories, dried over calcium hydride, then degassed by three freeze-pump-thaw cycles and vacuum-transferred prior to use.
Iodosobenzene was prepared according to literature procedures.39 LMn3(OAc)3 and[LMn3O(OAc)3]2Ca(OTf)2 were prepared according to previously published procedures.21,23 Caution! Iodosobenzene is potentially explosive and should be used only in small quantities.
The syntheses of all reported compounds, characterization methods and equipment are described in the Supplementary Information.
Synthesis of [1-Ca(DME)(OTf)][OTf]2
In the glovebox, a round-bottom flask equipped with a stir bar was charged with LMn3(OAc)3 (2.0 g, 1.67 mmol) and Ca(OTf)2 (0.90 g, 2.67 mmol, 1.6 equiv). DME (200 mL) was added, and the yellow suspension was stirred at room temperature for 5 min. Iodosobenzene (0.81 g, 3.68 mmol, 2.2 equiv) was added as a solid, and the mixture was stirred at room temperature for 4 h, turning from yellow to purple. The purple solid was collected via filtration, washed with DME, then extracted with dichloromethane. The red-purple solution was concentrated in vacuo to yield the product as a red-purple solid (2.45 g, 84%). 1H NMR (CD2Cl2, 300 MHz): δ 77.8, 76.5, 72.3, 69.6, 60.5, 53.7, 48.4, 38.6, 37.0, 24.2, 19.9, 17.5, 15.9, 8.1, 3.3, 2.9, −19.3, −23.5, −24.9, −26.4, −29.1 ppm. 19F NMR (CD2Cl2): δ−74.4 ppm. UV-Vis (CH2Cl2, λmax (ε)): 498 (1410 M−1 cm−1), 846 (640 M−1 cm−1) nm. Anal. Calcd. For C68H55CaF9Mn3N6O20S3: C, 46.72; H, 3.17; N, 4.81. Found: C, 46.92; H, 3.27; N, 4.89.
Synthesis of [2-Ca(DME)(OTf)][OTf]
In the glovebox, a round-bottom flask equipped with a stir bar was charged with [1-Ca(DME)(OTf)]2+ (0.750 g, 0.429 mmol) and decamethylferrocene (0.140 g, 0.429 mmol, 1 equiv). DME (30 mL) was added, and the purple mixture was stirred at room temperature over 1 h. The gray-purple precipitate was collected on a fritted glass funnel and washed with DME, then extracted with cold THF (40 mL). The purple filtrate was concentrated to ca. 20 mL in vacuo, then cooled to −35 °C to precipitate out more Cp*2Fe+, which was filtered off over Celite. The purple filtrate was concentrated in vacuo to a purple solid, then recrystallized from DME/CH2Cl2/hexanes to yield the product as a purple solid (0.405 g, 59%). 1H NMR (CD2Cl2, 300 MHz): δ 65.1, 57.8, 50.7, 41.6, 35.3, 29.6, 15.7, 15.0, 14.2, 8.2, 3.0, −9.6, −12.6, −17.0, −17.9 ppm. 19F NMR (CD2Cl2): δ−74.6 ppm. UV-Vis (CH2Cl2, λmax (ε)): 495 (710 M−1 cm−1), 860 (310 M−1 cm−1) nm. Anal. Calcd. for C67H55CaF6Mn3N6O17S2: C, 50.32; H, 3.47; N, 5.26. Found: C, 50.04; H, 3.63; N, 5.06.
Electrochemical measurements were recorded using a Pine Instrument Company AFCBP1 bipotentiostat using the AfterMath software package. Cyclic voltammograms were recorded on ca. 1 mM solutions of the relevant complexes in the glovebox at 20 °C with an auxiliary Pt-coil electrode, a Ag/Ag+ reference electrode (0.01 M AgNO3, 0.1 M nBu4NPF6 in CH3CN), and a 3.0 mm glassy carbon electrode disc (BASI). The electrolyte solutions were 0.1 M nBu4NPF6 in CH2Cl2 ([1-Na]24+) or 10:1 CH2Cl2/DME ([1-Ca(DME)(OTf)]2+, [1-Sr(DME)(OTf)]2+, [1-Zn(CH3CN)]3+, and [2-Y(DME)(OTf)]2+). For [1-Ca(DME)(OTf)]2+, [1-Sr(DME)(OTf)]2+, and [1-Na]24+, CVs were also recorded in an electrolyte solution of 0.1 M nBu4NPF6 in CH2Cl2 using a hanging mercury drop electrode (BASI CGME). Using the average mass of the mercury drop (0.037 g), the surface area of the drop (assumed to be a sphere) was calculated to be 0.095 cm2. All reported values are referenced to an internal ferrocene/ferrocenium couple.
Supplementary Material
Acknowledgments
This work was supported by the California Institute of Technology, the Searle Scholars Program, the NSF CAREER CHE-1151918 (T.A.), and the NSF Graduate Research Fellowship Program (E.Y.T.). We thank Lawrence M. Henling and David E. Herbert for assistance with crystallography, and Po-Heng Lin for assistance with magnetic susceptibility studies. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF Chemistry Research Instrumentation award to Caltech (CHE-0639094). The X-ray spectroscopy work was supported by NIH Grant F32GM100595 (R.T.), and by the Director of the Office of Basic Energy Science, OBES, Division of Chemical Sciences, Geosciences, and Biosciences, DOE, under Contract DE-AC02-05CH11231 (J.Y.). Synchrotron facilities were provided by the Stanford Synchrotron Radiation Lightsource (SSRL) operated by the DOE, OBES.
Footnotes
Author Contributions
E.Y.T. and T.A. designed research. E.Y.T. and R.T. performed experiments. R.T. and J.Y. provided XANES characterization. E.Y.T., R.T., J.Y., and T.A. analyzed data. E.Y.T. and T.A. wrote the paper.
The authors declare no competing financial interests.
Supplementary information and chemical compound information are available in the online version of the paper.
Reprints and permission information is available online at http://www.nature.com/reprints.
References
- 1.Fukuzumi S, Ohkubo K. Metal ion-coupled and decoupled electron transfer. Coord Chem Rev. 2010;254:372–385. [Google Scholar]
- 2.Fukuzumi S. Progress in Inorganic Chemistry. In: Karlin KD, editor. Progress in Inorganic Chemistry. 56 . John Wiley & Sons Inc; 2009. pp. 49–153. [Google Scholar]
- 3.Yachandra VK, Yano J. Calcium in the oxygen-evolving complex: Structural and mechanistic role determined by X-ray spectroscopy. J Photochem Photobiol B: Biol. 2011;104:51–59. doi: 10.1016/j.jphotobiol.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yocum CF. The calcium and chloride requirements of the O2 evolving complex. Coord Chem Rev. 2008;252:296–305. [Google Scholar]
- 5.McEvoy JP, Brudvig GW. Water-splitting chemistry of photosystem II. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 6.Park J, et al. Scandium ion-enhanced oxidative dimerization and N-demethylation of N,N-dimethylanilines by a non-heme iron(IV)-oxo Complex. Inorg Chem. 2011;50:11612–11622. doi: 10.1021/ic201545a. [DOI] [PubMed] [Google Scholar]
- 7.Fukuzumi S, et al. Crystal structure of a metal ion-bound oxoiron(IV) complex and implications for biological electron transfer. Nat Chem. 2010;2:756–759. doi: 10.1038/nchem.731. [DOI] [PubMed] [Google Scholar]
- 8.Morimoto Y, et al. Metal ion-coupled electron transfer of a nonheme oxoiron(IV) complex: remarkable enhancement of electron-transfer rates by Sc3+ J Am Chem Soc. 2011;133:403–405. doi: 10.1021/ja109056x. [DOI] [PubMed] [Google Scholar]
- 9.Park YJ, Ziller JW, Borovik AS. The effects of redox-inactive metal ions on the activation of dioxygen: Isolation and characterization of a heterobimetallic complex containing a MnIII-(μ-OH)-CaII core. J Am Chem Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park YJ, et al. Heterobimetallic complexes with MIII-(μ-OH)-MII cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): structural, kinetic, and redox properties. Chem Sci. 2013 doi: 10.1039/C2SC21400H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller CG, et al. A method for driving O-atom transfer: Secondary ion binding to a tetraamide macrocyclic ligand. J Am Chem Soc. 1998;120:11540–11541. [Google Scholar]
- 12.Risch M, et al. Water oxidation by electrodeposited cobalt oxides - role of anions and redox-inert cations in structure and function of the amorphous catalyst. ChemSusChem. 2012;5:542–549. doi: 10.1002/cssc.201100574. [DOI] [PubMed] [Google Scholar]
- 13.Kanan MW, et al. Structure and valency of a cobalt-phosphate water oxidation catalyst determined by in situ X-ray spectroscopy. J Am Chem Soc. 2010;132:13692–13701. doi: 10.1021/ja1023767. [DOI] [PubMed] [Google Scholar]
- 14.Kanan MW, Nocera DG. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+ Science. 2008;321:1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- 15.Wiechen M, Zaharieva I, Dau H, Kurz P. Layered manganese oxides for water oxidation: Alkaline earth cations influence catalytic activity in a photosystem II-like fashion. Chem Sci. 2012;3:2330–2339. [Google Scholar]
- 16.Najafpour MM, Ehrenberg T, Wiechen M, Kurz P. Calcium manganese(III) oxides (CaMn2O4•xH2O) as biomimetic oxygen-evolving catalysts. Angew Chem Int Ed. 2010;49:2233–2237. doi: 10.1002/anie.200906745. [DOI] [PubMed] [Google Scholar]
- 17.Najafpour MM, Pashaei B, Nayeri S. Calcium manganese(IV) oxides: Biomimetic and efficient catalysts for water oxidation. Dalton Trans. 2012;41:4799–4805. doi: 10.1039/c2dt12189a. [DOI] [PubMed] [Google Scholar]
- 18.Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–U65. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 19.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 20.Yamada Y, Yano K, Hong D, Fukuzumi S. LaCoO3 acting as an efficient and robust catalyst for photocatalytic water oxidation with persulfate. Phys Chem Chem Phys. 2012;14:5753–5760. doi: 10.1039/c2cp00022a. [DOI] [PubMed] [Google Scholar]
- 21.Kanady JS, Tsui EY, Day MW, Agapie T. A synthetic model of the Mn3Ca subsite of the oxygen-evolving complex in photosystem II. Science. 2011;333:733–736. doi: 10.1126/science.1206036. [DOI] [PubMed] [Google Scholar]
- 22.Tsui EY, Day MW, Agapie T. Trinucleating copper: Synthesis and magnetostructural characterization of complexes supported by a hexapyridyl 1,3,5-triarylbenzene ligand. Angew Chem Int Ed. 2011;50:1668–1672. doi: 10.1002/anie.201005232. [DOI] [PubMed] [Google Scholar]
- 23.Tsui EY, Kanady JS, Day MW, Agapie T. Trinuclear first row transition metal complexes of a hexapyridyl, trialkoxy 1,3,5-triarylbenzene ligand. Chem Commun. 2011 doi: 10.1039/c0cc05608a. [DOI] [PubMed] [Google Scholar]
- 24.Lacy DC, Park YJ, Ziller JW, Yano J, Borovik AS. Assembly and Properties of Heterobimetallic CoII/III/CaII Complexes with Aquo and Hydroxo Ligands. J Am Chem Soc. 2012 doi: 10.1021/ja304525n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukherjee S, et al. Synthetic model of the asymmetric [Mn3CaO4] cubane core of the oxygen-evolving complex of photosystem II. Proc Natl Acad Sci USA. 2012;109:2257–2262. doi: 10.1073/pnas.1115290109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishra A, et al. Heteronuclear Mn-Ca/Sr complexes, and Ca/Sr EXAFS spectral comparisons with the oxygen-evolving complex of photosystem II. Chem Commun. 2007:1538–1540. doi: 10.1039/b701355h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra A, Wernsdorfer W, Abboud KA, Christou G. The first high oxidation state manganese-calcium cluster: relevance to the water oxidizing complex of photosynthesis. Chem Commun. 2005:54–56. doi: 10.1039/b413680b. [DOI] [PubMed] [Google Scholar]
- 28.Kotzabasaki V, Siczek M, Lis T, Milios CJ. The first heterometallic Mn-Ca cluster containing exclusively Mn(III) centers. Inorg Chem Commun. 2011;14:213–216. [Google Scholar]
- 29.Hewitt IJ, et al. A series of new structural models for the OEC in photosystem II. Chem Commun. 2006:2650–2652. doi: 10.1039/b518026k. [DOI] [PubMed] [Google Scholar]
- 30.Nayak S, Nayek HP, Dehnen S, Powell AK, Reedijk J. Trigonal propeller-shaped [MnIII3MIINa] complexes (M = Mn, Ca): structural and functional models for the dioxygen evolving centre of PSII. Dalton Trans. 2011;40:2699–2702. doi: 10.1039/c0dt01463j. [DOI] [PubMed] [Google Scholar]
- 31.Visser H, et al. Mn K-Edge XANES and Kβ XES Studies of Two Mn-Oxo Binuclear Complexes: Investigation of Three Different Oxidation States Relevant to the Oxygen-Evolving Complex of Photosystem II. J Am Chem Soc. 2001;123:7031–7039. doi: 10.1021/ja004306h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leeladee P, et al. Valence Tautomerism in a High-Valent Manganese–Oxo Porphyrinoid Complex Induced by a Lewis Acid. J Am Chem Soc. 2012;134:10397–10400. doi: 10.1021/ja304609n. [DOI] [PubMed] [Google Scholar]
- 33.Horwitz CP, Ciringh Y. Synthesis and electrochemical properties of oxo-bridged manganese dimers incorporating alkali and alkaline earth cations. Inorg Chim Acta. 1994;225:191–200. [Google Scholar]
- 34.Horwitz CP, Ciringh Y, Weintraub ST. Formation pathway of a Mn(IV),Mn(IV) bis(μ-oxo) dimer that incorporates alkali and alkaline earth cations and electron transfer properties of the dimer. Inorg Chim Acta. 1999;294:133–139. [Google Scholar]
- 35.Fukuzumi S, Ohkubo K. Quantitative evaluation of Lewis acidity of metal ions derived from the g values of ESR spectra of superoxide: Metal ion complexes in relation to the promoting effects in electron transfer reactions. Chem Eur J. 2000;6:4532–4535. doi: 10.1002/1521-3765(20001215)6:24<4532::aid-chem4532>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 36.Perrin DD. Ionisation Constants of Inorganic Acids and Bases in Aqueous Solution. Pergamon Press; 1982. [Google Scholar]
- 37.Cox N, et al. Effect of Ca2+/Sr2+ substitution on the electronic structure of the oxygen-evolving complex of photosystem II: A combined multifrequency EPR, 55Mn-ENDOR, and DFT study of the S2 State. J Am Chem Soc. 2011;133:3635–3648. doi: 10.1021/ja110145v. [DOI] [PubMed] [Google Scholar]
- 38.Pecoraro VL, Baldwin MJ, Caudle MT, Hsieh WY, Law NA. A proposal for water oxidation in photosystem II. Pure Appl Chem. 1998;70:925–929. [Google Scholar]
- 39.Saltzman H, Sharefkin JG Iodosobenzene. Org Synth. 1973;43 [Google Scholar]
- 40.Sheldrick G. A short history of SHELX. Acta Crysallogr Sect A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 41.Yano J, et al. Where Water Is Oxidized to Dioxygen: Structure of the Photosynthetic Mn4Ca Cluster. Science. 2006;314:821–825. doi: 10.1126/science.1128186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peloquin JM, et al. 55Mn ENDOR of the S2-state multiline EPR signal of photosystem II: implications on the structure of the tetranuclear Mn cluster. J Am Chem Soc. 2000;122:10926–10942. [Google Scholar]
- 43.Ames W, et al. Theoretical Evaluation of Structural Models of the S2 State in the Oxygen Evolving Complex of Photosystem II: Protonation States and Magnetic Interactions. J Am Chem Soc. 2011;133:19743–19757. doi: 10.1021/ja2041805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.