

Abstract
The bacterial type IV secretion systems (T4SSs) deliver DNA and protein substrates to bacterial and eukaryotic target cells generally by a mechanism requiring direct contact between donor and target cells. Recent advances in defining the architectures of T4SSs have been made through isolation of machine sub-assemblies for further biochemical and ultrastructural analysis. Here, we describe a protocol for isolation and characterization of VirB protein complexes from the paradigmatic VirB/VirD4 T4SS of Agrobacterium tumefaciens. This protocol can be adapted for isolation of T4SS subassemblies from other gram-negative bacteria as well as gram-positive bacteria. The biological importance of isolated T4SS subcomplexes can be assessed by assaying for copurification of trapped or cross-linked substrates. This can be achieved with a modified form of the chromatin immunoprecipitation (ChIP) assay termed transfer DNA immunoprecipitation (TrIP). Here, a TrIP protocol is described for recovery of formaldehyde-cross-linked DNA substrate–channel subunit complexes from cells employing T4SSs for conjugative DNA transfer.
Keywords: Type IV secretion, Core complex, Affinity chromatography, DNA conjugation, Protein translocation, Transport, VirB/VirD4, TrIP, ChIP
1. Introduction
The Type IV Secretion Systems (T4SSs) are found in many species of gram-negative and -positive bacteria, as well as some species of Archaea (1). The largest and most widely distributed T4SS subfamily are the conjugation machines; these machines mediate the formation of direct cell-to-cell contact and then transfer of mobile DNA elements from bacterial donor to recipient cells. A second T4SS subfamily, termed the effector translocators, deliver protein effectors or other macromolecules from bacterial pathogens to eukaryotic cells during infection. A third, small subfamily of T4SSs promote transfer of DNA across the gram-negative cell envelope independently of any target cell contact; specifically, a Neisseria gonorrheae T4SS delivers fragments of chromosomal DNA to the extracellular milieu and a Helicobacter pylori T4SS coordinates with competence proteins to import DNA from the milieu.
Although the bacterial conjugation systems have been subjects of intensive study for many years, isolation of the translocation channels for biochemical and structural analysis has proven challenging. Recent advances in biochemical fractionation have enabled progress in development of structural models of T4SS channels along with correlative functional analyses of structural motifs. Biochemical fractionation and mutational studies have identified T4SS subunits or domains required for elaboration of stable machine subassemblies or extracellular pili (2–13). T4SS machine subunits or soluble domains also have been purified and crystallized. Using a nomenclature devised for the paradigmatic Agrobacterium tumefaciens VirB/VirD4 T4SS, X-ray structures have been solved for homologs of two hexameric ATPases termed VirD4 and VirB11 (14–16). Other solved structures exist for a pilus-associated subunit VirB5 (17), a complex of two outer membrane-associated subunits, lipoprotein VirB7 and VirB9 (18) and periplasmic domains of VirB8 and VirB10 (19, 20). Recently, a large subassembly termed a core complex consisting of homologs of VirB7, VirB9, and VirB10 subunits was isolated and structurally analyzed by cryoelectron microscopy and X-ray crystallography (21, 22).
In parallel with biochemical enrichment of T4SS machine subassemblies, an in vivo formaldehyde (FA) cross-linking procedure adapted from the chromatin immunoprecipitation (ChIP) assay was developed for identifying close interactions between translocating substrate DNA and subunits of the T4SS channel (23). Termed transfer DNA immunoprecipitation or TrIP, this assay supplied evidence that the DNA forms close interactions with the A. tumefaciens VirD4 and VirB11 ATPases located predominantly at the cytoplasmic entrance to the secretion channel, polytopic VirB6 and bitopic VirB8 at the inner membrane, and VirB2 pilin and VirB9 in the periplasm and outer membrane (23–25). Together with the recent structural data, results of the TrIP studies present a view of how DNA substrates pass through the cell-envelope-spanning T4SS channel (26).
Central to the developing understanding of T4SS architecture and function is the isolation of machine subassemblies without and with trapped substrate. The following protocols are described for (1) use of affinity tags for isolation and enrichment of VirD4/VirB T4SS subassemblies from A. tumefaciens, and (2) FA-cross-linking of DNA substrates for recovery of DNA–channel subunit complexes with the TrIP assay.
2. Materials
Prepare all solutions using nanopure water (which is done by purifying deionized water to attain a sensitivity of 18 MΩ cm at 25°C). Prepare and store all reagents at room temperature (unless otherwise indicated). Waste disposal regulations should be carefully followed when disposing of waste materials.
2.1. Induction of A. tumefaciens vir Genes
Incubator shaker.
Spectrophotometer.
MG/L agar: Agar (15 g) in 1 l of MG/L medium. Autoclave and store at room temperature.
MG/L medium: Dissolve 30 g Tryptone, 30 g NaCl, 15 g Yeast extract, 30 g Mannitol, 7 g Sodium glutamate, 1.5 g K2HPO4, 0.6 g MgSO4·7H2O, and 12 μg Biotin in 6 l of water. Adjust the pH to 7.2 with 1 M NaOH. Autoclave and store at room temperature.
Agrobacterium AB buffer: Dissolve 4 g MES (2-(N-morpholino) ethanesulfonic acid) and 10 g Glucose 10 g in 900 ml of water, adjust the pH to 5.5 with 12 N HCl, and bring to 1 l. Autoclave and store at room temperature.
Phosphate buffer: Dissolve 60 g K2HPO4 and 20 g Na2HPO4 in 900 ml of water. Adjust the pH to 7.0 and bring to 1 l. Autoclave and store at room temperature.
20× AB salts: Dissolve 20 g NH4Cl, 6 g MgSO4·7H2O, 3 g KCl, 0.2 g CaCl2, and 50 mg FeSO4·7H2O in 1 l of water. Autoclave and store at room temperature.
2% Yeast extract: Dissolve 2 g of yeast extract in 100 ml of water and autoclave.
100 mM Acetosyringone (4′-Hydroxy-3′,5′-dimethoxyacetophenone): Dissolve 0.196 g of acetosyringone in 10 ml of Dimethyl sulfoxide (DMSO). Store in the dark at −20°C.
ABIM (AB induction media): Add 5 ml 20× AB salts, 200 μl Phosphate buffer, 1 ml of 2% yeast extract, and 200 μl of 100 mM acetosyringone per 100 ml of Agrobacterium AB buffer.
Antibiotics for plasmid maintenance.
2.2. Cell Lysis and Membrane Solubilization
Centrifuge and associated rotor.
Ultracentrifuge and associated rotor.
Cell Lysis Buffer (500 ml): 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% Triton-X.
Lysate Buffer: 10 mM Tris–Cl (pH 8.0), 0.5 M NaCl, 5 mM EDTA, 0.5% LDAO.
Lysozyme solution: Add 300 mg of lysozyme to 10 ml of water and filter-sterilize.
DNase I solution: Add 100 mg DNase I to 10 ml of water and filter-sterilize.
5 M NaCl: make 500 ml. Autoclave.
1 M MgCl2: make 500 ml. Autoclave.
EDTA-free protease inhibitor tablet.
10× Cell Lytic B detergent mix.
10% DDM (n-Dodecyl-β-D-Maltopyranoside) in water.
10% LDAO (n-Dodecyl-N,N-Dimethylamine-N-Oxide) in water.
2.3. Isolation of Subcomplexes by Affinity Pull-Down
Tube rotator.
Eppendorf tubes.
Strep-Tactin Sepharose: immobilized streptavidin resin used for purification of Strep-tag II fusion proteins. Store at 4°C.
Anti-FLAG M2 Agarose beads: Anti-FLAG resin used for purification of FLAG tagged fusion proteins. Store at −20°C.
D-Desthiobiotin: 50 mM stock solution in NH4OH. Store at 4°C. This is used for elution of Strep-tag II fusion proteins.
Lysate buffer: see item 4 in Subheading 2.2.
Elution buffer 1: 2.5 mM desthiobiotin in lysate buffer.
2× SDS Sample loading buffer (Laemmli’s buffer): dissolve 20 mg bromophenol blue in 32.5 ml of water, 20 ml of glycerol, 5 ml of β-mercaptoethanol, 30 ml of 10% SDS, and 12.5 ml of 4× Resolving Gel buffer, and bring the volume to 100 ml with water. The sample loading buffer is aliquoted in eppendorf tubes and stored at −20°C.
Tris buffered saline (TBS; 10×): 1.5 M NaCl, 0.1 M Tris–HCl, pH 7.4.
3× FLAG peptide: 5 μg/μl in TBS. This is used for elution of FLAG tagged fusion proteins.
Elution buffer 2: dilute threefold the 3× FLAG Peptide into lysate buffer.
Microcentrifuge.
Syringe and needle.
2.4. Further Enrichment by CsCl Density Gradient Centrifugation
1 CsCl: 76% (w/v) dissolved in water.
Lysate buffer: see item 4 in Subheading 2.2.
Beckman ultracentrifuge.
Beckman SW55 Ti. Swinging bucket rotor.
2.5. SDS-PAGE and Staining
2.5.1. SDS-PAGE
Acrylamide/bisacrylamide (30%/0.8%): 30% (w/v) acrylamide, 0.8% (w/v) bis(N,N′-methylene) acrylamide. Store at 4°C (see Note 1).
SDS: 10% SDS (w/v).
Resolving Gel Buffer (4×): 1.5 M Tris–HCl, pH 8.8, filter and store at room temperature.
Stacking Gel Buffer (4×): 0.5 M Tris–HCl, pH 6.8, filter and store at room temperature.
Ammonium persulfate (AP): Prepare a 10% (w/v) solution in water before use.
N,N,N′,N′-tetramethyl-ethylenediamine (TEMED). Store at 4°C.
10× Tris–Glycine: 30 g Tris base, 144 g Glycine in 1 l of water.
SDS-PAGE running buffer: 200 ml of 10× Tris–glycine, 20 ml of 10% SDS, 1.78 l water. Store at room temperature.
Separating gel: prepare 40 ml (for four gels) of 12.5% poly-acrylamide separating gel by mixing 12.7 ml of water, 10.0 ml of 4× resolving gel buffer, 16.7 ml of acrylamide/bisacrylamide (30%/0.8%), 0.6 ml of 10% AP, and 20 μl of TEMED.
Stacking gel: prepare 12.75 ml (for four gels) of stacking gel by mixing 2.5 ml of water, 6.35 ml of water, 2.50 ml of stacking gel buffer, 1.25 ml of acrylamide/bisacrylamide (30/0.8%), 150 μl of 10% AP, and 10 μl of TEMED.
Prestained molecular weight marker.
SDS-PAGE apparatus.
2.5.2. Coomassie Staining
Coomassie stain solution: dissolve 100 mg of Coomassie Brilliant Blue R250 in 50 ml methanol, 10 ml glacial acetic acid, and water to a final volume of 100 ml.
Destaining solution: 45 ml ethanol, 10 ml glacial acetic acid, 45 ml of water.
Platform shaker.
2.5.3. Western Blotting and Immunostaining
Nitrocellulose membranes.
Western blot transfer buffer: 100 ml of 10× Tris–Glycine (see item 7 Subheading 2.5.1), 200 ml of methanol, 700 ml of water.
Methanol.
Aqueous transfer apparatus.
Tris buffered saline (TBS; 10×): 1.5 M NaCl, 0.1 M Tris–HCl, pH 7.4.
TTBS: 0.05% Tween-20 in 1× TBS.
Blocking solution: 5% Nonfat milk in 1× TBS. Store at 4°C.
Diluent solution: 5% Nonfat milk in TBST. Store at 4°C.
Whatman blotting paper.
Carbonate buffer: 8.40 g NaHCO3 and 0.203 g MgCl2 dissolved in 900 ml of water. Adjust the pH to 9.81 with 1 M NaOH and bring the volume to 1 l with water.
NBT solution: Dissolve 30 mg of Nitroblue tetrazolium (NBT) in 1 ml of 70% dimethylformamide (DMF).
BCIP solution: Dissolve 15 mg of 5-bromo-4-chloro-3-indolyl phosphate (BCIP) in 1 ml of 100% DMF.
Alkaline phosphatase staining solution: Add 100 μl of NBT solution and 100 μl of BCIP solution to 10 ml of carbonate buffer, just prior to adding to membrane.
Platform shaker.
Primary antibody.
Secondary antibody: conjugated to alkaline phosphatase.
2.6. Transfer Immunoprecipitation (TrIP) Assay
2.6.1. In Vivo Formaldehyde Cross-linking
MG/L medium: see item 4 in Subheading 2.1.
ABIM: see item 10 in Subheading 2.1.
Centrifuge and associated rotor.
Spectrophotometer.
20 mM sodium phosphate buffer, pH 6.8.
Cross-linking buffer: 0.1% (v/v) formaldehyde (FA) in 20 mM sodium phosphate buffer (pH 6.8). Prepare fresh before each use.
Formaldehyde: 37% w/v (see Note 2).
Buffer A: 50 mM Tris–HCl (pH 6.8), 2 mM EDTA, 1% β-mercaptoethanol, 1% SDS.
Buffer B: 150 mM Tris–HCl (pH 8.0), 0.5 M sucrose, 10 mM EDTA.
Lysozyme solution: 1 mg/ml of lysozyme in Buffer B. Prepare fresh before each use.
Triton X-100.
1 M Glycine: Dissolve 18.8 g glycine in water (may require gentle heating) and bring up to 250 ml with water.
Protease inhibitors cocktail: 5× solution, EDTA-free, in 25 mM MgCl2.
Rotating wheel.
2.6.2. Immunoprecipitation
Protein A-Sepharose CL4B.
Anti-VirB polyclonal antibodies.
Centrifuge and associated rotor.
Buffer B: see item 9 in Subheading 2.6.1.
Buffer B supplemented with 1% Triton X-100.
Buffer B supplemented with 0.1% Triton X-100.
10 mM Tris–Cl, pH 6.8.
30% (w/v) Trichloroacetic acid (TCA): store at 4°C.
Acetone: Store at −20°C.
Speed Vac.
4× Resolving Gel buffer: see item 3 Subheading 2.5.1.
2× SDS Sample loading buffer: see item 8 Subheading 2.3.
2.6.3. PCR Amplification
Primers for PCR amplification of the desired transferred DNA (T-DNA) or chromosomal DNA fragments.
TAQ Polymerase.
dNTPs: 2.5 mM stock.
Thermocycler.
Agarose.
Ethidium bromide.
Agarose gel electrophoresis apparatus.
3. Methods
The following protocols have been developed for (1) isolation of subassemblies of the A. tumefaciens VirB/VirD4 T4SS and (2) recovery of formaldehyde-cross-linked complexes of translocating T-DNA substrate and channel subunits. The fractionation procedure is adapted from protocols described for isolation of type III secretion needle complexes from Salmonella typhimurium by the Galan laboratory (27, 28) and the T4SS “core” complex encoded by the E. coli pKM101 conjugation system by the Waksman laboratory (21, 22). By use of this fractionation procedure, we enrich VirB subcomplexes composed minimally of the VirB4, VirB7, VirB9, and VirB10 subunits that assemble as ring-shaped structures when analyzed by negative-stain electron microscopy. These structures closely resemble ring-shaped “core” complexes of the E. coli pKM101 conjugation machine; these are composed of TraN, TraO, and TraF, which are homologs of the A. tumefaciens VirB7, VirB9, and VirB10 subunits, respectively (21, 22). By use of the FA-cross-linking protocol termed TrIP (Transfer DNA ImmunoPrecipitation) (23), we identify close contacts between the DNA substrate and six of the A. tumefaciens T4SS subunits - VirD4, VirB11, VirB6, VirB8, VirB2, and VirB9. Our current model is that these subunits assemble as the translocation channel within the ring-shaped VirB7/VirB9/VirB10 “core” complex (26).
3.1. Induction of A. tumefaciens vir Genes
Streak A. tumefaciens strains from −80°C stocks onto MG/L agar containing appropriate antibiotics for plasmid maintenance. Incubate for 1–2 days at 18°C. Inoculate A. tumefaciens strains producing Strept-tag II- or FLAG-tagged VirB proteins of interest from MG/L agar plates into 5 ml of MG/L medium containing appropriate antibiotics.
Grow culture overnight with shaking at 28°C, then inoculate culture into 400 ml of MG/L medium and continue incubating with shaking to an OD600 of 0.5–0.8.
Harvest cells by centrifugation and resuspend in 2 l ABIM to an OD600 of 0.1–0.2.
Incubate with shaking for 12–14 h at 20°C (see Notes 3 and 4).
3.2. Cell Lysis and Membrane Solubilization
Spin cells down and resuspend in 12.5 ml Cell Lysis buffer (see Note 5).
Stir slowly using magnetic stirrer at room temperature, and add one complete EDTA-free protease inhibitor tablet, drop by drop of 250 μl of lysozyme solution, and drop by drop of 250 μl of DNAse I solution in that order.
Place in an ice water bath and incubate slowly with stirring for 1 h.
Add 650 μl of 10× Cell Lytic B detergent mix, 500 μl of 10% LDAO, and 500 μl of 10% DDM and incubate at 37°C for 30 min.
Add 150 μl of 1 M MgCl2 slowly with stirring, incubate with stirring for 15 min.
Add 1.6 ml of 5 M NaCl slowly under stirring, incubate with stirring for 1 h at 37°C (see Note 6).
Pellet debris by centrifugation at 12,000 × g at 4°C for 30 min. Repeat if lysate appears cloudy (see Note 7).
Recover high molecular weight complexes from clarified lysates by ultracentrifugation at 181,000 × g for 2 h at 4°C. (see Note 8).
Resuspend pelleted material in 1 ml of Lysate Buffer. Suspension of all material can take several hours. Pool all suspended material (see Note 9).
3.3. Isolation of Subcomplexes by Affinity Pull-Down
We use the FLAG tag (29) or Strep-tag II (engineered from a short sequence of eight amino acids that bind to the biotin binding pocket of Streptavidin) (30) for isolation of VirB subassemblies by affinity chromatography. The tagged proteins are initially assayed for effects on T4SS machine function, as monitored by virulence and pilus production. We use only functional tagged proteins for enrichments of the VirB subassemblies.
-
Perform an affinity pulldown of the tagged subunits/complexes in the following way.
For affinity pulldown of Strep-tag II-tagged subunits/complexes: Mix 100 μl of Strep-tactin sepharose (50% suspension) with 1 ml of suspended material from step 9 Subheading 3.2 in an eppendorf tube. Incubate overnight at 4°C with gentle tube inversion/mixing. Elute Strep-tag II tagged subunit/complexes from resin by adding 0.8 ml of Elution Buffer 1. Mix by inversion at room temperature for 1 h.
For affinity pulldown of FLAG-tagged subunits/complexes: Mix 100 μl anti-FLAG M2 agarose beads with 1 ml of the sample from step 9 Subheading 3.2 in an eppendorf tube. Incubate overnight at 4°C with gentle tube inversion/mixing. Elute the FLAG-tagged fusion proteins from anti-FLAG M2 agarose beads by adding 500 μl of elution buffer 2. Mix by inversion at room temperature for 1 h.
Remove Strep-tag II- and FLAG-beads by centrifugation at 16,000 × g for 1 min in a microcentrifuge to pellet resin. Collect resin-free supernatant using a syringe needle.
At this point, eluted proteins should be analyzed by SDS-PAGE and Coomassie staining, and development of western blots with anti-VirB antibodies (see Subheading 3.5).
3.4. Further Enrichment by CsCl Density Gradient Centrifugation
Complexes recovered by affinity chromatography are next subjected to further enrichment by one or two rounds of CsCl density centrifugation. Additional enrichment steps can include centrifugation through sucrose density gradients or gel filtration chromatography; development of further purification steps is empirical and not described in detail here (see Note 10).
Material recovered in the eluates from the FLAG or Streptactin resins is added to ultracentrifuge tubes containing 1.63 ml of 76% CsCl. Make sample up to 2.85 ml with Lysate Buffer if there is a shortage of sample volume. Mix by inversion and then centrifuge in a Beckman SW55 Ti swing-bucket rotor at 340,000 × g for 12 h at 4°C.
Fractionate tubes into ~0.25 ml fractions and analyze by SDS-PAGE and immunostaining (see Subheading 3.5 below).
To remove the CsCl and concentrate the sample, pool the desired fractions, dilute eight to tenfold with Lysate Buffer, ultracentrifuge at 181,000 × g and resuspend in ~200 μl of Lysate buffer.
3.5. SDS-PAGE and Staining
For 12.5% SDS-PAGE gel electrophoresis, carry out the following procedures at room temperature, unless otherwise specified. Five SDS-polyacrylamide gels are prepared by this method. The gels can be stored for several days wrapped in cellophane at 4°C.
3.5.1. SDS-PAGE
Cast a separating acrylamide gel within an 8 cm × 10 cm × 1.5 mm gel cassette as specified in item 9 of Subheading 2.5.1. Allow space for the stacking gel and overlay with ethanol or water (see Note 11).
Remove the ethanol or water used for overlaying the resolving gel, wick dry with a piece of Whatman paper, being careful not to touch the gel, and then add the stacking gel. Insert a 10-well gel comb immediately without introducing air bubbles (see Note 12).
Place the gel cassette in a gel tank and fill it with SDS-PAGE running buffer. Heat the protein samples for 5 min in boiling water. Load about 20 μl of protein sample onto each well. Also load a protein standard in one of the wells (10 μl/well; 2 μg/marker/lane). Electrophorese at 15 mA when the sample is entering the stacking gel, and then continue at 25 mA when the sample enters the resolving gel and then continue at that current until the dye front has reached the bottom of the gel.
After electrophoresis, pry the gel plates open with the use of a blunt razor or spatula. The gel remains on one of the glass plates. Then proceed to Subheading 3.5.2 for Coomassie staining or Subheading 3.5.3 for Western blotting and immunostaining.
3.5.2. Coomassie Staining
Transfer the gel carefully to a container containing Coomassie stain and place it on a shaker at RT for 1 h.
Carefully discard the Coomassie stain and wash the gel once with water. Add destaining solution to the container, so that the gel is submerged and incubate for 15 min. Repeat this step three times.
You should be able to see distinct protein bands of your samples.
3.5.3. Western Blotting and Immunostaining
Transfer the gel to a container with Western blot transfer buffer.
Cut a nitrocellulose membrane to the size of the gel and immerse in methanol. Rinse once in distilled water and once in Western blot transfer buffer. Hydrate two Whatman blotting papers cut to the size of the gel in the transfer buffer.
To perform the protein transfer with an aqueous transfer apparatus, lay the nitrocellulose membrane over one Whatman paper. Place the gel on top of the membrane, and lay another Whatman paper on top of the gel. Roll over the “sandwiched” gel-nitrocellulose membrane using a pipette to make sure that there are no air bubbles trapped.
Place the sandwiched package within the transfer cassette and put it vertically in a holder within the transfer tank. The nitro-cellulose should face the positive (+ve) side in the transfer apparatus.
Perform the transfer for 2 h at 200 mA or overnight at 100–120 mA. Turn on the stir bar and water with a hose linked from the apparatus into the sink.
After transfer, take apart the apparatus. Discard the gel and the Whatman papers.
Place the nitrocellulose membrane in blocking solution. Incubate at room temperature with gentle shaking for 45 min.
Wash the membrane 3× with TTBS for 5 min each.
Add primary antibody at 1:1,000-fold dilution in diluent solution and incubate 2 h to overnight at room temperature with gentle shaking.
Wash the membrane as in step 8.
Add secondary antibody at 1:20,000- to 1:500,000- fold dilution in diluent solution and incubate for 1–2 h at room temperature with gentle shaking.
Wash the membrane as in step 8.
Wash the membrane 1× with TBS for 5 min.
Prepare the alkaline phosphatase staining solution as described in item 13 in Subheading 2.5.3, mix properly and then pour over the nitrocellulose membrane.
-
When the protein bands develop, discard the developing solution and wash the blot in water. Dry on paper towel.
Using the above fractionation protocol, we detect appreciable amounts of the VirB4, VirB7, VirB9, and VirB10 subunits in immunoblots developed with antisera raised against each of these subunits (see Fig. 1). Surprisingly, we do not detect other VirB subunits even though the A. tumefaciens strains subjected to analysis produce all of the T4SS subunits and are phenotypically wild type with respect to virulence, e.g., T-DNA transfer, and pilus biogenesis. When the isolated material is subjected to 1% uranyl acetate negative staining on a carbon-coated grid followed by transmission electron microscopy using a JEOL 1400 microscope, we detect ring-shaped complexes of a uniform size (see Fig. 2). These complexes resemble those of the TraN/O/F “core” complex purified from E. coli cells carrying pKM101 (21, 22).
Fig. 1.
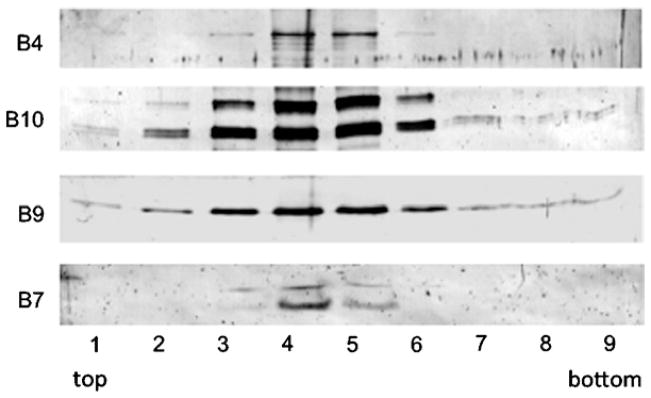
Detection of enriched VirB complexes. Western blot of VirB/VirD4 T4SS machine subassemblies from vir-induced A. tumefaciens cells enriched by successive Strep-tag II affinity pull-down and CsCl density gradient centrifugation. Complexes were isolated from A. tumefaciens strain PC1010, a nonpolar virB10 deletion mutant (31), engineered to produce N-terminally Strep-tagged VirB10. The western blot was developed successively with antisera specific for the VirB4, VirB7, VirB9, and VirB10 subunits. Lanes correspond to fractions recovered from the CsCl gradient. VirB subunits detected in the gradient fractions are listed at the left.
Fig. 2.
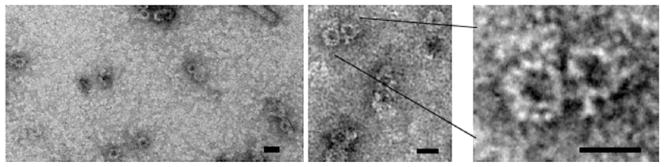
Visualization of ring-shaped VirB complexes. Complexes in peak CsCl fractions 4 and 5 from Fig. 1 were pelleted by ultracentrifugation and resuspended in Lysate buffer (see step 3 in Subheading 3.4). These samples were applied to a carbon-coated grid, stained with 1% uranyl acetate, and analyzed by transmission electron microscopy using a JEOL 1400 microscope. The ring-shaped complexes are similar in size and general architecture to the “core” complexes isolated from the E. coli pKM101 conjugation system by the Waksman laboratory (21, 22). Scale bar: 20 nm.
3.6. Transfer Immunoprecipitation (TrIP) Assay
This TrIP protocol results in recovery of DNA substrate—channel complexes by immunoprecipitation, as determined by PCR amplification of the immunoprecipitated material (see Fig. 3). In to our studies of the A. tumefaciens VirB/VirD4 T4SS, we have applied this protocol to characterize DNA substrate interactions with components of the Enterococcus faecalis pCF10 transfer system (32), and others have detected DNA substrate contacts with components of a conjugation machine in Bacteroides fragilis (33).
Fig. 3.
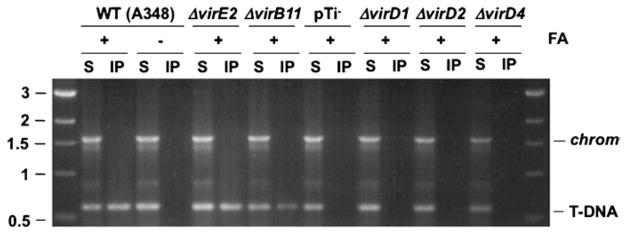
Isolation of substrate DNA–channel subunit complexes with the TrIP assay. The translocating T-DNA substrate is formaldehyde cross-linked to channel subunits as described in Subheading 3.6.1. The resulting complexes are recovered by immunoprecipitation with antisera against the channel subunits as described in Subheading 3.6.2. The precipitated DNA substrate is detected by PCR amplification as described in Subheading 3.6.3. Here, anti-VirD4 antisera was used to precipitate the VirD4 substrate receptor following FA-cross-linking and disruption of the strains shown. Total soluble (S) and immunoprecipitated (IP) material were assayed for the presence of substrate T-DNA (T-DNA) or the chvE gene (chrom) carried on the chromosome. Strains: WT strain A348; pTi-, A348 lacking the pTi plasmid; nonpolar deletion mutants - ΔvirE2 (virE2 codes for the exported effector VirE2; ΔvirD1 and ΔvirD2 (virD1 and virD2 code for relaxosomal subunits which are required for processing of the T-DNA substrate); ΔvirD4 (virD4 codes for the substrate receptor). A precipitable VirD4-T-DNA complex is recovered from FA-cross-linked WT cells and mutants lacking the VirE2 effector and VirB11 ATPase, but not from WT cells without prior FA cross-linking or mutants lacking the T-DNA processing factors VirD1 and VirD2 or the substrate receptor VirD4 (23).
The Transfer ImmunoPrecipitation (TrIP) assay was adapted from the Chromatin ImmunoPrecipitation (ChIP) assay. For more information, on ChIP, see refs. 34–36.
3.6.1. In Vivo Formaldehyde Cross-linking
Grow 1.5 ml of A. tumefaciens cells in MG/L medium to an OD600 = 0.5, pellet the cells by centrifugation, and resuspend in 6 ml of ABIM. Incubate for 14–16 h at 20°C with shaking.
Harvest the cells, wash with 6 ml of 20 mM sodium phosphate buffer pH 6.8, and suspend in 1 ml of cross-linking buffer.
Incubate the cells for 20 min at 18°C with shaking.
Add formaldehyde in 0.2% increments to reach a final concentration of 1% over a 15 min period, and then continue incubation for 40 min at room temperature without shaking.
Quench the formaldehyde by adding 141 μl of 1 M glycine per 1 ml of reaction mix (to reach a final concentration of 125 mM glycine) for 5–10 min at room temperature.
Pellet the cells and resuspend in 200 μl of Buffer A.
Incubate for 30 min at 37°C with shaking.
Add 900 μl of lysozyme solution, and continue incubating the mixture for 1–2 h on ice and then 30 min at 37°C with shaking.
Add Triton X-100 to a 4% final concentration and incubate the mixture for 15 min at room temperature rotating on a wheel.
Add the protease inhibitors cocktail to achieve a 5× dilution, and incubate the mixture with rocking for 15 min at 37°C and then for 2–3 h at 4°C on a wheel.
Add 3.2 ml of Buffer B and remove insoluble material by centrifugation for 15 min at 14,000 × g. Collect the supernatant.
For recovery of the VirD2-T-strand complexes with anti-VirD2 antibodies, treat the whole cells as described above in the absence of in vivo formaldehyde cross-linking.
3.6.2. Immunoprecipitation
For immunoprecipitation, first pre-incubate protein A-Sepharose CL4B (30 μl bed volume) with 1.1 ml of the detergent-solubilized, FA-cross-linked material from step 11 Subheading 3.6.1 for 60 min at RT (see Note 13).
Centrifuge at 5,000 × g and withdraw supernatant to another eppendorf tube. Discard pelleted Protein A-Sepharose and nonspecifically bound proteins.
Incubate the supernatant overnight at 4°C with 5 μl of antibody against a protein of interest and 30 μl of Protein A-Sepharose CL4B. Pellet the beads by centrifugation at 5,000 × g and carefully remove the supernatant (“S” fraction) to another eppendorf tube (see Note 14).
Wash the beads twice with buffer B supplemented with 1% Triton X-100 and once with buffer B supplemented with 0.1% Triton X-100.
Elute the material from the Sepharose beads by incubation for 20 min at 96°C in 20 μl of 10 mM Tris–Cl pH 6.8. This material is the “IP” fraction.
To resolve the proteins by SDS-PAGE, precipitate the material in the supernatant (S) fraction with 15% trichloroacetic acid. This is done by adding an equal volume of 30% TCA solution to the superanatant. Keep the sample on ice for at least 1 h. Then, centrifuge the samples for 15 min at 15,000 × g and 4°C. Remove the supernatant and wash the pellet twice with acetone. After each washing step, centrifuge the samples for 15 min at 15,000 × g and 4°C and remove the supernatant. Finally, dry the samples in the Speed Vac for 5 min, and then resuspend in sample loading buffer.
The immunoprecipitated (IP) material is resuspended directly in sample loading buffer.
Protein samples are boiled for 5 min prior to loading the gel.
3.6.3. PCR Amplification
Perform PCR in a 25 μl reaction volume containing 1/20 volume of the immunoprecipitates (IP fraction) or 1/100,000 vol. of the soluble (S) fraction.
Select the primers to amplify a ~300–500 bp fragment of the transferred T-DNA.
As a control, carry out PCR with a second set of primers designed to amplify a region of the chromosome.
-
Perform PCR amplification. An example of a typical reaction is as follows:
Immunoprecipitate (“IP” fraction) 1.25 μl Taq PCR buffer 5 μl dNTP’s (2.5 mM stock) 1.6 μl Forward primer (10 mM) 1 μl Reverse primer (10 mM) 1 μl Taq polymerase 0.2 μl Water 15 μl Reaction cycling conditions:
98°C for 45 s (Initial denaturation)
30 cycles of the following:
98°C for 10 s (denaturation)
45°C for 10 s (annealing)
72°C for 30 s (extension)
72°C for 10 min (final extension)
4°C or ice until the sample is visualized (see next step).
Separate the PCR products by electrophoresis through a 1.2% agarose gel and visualize by ethidium bromide staining (see Note 15).
Acknowledgments
We thank members of the Waksman, Galan, and Baron laboratories for sharing ideas regarding biochemical enrichments of T4SS sub-assemblies. We thank Drs. Sayyed Shah and Eric Cascales for initiating studies in this laboratory aimed at isolation of T4SS subassemblies and cross-linked DNA substrate–channel subunit complexes. We thank other members of the Christie laboratory for valuable discussions and technical expertise. This work was supported by NIH grant GM48746 to PJC.
Footnotes
Caution: Acrylamide is a neurotoxin and must be handled with gloves. Do not dispose of liquid acrylamide down the sink.
Caution: Formaldehyde is very toxic if inhaled, ingested, or absorbed through the skin.
Induction at 20°C is not required; however, efficient induction of the vir genes is best achieved at temperatures lower than ~23–25°C (37).
Induction of the vir genes in ABIM requires a minimum of 10 h incubation with vigorous aeration. Large scale-up of the cell culture without appropriate aeration, e.g., 1 l culture in a 2 l flask, is not advised as this will limit cell growth and diminish the yield of isolated machine subassemblies.
Cell pellets can be stored at −80°C for a few days prior to fractionation.
Cell lysis should be complete at this stage, and the cell culture should have a translucent appearance.
After addition of NaCl, the viscosity of the solution should considerably decrease.
We use a Beckman TLA100.3 centrifuge, with each centrifuge tube filled with ~3.5 ml of the cell lysate.
The suspension will become cloudy, but all flakes will not resuspend.
The use of sucrose gradients can be problematic for subsequent analysis by electron microscopy due to the presence of sucrose in the sample. The use of gel filtration chromatography offers promise for further purification, but in our experience requires scale-up of the starting culture to >10 l of induced cells for recovery of detectable levels of VirB subcomplexes upon fractionation.
The overlay prevents contact with atmospheric oxygen which inhibits acrylamide polymerization and also helps to level the resolving gel solution. The gel should polymerize within 15–30 min. If it has not polymerized within 1 h, prepare fresh AP solution and repeat the gel casting steps.
You need to be careful and efficient in quickly adding the stacking gel into the gel cassette. The gel combs need to be inserted immediately, as polymerization almost starts within a few minutes. Polymerization of stacking gel is complete within 15 min. After that, the combs can be taken out gently, and the wells washed gently and thoroughly for removing in air bubbles.
This step removes proteins that bind nonspecifically to the protein A-Sepharose beads.
The amount of antibody needed for immunoprecipitation of the T4SS subunits should be adjusted for each different batch of antibodies to achieve the best results. Too much antibody can cause background signal to occur.
This TrIP assay can generate either (1) no specific signal or (2) a high background level of nonspecific DNA, e.g., chromosomal DNA. When encountering either situation, we first verify that the antibodies successfully precipitate the target protein of interest. Although we typically employ polyclonal antibodies raised against the T4SS subunits, low signal or high background could be circumvented by use of monoclonal antibodies specific for precipitating epitope-tagged proteins of interest. If we encounter problems with the TrIP protocol, we first prepare or purchase new stocks of lysis buffer, formaldehyde, and reagents for PCR amplification. Further trouble shooting relies on strategies developed for enhancing specificity of DNA–protein cross-links recovered with the ChIP assay. These steps, described in detail elsewhere (see refs. 34–36), include (1) adjustment of formaldehyde concentration and cross-linking time, (2) more extensive washing of the immunoprecipitates to remove nonspecifically bound DNA, (3) fragmentation of DNA by sonication prior to immunoprecipitation, (4) removal of antibodies and DNA-associated proteins by proteinase K digestion, phenol–chloroform extraction, and ethanol precipitation prior to PCR amplification, and (5) altering PCR amplification parameters.
References
- 1.Alvarez-Martinez CE, Christie PJ. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev. 2009;73:775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandez D, Spudich GM, Zhou XR, Christie PJ. The Agrobacterium tumefaciens VirB7 lipoprotein is required for stabilization of VirB proteins during assembly of the T-complex transport apparatus. J Bacteriol. 1996;178:3168–3176. doi: 10.1128/jb.178.11.3168-3176.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hapfelmeier S, Domke N, Zambryski PC, Baron C. VirB6 is required for stabilization of VirB5 and VirB3 and formation of VirB7 homodimers in Agrobacterium tumefaciens. J Bacteriol. 2000;182:4505–4511. doi: 10.1128/jb.182.16.4505-4511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krall L, et al. Detergent extraction identifies different VirB protein subassemblies of the type IV secretion machinery in the membranes of Agrobacterium tumefaciens. Proc Natl Acad Sci U S A. 2002;99:11405–11410. doi: 10.1073/pnas.172390699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jakubowski SJ, Krishnamoorthy V, Christie PJ. Agrobacterium tumefaciens VirB6 protein participates in formation of VirB7 and VirB9 complexes required for type IV secretion. J Bacteriol. 2003;185:2867–2878. doi: 10.1128/JB.185.9.2867-2878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cascales E, Christie PJ. Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proc Natl Acad Sci U S A. 2004;101:17228–17233. doi: 10.1073/pnas.0405843101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan Q, et al. Identification of the VirB4-VirB8-VirB5-VirB2 pilus assembly sequence of type IV secretion systems. J Biol Chem. 2005;280:26349–26359. doi: 10.1074/jbc.M502347200. [DOI] [PubMed] [Google Scholar]
- 8.Paschos A, et al. Dimerization and interactions of Brucella suis VirB8 with VirB4 and VirB10 are required for its biological activity. Proc Natl Acad Sci U S A. 2006;103:7252–7257. doi: 10.1073/pnas.0600862103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakubowski SJ, et al. Agrobacterium VirB10 domain requirements for type IV secretion and T pilus biogenesis. Mol Microbiol. 2009;71:779–794. doi: 10.1111/j.1365-2958.2008.06565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerr JE, Christie PJ. Evidence for VirB4-mediated dislocation of membrane-integrated VirB2 pilin during biogenesis of the Agrobacterium VirB/VirD4 type IV secretion system. J Bacteriol. 2010;192:4923–4934. doi: 10.1128/JB.00557-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mossey P, Hudacek A, Das A. Agrobacterium tumefaciens type IV secretion protein VirB3 is an inner membrane protein and requires VirB4, VirB7, and VirB8 for stabilization. J Bacteriol. 2010;192:2830–2838. doi: 10.1128/JB.01331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banta LM, et al. An Agrobacterium VirB10 mutation conferring a type IV secretion system gating defect. J Bacteriol. 2011;193:2566–2574. doi: 10.1128/JB.00038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sivanesan D, Baron C. The dimer interface of Agrobacterium tumefaciens VirB8 is important for type IV secretion system function, stability, and association of VirB2 with the core complex. J Bacteriol. 2011;193:2097–2106. doi: 10.1128/JB.00907-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeo HJ, Savvides SN, Herr AB, Lanka E, Waksman G. Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV secretion system. Mol Cell. 2000;6:1461–1472. doi: 10.1016/s1097-2765(00)00142-8. [DOI] [PubMed] [Google Scholar]
- 15.Gomis-Ruth FX, et al. The bacterial conjugation protein TrwB resembles ring helicases and F1- ATPase. Nature. 2001;409:637–641. doi: 10.1038/35054586. [DOI] [PubMed] [Google Scholar]
- 16.Hare S, Bayliss R, Baron C, Waksman G. A large domain swap in the VirB11 ATPase of Brucella suis leaves the hexameric assembly intact. J Mol Biol. 2006;360:56–66. doi: 10.1016/j.jmb.2006.04.060. [DOI] [PubMed] [Google Scholar]
- 17.Yeo H-J, Yuan Q, Beck MR, Baron C, Waksman G. Structural and functional characterization of the VirB5 protein from the type IV secretion system encoded by the conjugative plasmid pKM101. Proc Natl Acad Sci U S A. 2003;100:15947–15952. doi: 10.1073/pnas.2535211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayliss R, et al. NMR structure of a complex between the VirB9/VirB7 interaction domains of the pKM101 type IV secretion system. Proc Natl Acad Sci U S A. 2007;104:1673–1678. doi: 10.1073/pnas.0609535104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terradot L, et al. Structures of two core subunits of the bacterial type IV secretion system, VirB8 from Brucella suis and ComB10 from Helicobacter pylori. Proc Natl Acad Sci U S A. 2005;102:4956–4961. doi: 10.1073/pnas.0408927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailey S, Ward D, Middleton R, Grossmann JG, Zambryski PC. Agrobacterium tumefaciens VirB8 structure reveals potential protein-protein interaction sites. Proc Natl Acad Sci U S A. 2006;103:2582–2587. doi: 10.1073/pnas.0511216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fronzes R, et al. Structure of a type IV secretion system core complex. Science. 2009;323:266–268. doi: 10.1126/science.1166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandran V, et al. Structure of the outer membrane complex of a type IV secretion system. Nature. 2009;462:1011–1015. doi: 10.1038/nature08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cascales E, Christie PJ. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science. 2004;304:1170–1173. doi: 10.1126/science.1095211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jakubowski SJ, Krishnamoorthy V, Cascales E, Christie PJ. Agrobacterium tumefaciens VirB6 domains direct the ordered export of a DNA substrate through a type IV secretion system. J Mol Biol. 2004;341:961–977. doi: 10.1016/j.jmb.2004.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakubowski SJ, Cascales E, Krishnamoorthy V, Christie PJ. Agrobacterium tumefaciens VirB9, an outer-membrane-associated component of a type IV secretion system, regulates substrate selection and T-pilus biogenesis. J Bacteriol. 2005;187:3486–3495. doi: 10.1128/JB.187.10.3486-3495.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christie PJ. Structural biology: translocation chamber’s secrets. Nature. 2009;462:992–994. doi: 10.1038/462992b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kubori T, et al. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science. 1998;280:602–605. doi: 10.1126/science.280.5363.602. [DOI] [PubMed] [Google Scholar]
- 28.Sukhan A, Kubori T, Wilson J, Galan JE. Genetic analysis of assembly of the Salmonella enterica serovar Typhimurium type III secretion-associated needle complex. J Bacteriol. 2001;183:1159–1167. doi: 10.1128/JB.183.4.1159-1167.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Einhauer A, Jungbauer A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. J Biochem Biophys Methods. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt TG, Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat Protoc. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 31.Berger BR, Christie PJ. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J Bacteriol. 1994;176:3646–3660. doi: 10.1128/jb.176.12.3646-3660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, et al. Enterococcus faecalis PcfC, a spatially localized substrate receptor for type IV secretion of the pCF10 transfer intermediate. J Bacteriol. 2008;190:3632–3645. doi: 10.1128/JB.01999-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas J, Hecht DW. Interaction of Bacteroides fragilis pLV22a relaxase and transfer DNA with Escherichia coli RP4-TraG coupling protein. Mol Microbiol. 2007;66:948–960. doi: 10.1111/j.1365-2958.2007.05967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- 35.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 36.Grably M, Engelberg D. A detailed protocol for chromatin immunoprecipitation in the yeast Saccharomyces cerevisiae. Methods Mol Biol. 2010;638:211–224. doi: 10.1007/978-1-60761-611-5_16. [DOI] [PubMed] [Google Scholar]
- 37.Baron C, Domke N, Beinhofer M, Hapfelmeier S. Elevated temperature differentially affects virulence, VirB protein accumulation, and T-Pilus formation in different Agrobacterium tumefaciens and Agrobacterium vitis strains. J Bacteriol. 2001;183:6852–6861. doi: 10.1128/JB.183.23.6852-6861.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]