

SUMMARY
STING is an essential signaling molecule for DNA and cyclic di-GMP (c-di-GMP)-mediated type I interferon (IFN) production via TANK-binding kinase 1 (TBK1) and Interferon regulatory factor 3 (IRF3) pathway. It contains an N-terminal transmembrane region and a cytosolic C-terminal domain (CTD). Here, we describe crystal structures of STING CTD alone and complexed with c-di-GMP in a unique binding mode. The strictly conserved AA153-173 region was shown to be cytosolic and participated in dimerization via hydrophobic interactions. The STING CTD functions as a dimer and the dimerization was independent of post-translational modifications. Binding of c-di-GMP enhanced interaction of a shorter construct of STING CTD (residues 139-344) with TBK1. This suggests an extra TBK1 binding site, other than Ser358. This study provides a glimpse into the unique architecture of STING and sheds new light on the mechanism of c-di-GMP-mediated TBK1 signaling.
INTRODUCTION
A stimulator of interferon genes (STING) (Ishikawa and Barber, 2008; Ishikawa et al., 2009), also known as MITA (Zhong et al., 2008; Zhong et al., 2009), ERIS (Chen et al., 2011; Sun et al., 2009), MPYS (Jin et al., 2011a; Jin et al., 2010; Jin et al., 2008; Jin et al., 2011b) and TMEM173, is known to play a vital role in the production of type I IFNs (Barber, 2011; Bowzard et al., 2009; Ishikawa and Barber, 2011; Nakhaei et al., 2010; Saitoh et al., 2010). The membrane protein STING was initially characterized as a plasma membrane tetraspanner associated with type II major histocompatibility complex (MHC-II) with a function to transduce apoptotic signals during antigen presentation (Jin et al., 2008). Subsequently, STING is shown to reside predominantly in the endoplasmic reticulum (ER) membrane where it plays a role in relaying the intracellular DNA-mediated innate signals to type I IFN (IFN-I) production (Ishikawa and Barber, 2008; Ishikawa et al., 2009). STING-deficient (Tmem173-/-) cells are defective in IFN-I induction triggered by viral, bacterial or synthetic DNA and STING-deficient mice are more sensitive than wild type (WT) controls when infected with DNA viruses such as HSV-1 (Ishikawa et al., 2009).
Despite the essential role of STING in DNA-mediated IFN-I induction, the mechanisms of its action are less clear and controversial in some cases. First, STING is believed to function as an adaptor molecule activated by cytoplasmic receptors after sensing DNA. One potential cytoplasmic DNA receptor, DDX41, has been shown to form a complex with STING and trigger STING-dependent IFN-I induction (Zhang et al., 2011). However, the nature of the interaction of STING with DDX41 is not known. Another recent report indicates that STING can directly interact with c-di-GMP (Burdette et al., 2011), a product released by bacteria such as Listeria monocytogene. In addition to ER localization, some reports indicate that STING is also located at the outer membrane of mitochondria while other studies show that STING is translocated from ER to mitochondria during viral infections (Zhong et al., 2008). Further, activated STING is believed to mediate IFN-I induction through recruiting cytosolic TANK-binding kinase 1 (TBK1), which phosphorylates and activates Interferon regulatory factor 3 (IRF3) (Sun et al., 2009). However, it is still not clear how STING is activated and how the activated STING triggers the recruitment and activation of TBK1. Interestingly, STING is also subject to a broad range of post-translational modifications like phosphorylation and ubiquitination by TBK1 and RNF5, respectively (Zhong et al., 2008). Phosphorylation of STING at Ser358 by TBK1 is increased after viral infection. This phosphorylation is not only critical for the interaction of STING with TBK1 but also helps STING evoke the immune response (Zhong et al., 2008). In addition, IFN-inducible TRIM56 interacts with and facilitates lysine 63-linked polyubiquitination at Lys150 of STING, which is proposed as a prerequisite for the recruitment and activation of TBK1 to STING and IFN-I induction following detection of a pathogen’s double-strand DNA (dsDNA) (Tsuchida et al., 2010). This finding raises an interesting question - how does TRIM56 gain access to STING Lys150? TRIM56 is found in the cytoplasm and is known to interact with the C-terminal domain (CTD) of STING spanning AA174-379 (Tsuchida et al., 2010). The AA153-173 region of STING has been predicted to be a transmembrane region; suggesting that Lys150 and the CTD of STING reside on opposite sides of the membrane. If this is the case, then how does TRIM56 associate with STING across the membrane and carry out ubiquitination? Another contradictory fact is that a K150R STING mutant is shown to be incapable of associating with TBK1 and unable to activate the production of IFN-I (Tsuchida et al., 2010). However, in a previous study the same STING K150R mutant is proposed to activate IFN-stimulation responsive element (ISRE) resulting in production of IFN-I (Zhong et al., 2009).
Thus, mounting evidence reveals a central role for STING in innate immune responses; prompting us to study its structure and function. Here, we describe a crystal structure of the STING CTD alone and in a complex with c-di-GMP refined to 2.45 Å and 2.15 Å resolution, respectively. Our structural, functional and mutagenesis studies defined the dimer interface of STING and characterized the structure of c-di-GMP bound to the STING CTD. New insights into the understanding of c-di-GMP-mediated TBK1 signaling are provided.
RESULTS
Crystallization and Structure Determination of STING CTD
Full length STING (AA1-379) could not be expressed as a soluble protein in E. coli. Our sequence analysis results showed that the AA153-173 region, reported as the last transmembrane domain (Table S1), is the most conserved region in all species examined (Figure 1A, Figure S1A and S1B), suggesting an important role for this region in the function of STING. After expression screening of several N-terminal deletion truncations (Figure 1B) in E. coli, truncations STINGΔ1-148 (AA149-379) and STINGΔ1-138 (AA139-379) could be expressed as highly soluble proteins and purified to homogeneity with excellent solution properties as stable homodimers (Figure 1C and S1C).
Figure 1. Sequence Conservation of STING CTD, STING Domain Organization and STING CTD Protein Characterization.
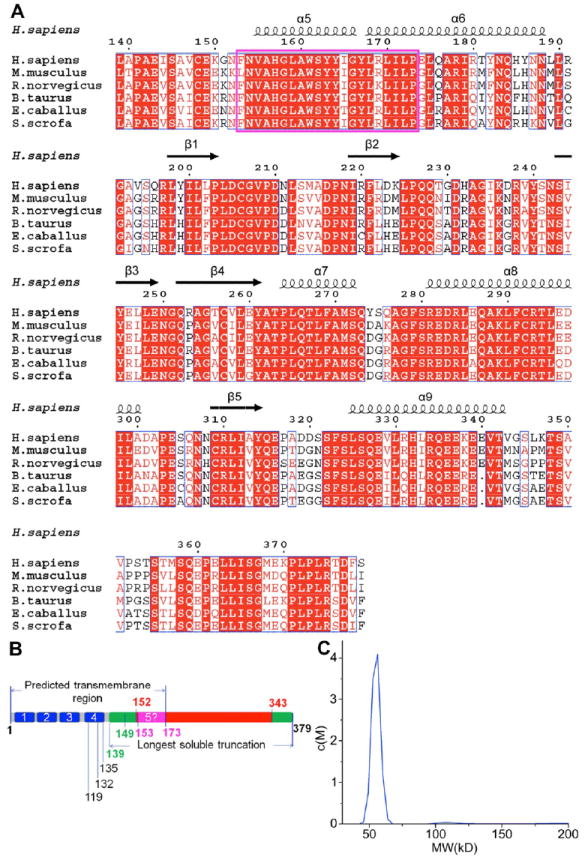
(A) Alignment of the STING CTD domain sequences of representative STINGs from human (NP_938023), mouse (NP_082537), rat (NP_001102592), cattle (NP_001039822), horse (XP_001504275) and pig (NP_001136310). Strictly conserved residues are boxed in white on a red background and highly conserved residues are boxed in red on a white background. The predicted last transmembrane domain is boxed in magenta. At the top of the sequences, a schematic representation of the secondary structure elements of unliganded STING CTD are shown, and every 10 residues are indicated with a dot (.). Alignment was generated by ClustalW (Thompson et al., 1994). The α-helix is depicted by a coil, and β-strand by an arrow. The figure was generated by ESPript (Gouet et al., 1999).
(B) The schematic of human STING domain organization. The numbers indicate residues at the domain boundaries. AA1-173 contains the predicted 5 transmembrane domains of STING. The crystals were generated from AA139-379 (green) and AA152-343 (red) was observed in the final structure.
(C) His-tag removed STING CTD analysed by analytical ultracentrifugation shows dimeric STING CTD (55 kD) in solution.
STINGΔ1-138 (AA139-379) (referred to as STING CTD hereafter) crystallized in space group C2221. Se-Met derivative of the proteins were used to determine the phases by Se-SAD method using PHENIX (Adams et al., 2010) (Figure S2). The phases were transferred to native data set (program CAD) and followed by density modification (program DM) and the model was automatically traced by program Warp (Winn et al., 2011). The best model derived from the native data was refined to 2.45 Å resolution with PHENIX and Refmac (Adams et al., 2010; Winn et al., 2011). All additional crystallographic statistics were summarized in Table 1. Except for residues 139-151, 227-239, 319-320 and 344-370, the electron density was clear and permitted unambiguous placement of residues. These missing residues were located in the loop regions, suggesting they are mobile in the constructs used for this study. To locate the missing residues, B-factor sharpening method was attempted (DeLaBarre and Brunger, 2003; Su et al., 2010). However, the weak electron densities could not be improved any further.
Table 1.
Data Collection and Refinement Statistics
| Crystal | STING CTD Se-Met | STING CTD Native | STING CTD:c-di-GMP complex |
|---|---|---|---|
| Data collection | |||
| X-ray source | BL17U1, SSRF | BL17U1, SSRF | BL5.0.1, ALS |
| Detector | ADSC 315 | ADSC 315 | ADSC 315 |
| Crystal to detector distance (mm) | 350.0 | 300.0 | 290.0 |
| Number of images | 260 | 612 | 180 |
| Oscillation width (°) | 1.0 | 0.5 | 1.0 |
| Wavelength (Å) | 0.9793 | 0.9793 | 0.9794 |
| Space group | C2221 | C2221 | P21 |
| Unit cell dimensions | |||
| a, b, c (Å) | 78.84, 88.53,74.43 | 81.04, 90.36, 73.32 | 61.38, 72.84, 62.44 |
| α, β, γ(°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 97.03, 90.00 |
| Resolution range(Å) | 50.00-3.10 (3.21-3.10) | 50.00-2.45 (2.54-2.45) | 50.00-2.15 (2.23-2.15) |
| Rsym (%) | 10.8 (52.4) | 7.1 (49.6) | 5.5 (39.5) |
| Mean I/σI (I) | 39.2 (9.5) | 56.9 (5.0) | 23.5 (2.3) |
| Completeness (%) | 99.9 (100.0) | 99.9 (99.2) | 96.9 (80.7) |
| Redundancy | 9.8 (10.2) | 11.3 (9.7) | 3.6 (3.2) |
| Phasing | |||
| Phasing resolution (Å) | 50.00 – 3.10 | ||
| Rano/Rp.i.m *(50.00 – 3.10 Å) | 1.16 | ||
| Number of Se located (Occupancy) | 1 (2.33) | ||
| Mean FOM | 0.38 | ||
| Refinement | |||
|
| |||
| Resolution (Å) | 45.18-2.45 | 41.01-2.15 | |
| No. reflections | 10182 | 29026 | |
| Rwork/ Rfree (%) | 20.43/ 27.59 | 20.92/25.44 | |
| No. atoms | |||
| Protein | 1455 | 2937 | |
| Water | 48 | 106 | |
| Ligand | c-di-GMP | ||
| Mean B value | 68.59 | 56.69 | |
| Mean B value of protein (Å2) | 68.57 | 57.22 | |
| Mean B value of c-di-GMP (Å2) | 47.36 | ||
| R.m.s deviations | |||
| Bond lengths (Å) | 0.007 | 0.008 | |
| Bond angles (°) | 1.081 | 1.157 | |
| Ramachandran analysis | |||
| Favored region (%) | 155 (92.26) | 316 (93.77) | |
| Allowed region (%) | 10 (5.95) | 12 (3.56) | |
| Outliers (%) | 3 (1.79) | 9 (2.67) | |
Thus, construct boundaries based on the conserved regions suggested by sequence alignment, rather than those based on previously predicted transmembrane regions, resulted in protein suitable for structure determination.
Overall Structure of STING CTD
Amino acids 152-343 folded into a single domain containing 5 helices and 5 strands (Figure 2A). Since the N-terminal is predicted to fold into 4 transmembrane helices, the numbering of helices in the STING AA152-343 structure starts with helix α5 (Figure 2A). Helices α5 and α6 (Val155-Asn188) form a long extended helix with only a tyrosine (Thr167) separating them. Helix α6 is long and bends at Pro173 along the helical axis resulting in a change in the direction of the helix (Figure 2A). Amino acids from helices α5 and α7 are involved in intermolecular hydrophobic interactions. STING CTD contains a single curved sheet made up of 5 strands (Figure 2A). Helices α7 and α8 are stacked against the sheet and are inserted between helix α5 and the sheet. Interestingly, part of helix α9 is buried in the concave cavity formed by the sheet (Figure 2A). A number of salt bridges formed between 3 glutamic acid and 2 arginine residues hold the helix α9 firmly inside the cavity. Electron density for the C-terminal end of the protein, AA344-379, positioned at the end of helix α9 was missing, suggesting that this part of the protein is flexible and probably swings around the tip of helix α9 protruding out of the concave cavity of the sheet.
Figure 2. Crystal Structure of STING CTD Dimer.
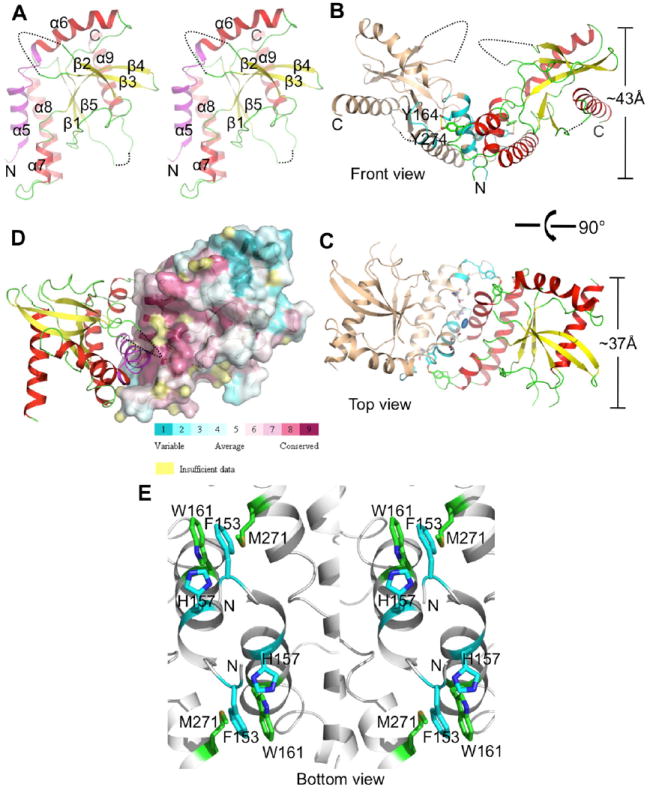
(A) Stereo view of STING CTD. Side view (parallel to the membrane) of cartoon representation of the overall structure of STING CTD monomer. β-strands and α-helices are numbered. The conserved hydrophobic dimeric interface (AA153-173), predicted as last transmembrane helix, is shown in magenta. The N- and C-termini of each chain are labeled with letters. The missing residues are shown in dashed lines.
(B) Side view of the STING dimer. The yellow dashed line shows the hydroxyl group of Tyr274 interacting with the hydroxyl oxygen of Tyr164. Tyr164 and Tyr274 from two monomers are shown as sticks. Interfacing residues identified by PISA (Krissinel and Henrick, 2007) are shown in cyan.
(C) Top view (perpendicular to membrane) of the STING dimer. Among the interfacing residues, highly conserved residues are shown as gray sticks. The 2-fold symmetric axis, located in the middle of STING CTD dimer, is represented by oval dot.
(D) Residues (amaranth) of the dimer interface are highly conserved. Amino acid conservation of STING CTD using 17 homologs in Figure S1 displayed on a 3D structure using ConSurf (Ashkenazy et al., 2010).
(E) A stereo view of the dimer interface from bottom view showing symmetry related π-π stacking between Phe153 and His157 (cyan color) and the aryl-sulfur interaction between Trp161 and Met271 (green color). N-termini is marked N.
A Dali (Holm and Rosenstrom, 2010) analysis retrieved a very low structural match with leucine-rich repeat kinase 2 (PDB code 2ZEJ, Z score – 4.4, r.m.s. deviation of 4.0 Å for 162 matching residues with 9% sequence identity). Similarly, a ProFunc (Krissinel and Henrick, 2004) analysis retrieved matches for a small portion of the protein with a number of GTP and DNA binding proteins. However, the overall structure of STING CTD is unique and does not resemble any structures deposited in PDB.
Interestingly, previous studies propose that the STING AA153-173 hydrophobic region constitutes the last transmembrane domain (summarized in Table S1). From the structure of STING CTD (AA152-343), this region (AA153-173) of STING is actually not a transmembrane helix (Figure 2A). Instead, our studies suggest that the STING AA1-138 region is transmembrane with the secondary structural elements threading the membrane 4 times (Figure S1A).
STING CTD Forms a Unique Dimeric Structure
Although the asymmetric unit contained one molecule of STING CTD, analysis of the symmetry mates revealed that unliganded STING CTD had crystallized as a dimer, measuring ~43 Å in height and ~37 Å in diameter (Figure 2B, 2C). Dimerization occurs via an extensive hydrophobic interface and the major contact area spans 916.2 Å2 (9.2%) of surface area per monomer with calculated solvation free energy gain upon formation of the interface (ΔiG) of 13.4 kcal/mol as determined by PISA (Krissinel and Henrick, 2007). To rule out the possibility that dimerization might have occurred as an artifact of crystallization, gel filtration and analytical ultracentrifugation analysis were performed to determine the oligomerization state of the protein. Results of both the analyses showed that unliganded STING CTD exists as a dimer in solution, which was consistent with the crystal packing analysis (Figure 1C, 2B and 2C). Thus, we conclude that STING CTD exists as a homodimer in solution.
Within the dimer, STING CTD monomers were related by a crystallographic 2-fold symmetry axis (Figure 2C). The 2-fold axis was located near the junction of the two N-terminal α5 helices. STING CTD dimerizes via helix-helix interactions, with helices α5 and α7 predominantly involved in the intermolecular interactions. Interestingly, residues from the helix α5 are the most conserved (Figure 2D), suggesting that the mode of dimerization might be very similar across species. Hydrophobic amino acids constituted about 65.5% of the total residues involved in dimerization (Table S2). A symmetry related stacking interaction between Phe153 of one chain with the His157 from the other chain, seems to stabilize the dimer (Figure 2E). In addition, a symmetry related aryl-sulfur interaction between the π-electron cloud of the indole ring of Trp161 from helix α5 and the side chain of Met271 from helix α6 further contributed to the stabilization of the dimer (Figure 2E). Interestingly, these amino acids are absolutely conserved in STING from different organisms (Figure 1A and Figure S1A). A number of weak interactions were also observed between helices α5 and α7. The dimer interface of STING CTD was dominated by hydrophobic interactions with no salt bridges participating in the dimerization. Thus, based on several lines of evidence, we propose that STING CTD exists as a V-shaped dimer even in the absence of a ligand.
C-di-GMP Binds into the Trough at the Dimer Interface
The structure of the binary complex of STING CTD with c-di-GMP was determined in P21 space group by co-crystallizing STING CTD with the nucleotide analog (Figure 3A). Clear electron density for the c-di-GMP permitted unambiguous modeling of the nucleotide into the structure of STING CTD (Figure 3B). Similar to the structure of unliganded STING CTD, several residues at the N-terminus, the entire C-terminal region from AA344-379, the loops connecting strand β2 with β3 and β5 with helix α9 were disordered (Figure 3A). One molecule of c-di-GMP bound a dimer of STING CTD. Thus, a dimer seems to be the minimal functional unit of STING as shown in Figure S3. Overall, the structure of the c-di-GMP bound STING CTD closely mirrored the unliganded structure (with RMSD 0.64 Å over main chain atoms between one monomer) with an almost identical dimer interface (Figure S4A). One molecule of c-di-GMP bound into the trough formed at the junction of dimerization where its innate 2-fold symmetry axis was coaxial with the non-crystallographic 2-fold symmetry axis of the STING CTD dimer (Figure 3A). This resulted in nearly symmetrical interactions for the ribose-phosphate ring with the STING CTD dimer (Figure 3B). Interestingly, the loop connecting strand β2 with β3 remained disordered after binding of c-di-GMP (Figure 3A). Visual inspection of the superimposition of the unliganded and c-di-GMP complex structures revealed that except for some minor changes, there were no obvious structural re-arrangements induced as a result of binding of c-di-GMP. Positions of the loop connecting helix α6 with strand β1 (residues Ala302 - Asn307) and the region connecting helix α8 with strand β5 (residues Asn183 - Ala192) seemed to have deviated slightly in the binary complex. Further, strands β2 and β3 (residues Phe221 - Gln227 and Ser241 - Tyr245) together with the disordered loop connecting them had moved towards c-di-GMP upon its binding to STING CTD. Intriguingly, the movements of those two β strands were asymmetrical between monomer A and monomer B. The two β strands in monomer A were closer to the guanine moiety than that seen in monomer B when monomer B was superimposed onto monomer A as shown in Figure S4B. We still do not know why and how c-di-GMP induces such asymmetry, albeit minor, between the two monomers and what is the physiological significance of such a deviation observed in the structure. Interestingly, such asymmetry has been reported previously for a c-di-GMP riboswitch (Smith et al., 2009).
Figure 3. C-di-GMP Binds into the Trough at the Dimer Interface.
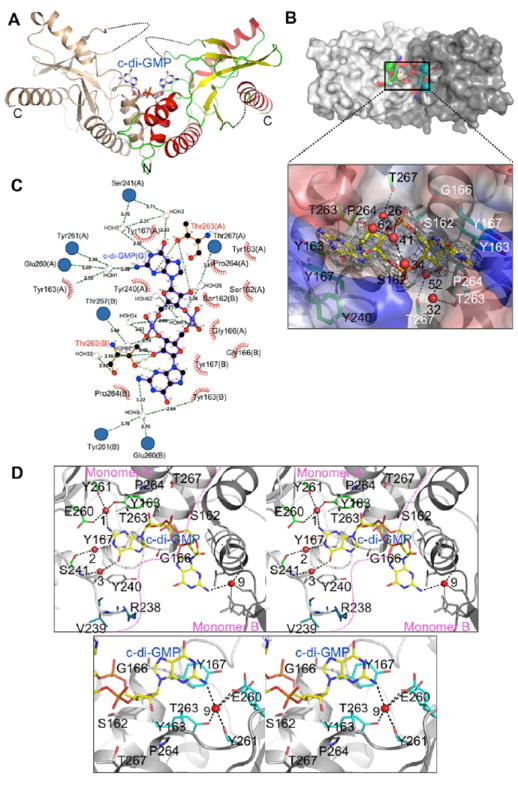
(A) The binary complex structure of STING CTD with c-di-GMP. C-di-GMP is shown as gray sticks. The STING dimer is shown in similar orientation as in Figure 2B.
(B) Close-up view of specific recognition of c-di-GMP by STING and details of hydrogen bonds ≤ 3.50 Å. Interactions between ribose-phosphate of GMP and waters, as well as c-di-GMP and Thr263 from STING monomer A and B, respectively. Hydrogen bonding atoms between c-di-GMP and residues of STING are connected by red dashed lines. Hydrogen bonds between c-di-GMP and waters are shown as black dashed lines. Residues from STING monomer A and monomer B that interact with c-di-GMP are shown as green (with black labels) and cyan (with white labels) sticks, respectively. C-di-GMP is shown as yellow sticks. Electron density of a SA (simulated annealing) Fo-Fc omit map for c-di-GMP contoured at 3.0 σ. Water molecules are shown as red spheres with numbers labeled.
(C) LIGPLOT representation of the STING CTD:c-di-GMP complex revealing bonding atoms and bonding lengths (Wallace et al., 1995). Interacting atoms are connected by green dashed lines with bonding lengths indicated (in Å). Non-ligand residues involved in direct hydrophobic contacts with c-di-GMP are shown as red semicircles with radiating spokes, while those indirectly involved are shown as blue circles. Water atoms are shown in gray spheres with numbers labeled.
(D) Stereographic close-up view of the c-di-GMP binding site and details of hydrogen bonds between c-di-GMP and waters, as well as c-di-GMP and residues from STING monomer A (top panel) and monomer B (bottom panel), respectively.
The c-di-GMP was seen anchored to the protein mainly by several hydrogen bonding and hydrophobic interactions (Figure 3C, Table S3). The interaction between ribose-phosphate of GMP with STING CTD was more symmetrical than that of the guanine with STING CTD (compare Figure 3B and Figure 3D). There were 4 hydrogen bonds (≤ 3.5 Å) between the ribose-phosphate ring and STING CTD: two pairs formed between O2 of ribose and OG1 of Thr263, two pairs formed between O3 of ribose and OG1 of Thr267 via a water molecule (water 26 and 52, respectively) (Figure 3B). Interestingly, the phosphates were neither involved in hydrogen bonding nor were anchored by any basic side chains (Figure 3B). The interactions of the two guanines with STING CTD monomers were more deviated from the 2-fold symmetry axis. Both purine rings of c-di-GMP were stacked against the aromatic ring of Tyr167 (Figure 3B and 3D). OG1 atoms of Thr263 from two monomers were interacting with N3 and N31 atoms of c-di-GMP via hydrogen bonds, 3.02 and 2.96 Å, respectively (Figure 3B). Both amine nitrogen atoms (N21 and N2) of GMP were anchored by hydrogen bonds with two water molecules (water 1 and 9) which formed hydrogen bonds with residues Tyr163, Tyr261 and Glu260 on the other side (Figure 3C and 3D). These two water molecules were observed in unliganded STING CTD crystal structure as well. These water-mediated hydrogen bonds probably make STING prefer c-di-GMP over c-di-AMP because c-di-AMP does not have the amine group of GMP (Figure S4C). Intriguingly, two extra hydrogen bonds between guanine and two water molecules (water 2 and 3) which were mediated through hydrogen bonds by main chain nitrogen and oxygen atoms of Ser241 of monomer A were observed (Figure 3D top panel). Such extra hydrogen bonds were not observed in monomer B (Figure 3C, 3D bottom panel and Table S3). Further, 2 additional residues (residues: Arg238 and Val239) from monomer A became ordered due to the c-di-GMP binding (Figure 3D top panel). In addition, Tyr240’s side chain swung towards c-di-GMP as shown in Figure 3D top panel. These changes upon binding of c-di-GMP were not observed in monomer B. Thus, binding of c-di-GMP does not induce large conformational changes in STING CTD. Upon close inspection of the c-di-GMP bound STING, some asymmetrical interactions of STING with the purine rings of c-di-GMP were observed. C-di-GMP was anchored on STING using a combination of hydrophilic, hydrophobic and water mediated contacts.
Requirement of STING Self-association for IFN-β Induction
It has been reported that STING dimerization is responsible for self-activation and subsequent downstream signaling (Sun et al., 2009). However, the nature of the intermolecular interactions and whether STING dimerization is required for IFN-I induction remains unknown. To address this issue, we used our crystal structure to first identify the key residues involved in dimerization and mutated them to disrupt the dimerization of STING. The following mutations were selected - V155R, G158L, W161A, Y164A, and I165R. All the mutants were expressed in E. coli with an N-terminal 6His tag. Mutants V155R, W161A and Y164A were insoluble even when salvaged with an N-terminal GST tag (Figure S5A) suggesting that these mutations probably disrupt the dimerization interface and expose the hydrophobic patches resulting in precipitation of the proteins. The soluble G158L and I165R mutants were further purified by Ni-affinity followed by gel filtration chromatography. The size exclusion chromatography (SEC) profile of the mutants indicated a highly aggregated protein when compared to the STING CTD WT. Interestingly, part of the G158L mutant eluted similar to the STING CTD WT suggesting that a small portion of G158L mutant remained dimeric (Figure 4A).
Figure 4. The Screening of Dimerization Deficient Mutants.
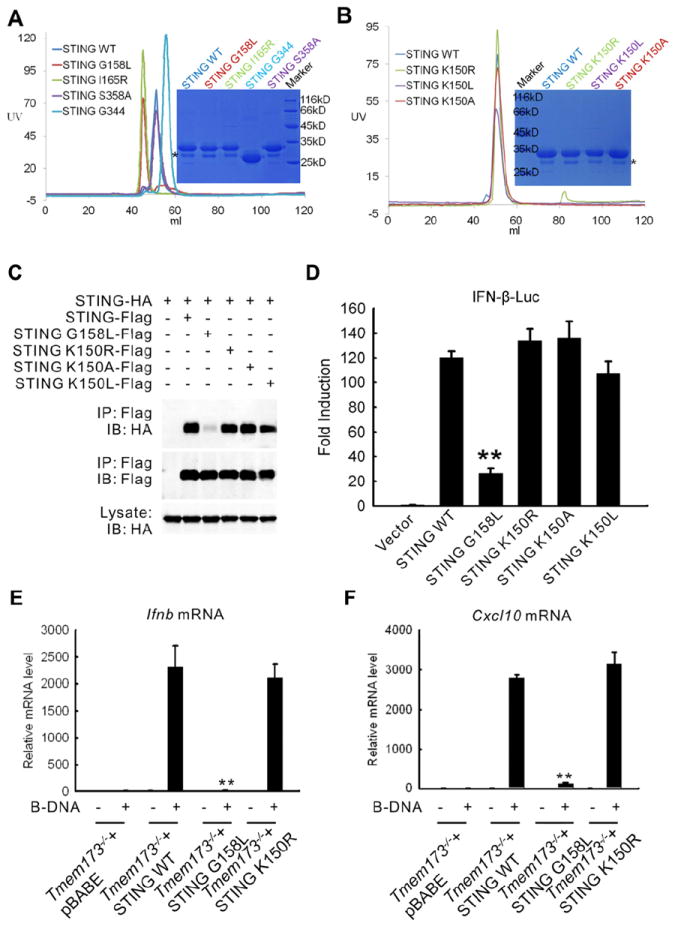
(A) Gel-filtration chromatography of STING CTD WT, G158L and I165R mutants. STING CTD mutants S358A and G344 (AA139-344) were used as control. The profile of STING CTD WT is used as the marker. The proteins were analyzed by Superdex G75 (120 ml) gel-filtration chromatography. The calculated molecular masses of the monomeric human STING WT and G344 mutant with His-tag are 30 kD and 26 kD, respectively.
(B) Gel-filtration chromatography of STING WT and K150R, K150L, K150A. The proteins were analyzed by Superdex G75 (120 ml) gel-filtration chromatography. Asterisk: degraded STING bands (A, B).
(C) Co-immunoprecipitation of cell lysates from HEK 293T cells expressing STING-Flag and STING-HA or STING G158L-HA. Lysates were incubated with anti-Flag M2 beads and STING self-association was detected by immunoblot analysis against HA.
(D) IFN-β reporter activity of 293T cells expressing STING WT or STING G158L.
(E) Quantitative real-time PCR analysis of relative Ifnb mRNA after transfection of B-DNA in WT MEFs (WT), Tmem173-/- MEFs, Tmem173-/- MEFs reconstituted with STING (Tmem173-/- + STING WT), or Tmem173-/- MEFs reconstituted with STING G158L (Tmem173-/- + STING G158L).
(F) Quantitative real-time PCR analysis of relative Cxcl10 mRNA after transfection of B-DNA in WT, Tmem173-/-, Tmem173-/- + STING WT, or Tmem173-/- + STING G158L MEFs. Double asterisks (**) indicate significant difference (P < 0.01) as determined by Student’s t-test. Error bars indicate SEM.
To test whether the integrity of the dimer of STING was essential for its function, we performed luciferase reporter assay on the full length mutants and the STING WT in 293T cells. The V155R, W161A and Y164A mutations of the full length STING showed no activity as expected. Further, the I165R mutant that could be expressed as soluble protein in E. coli was inactive. Interestingly, the G158L mutation showed partial activity when compared to the STING WT (Figure S5B). To further investigate, we performed co-immunoprecipitation assay in 293T cells co-expressing Flag-tagged STING and HA-tagged STING or HA-tagged STING G158L. While STING WT interacted strongly with each other, the STING G158L mutant dramatically lost its ability to interact with WT STING and with G158L mutant itself (Figure S5C), corroborating the results of the gel filtration chromatography and the luciferase reporter assay experiments.
A recent study shows that an IFN-inducible tripartite-motif protein - E3 ligase TRIM56 - carries out ubiquitination of STING when stimulated by poly (dA:dT). Ubiquitination of STING at Lys150 results in the dimerization of STING, which is a prerequisite for the recruitment and activation of TBK1 leading to the induction of IFN-I (Tsuchida et al., 2010). However, based on our structure of the STING dimer, Lys150 may not play a major role in the dimerization of STING. To test this, we expressed and purified the STING CTD K150A, K150L and K150R mutants and carried out SEC to determine the oligomerization state of the protein. The results revealed that similar to the STING CTD WT, the mutants existed as dimers in solution (Figure 4B). Next, to characterize the importance of Lys150 for STING dimerization in vivo, we generated K150A, K150L and K150R mutants of full-length STING in 293T cells. As shown in Figure 4C, all the three mutant Lys150 proteins could bind STING-HA as strongly as STING-Flag. These results demonstrate that Lys150 may not be essential for dimerization of STING.
To determine whether the dimerization-deficient mutant had a defect in triggering downstream signaling, we performed luciferase reporter assay in 293T cells expressing STING WT, STING G158L and K150A, K150L, K150R mutants. While STING WT and STING K150A, K150L, K150R mutants induced strong activation of the IFN promoter (IFN-β-Luc), the activation of IFN-β-Luc by STING G158L was significantly reduced (Figure 4D, P<0.01). To confirm STING dimerization was required for induction of IFN-I, we generated STING-deficient (Tmem173−/−) murine embryonic fibroblasts (MEFs) reconstituted with vector, STING WT, STING G158L and K150R mutants. After transfection of B-DNA, IFN-β production in MEFs was examined by quantitative real-time PCR (Q-PCR). STING deficiency caused a dramatic loss of IFN-β production compared to WT MEFs (Figure 4E, P<0.01). Reconstitution of STING-deficient MEFs with STING WT and K150R rescued B-DNA-induced IFN-β production, while reconstitution with STING G158L mutant failed to do so (Figure 4E, P<0.01). Similar results were obtained for Cxcl10 (Figure 4F, P<0.01).
Next, we carried out GST pull-down assays to test the association of STING CTD K150R, K150L and K150A, with TBK1. In addition, we also tested whether G158L, S358A, and a truncated STING containing 139-344 amino acids could associate with TBK1. Both STING CTD WT and the three STING CTD Lys150 mutants could interact directly with TBK1 (Figure 5A). To confirm the interaction in cells, we performed co-immunoprecipitation assay in 293T cells expressing HA-tagged STING and Flag-tagged TBK1 and showed that TBK1 immunoprecipitated together with STING (Figure 5B). The other two kinases involved in DNA virus-induced IFN-β production, IKKα and NIK did not interact with STING (Figure 5B). Surprisingly, the 3 mutants K150A, K150L, and K150R and the dimerization-deficient G158L, interacted with TBK1 in a manner similar to the interaction of STING WT with TBK1 (Figure 5C), suggesting that dimerization of STING may not be absolutely required for interaction with TBK1. Further, the truncated STING (AA139-344) and the S358A mutant showed comparatively weaker interaction with TBK1 (Figure 5A). More importantly, the inability of dimerization-deficient STING G158L to induce IFN production (Figure 4E) in spite of retaining its TBK1 binding ability (Figure 5C) suggests that only the dimerized STING-TBK1 interaction can induce IFN-β production.
Figure 5. Requirement of STING Self-association for IFN-β Induction.
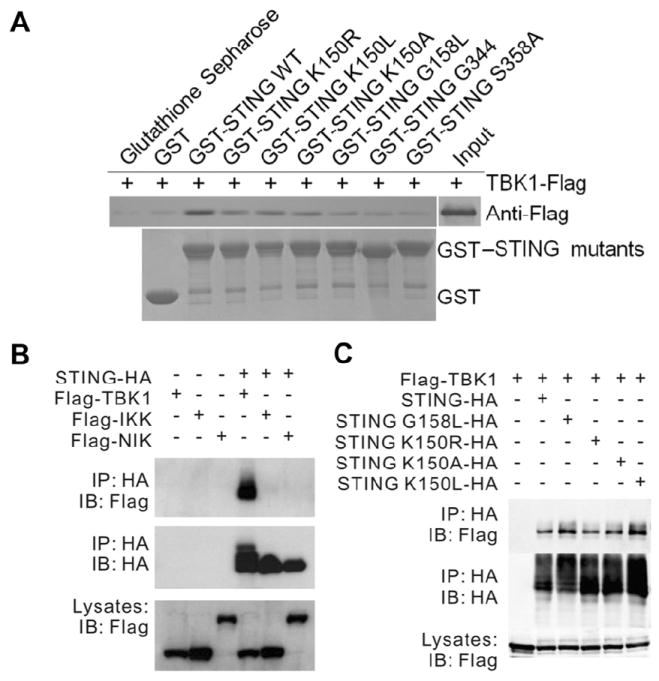
(A) Interaction of STING with TBK1 by GST pull-down. GST and GST fusion protein (STING CTD mutants) were expressed in E. coli and conjugated to glutathione-agarose beads and incubated with 1.65 μg of recombinant TBK1 in a final volume of 1.0 ml of PBS (137.0 mM NaCl, 2.7 mM KCl, 50.0 mM Na2HPO4, 10.0 mM KH2PO4, pH 7.4) followed by 5 times wash and immunoblotting with anti-Flag antibodies.
(B) Interaction of STING-HA with Flag-TBK1, Flag-IKKα, or Flag-NIK in 293T cells. Cell lysates were immunoprecipiated with anti-HA beads followed by immunoblotting with indicated antibodies.
(C) Interaction of Flag-TBK1 with STING or STING G158L in 293T cells. Cell lysates were immunoprecipiated with anti-HA beads followed by immunoblotting with indicated antibodies.
Human STING CTD Binds c-di-GMP, Resulting in Enhanced Recruitment of TBK1
Recently, the mouse STING was shown to bind c-di-GMP, resulting in the induction of IFN (Burdette et al., 2011). Based on this result, STING is proposed to function as the sensor in inducing a STING-dependent IFN-I response. Human STING shares about 70% sequence homology with its counterpart from mouse (Figure 1A and S1A). To test if human STING could interact with c-di-GMP, we performed binding studies. ITC and thermal shift assay results showed that human STING bound c-di-GMP with an affinity of ~4.4 μM (Figure 6A). Both ITC and thermal shift assays could not detect binding of c-di-AMP with STING (Figure 6B, Figure S6A and Table S4). In addition, human STING did not bind GTP or dGTP (Figure 6B). Interestingly, unlike phosphodiesterase (PDE) (Christen et al., 2005; Minasov et al., 2009), cations, such as Mg2+, were not required for c-di-GMP binding to STING (Figure 6B), which was consistent with the fact that we could not detect any well-ordered cations associated with c-di-GMP in the complex structure. Further, STING mutants K150R, K150L, K150A, S358A and a shorter truncation AA139-344 (G344) bound c-di-GMP similar to STING WT. However, G158L and I165R mutants lost their c-di-GMP binding ability as suggested by thermal shift assays (Figure S6B and Table S4). These mutagenesis results can be explained by inspecting the STING CTD-c-di-GMP complex structure. K150 and S358 are located far away from the dimer interface and c-di-GMP. On the other hand, G158 and I165 are part of the dimer interface and are essential for the formation of the c-di-GMP binding pocket.
Figure 6. STING CTD Binds c-di-GMP and Enhances Recruitment of TBK1.
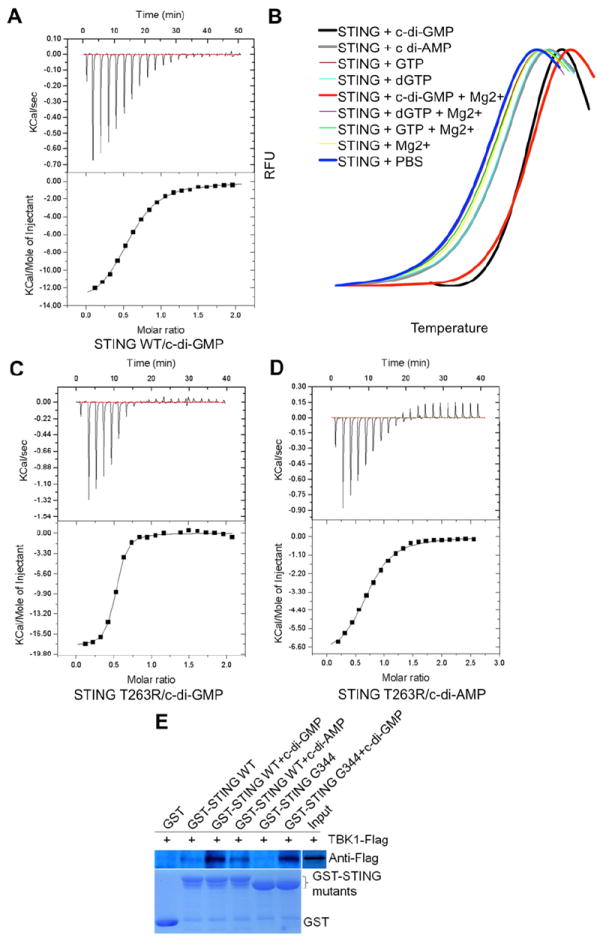
(A) ITC titration curves of STING with c-di-GMP. Top panel, raw data of heat changes upon addition of c-di-GMP (500 μM) into the cell containing 50 μM of STING CTD protein. Bottom panel, processed data corresponding to the heat of each injection plotted against the molar ratio of total c-di-GMP to total STING CTD after subtraction of the heat of control. The affinity constant (KD=4.42 ± 0.02 μM) was derived at 2:1 fixed stoichiometry.
(B) The interaction of STING with c-di-GMP and c-di-AMP detected by thermal shift assay. GTP and dGTP were used as negative controls.
(C) The affinity of STING T263R with c-di-GMP is about 10 times higher than that of STING WT with c-di-GMP as estimated by ITC.
(D) The affinity of STING T263R and c-di-AMP increases to about one half of WT STING and c-di-GMP as estimated by ITC.
(E) Interaction of STING with TBK1 enhanced by c-di-GMP binding to STING. GST pull-down assays were performed in the same way as in Figure 5A.
Next, based on the structure of the binary complex we mutated residues surrounding c-di-GMP to perturb the binding of the nucleotide analog. C-di-GMP binding ability of STING could be completely abolished by mutating Ser162 to a tyrosine or glutamic acid (Figure S6C and S6D). Ser162 was located below the ribose ring, in proximity to the phosphate group of c-di-GMP. A longer or wider side chain probably obstructs the docking of c-di-GMP into the binding pocket (Figure S6E). Interestingly, when over expressed in 293T cells, the STING S162Y mutant induced much lower levels of IFN-β than STING WT, highlighting the importance of this residue in STING-mediated type I interferon production (Figure S6F). In contrast, mutating Thr263 to arginine increased the c-di-GMP binding affinity (0.461 ± 0.053 μM) by about 10-fold (compare Figure 6A and 6C). Interestingly, this mutation increased the c-di-AMP binding affinity of STING to 8.20 ± 0.553 μM, which was about one half of the affinity of STING WT for c-di-GMP (Figure 6D). The side chain of Thr263 pointed towards one side of guanine, while the other side was stacked against the aromatic ring of Tyr167 (Figure 3B and 3D). The mutated longer arginine side chain, together with Tyr167, sandwiched guanine in the middle, which probably increased STING CTD’s affinity for c-di-GMP or c-di-AMP. Mutating Tyr167 (seen stacked against the guanine ring) to serine resulted in an insoluble protein. I200N mutant of STING has been previously shown to be unable to bind c-di-GMP (Burdette et al., 2011; Sauer et al., 2011). We attempted to bacterially express STING I200N using different strategies without success; which was consistent with the prediction by SDM server that this mutant was highly unstable (Worth et al., 2011). Mutating Tyr240 to alanine, histidine, phenylalanine or tryptophan had no obvious effect on c-di-GMP binding. Thus, structure based mutagenesis confirmed an essential role for highly conserved residues like Ser162, Tyr167 and Thr263 in binding c-di-GMP.
How does binding of c-di-GMP elicit IFN production by STING is currently unknown. Since TBK1 is central to the activation of NF-κB and IRF3 signaling pathways, we studied the binding of TBK1 with STING in presence of c-di-GMP using GST pull-down assays. Interestingly, STING interacted more strongly with TBK1 in presence of c-di-GMP than with STING alone (Figure 6E). Although c-di-AMP also increased the association of TBK1 with STING, its effectiveness was much lower than that of c-di-GMP. TBK1 is known to phosphorylate STING at Ser358. To find out if there were additional TBK1 binding sites on STING, we carried out GST pull-down assays using truncated STING containing residues 139-344 only. Surprisingly, the truncated STING bound TBK1 strongly in presence of c-di-GMP suggesting the presence of additional TBK1 binding sites on STING (Figure 6E). Taken together, these results showed that upon binding c-di-GMP, recruitment of TBK1 was enhanced. TBK1 was recruited to a new site which did not involve Ser358. Thus, phosphorylation of Ser358 is probably not required for IFN-I production elicited by c-di-GMP.
DISCUSSION
Several aspects of the structure-function studies on STING produced unexpected results. Our initial attempts to express truncations of STING around the previously predicted soluble domain (AA174-379) in E. coli as stable soluble protein failed. Surprisingly, when the highly conserved, predicted last transmembrane domain (AA153-173) was included in the truncations, a highly soluble and stable STING CTD could be produced. Purified STING CTD preferred to stay dimeric in solution. This result provided the first clue that STING might exert its function via homotypic interactions in a manner similar to the symmetric dimerization of TIR domains of TLR10 and MyD88 (Monie et al., 2009). The structure of STING CTD provided evidence of a symmetric homotypic interaction and defined the interface of the interaction. The structure of STING CTD does not resemble any known adaptor or proteins involved in innate immune responses, and has no structural homologues in PDB.
Although there is a general consensus on the presence of a transmembrane region at the N-terminus of STING, the boundaries and the number of transmembrane passes reported for STING are variable. Interestingly, all the reports are in agreement with the proposition that the region encompassing AA153-173 is the last transmembrane domain. Surprisingly, our structure of STING CTD reveals that this region is cytosolic and part of the dimerization interface. Thus, our structure of STING CTD redefines the boundaries of the transmembrane region of STING. However, it is unclear whether the current structure can formally exclude the possibility that the α5 helix could insert into the membrane in the full-length STING protein, and/or mediating STING dimerization in the membrane; although such dramatic conformational changes may not be thermodynamically favorable.
The purified STING K150A, K150L and K150R mutants exhibited a dimeric assembly in solution and retained their wild type ability to bind TBK1 and stimulate IFN production as indicated by GST pull-down studies and luciferase reporter assay results. Thus, from our structural and functional data it seems like the minimum physical and functional unit of STING is a dimer because of the presence of a large hydrophobic patch that can be solvent excluded only via dimerization. Further, although a majority of the STING homologues have K at position 150, some of the homologues, for example the horse STING does not have K in this position, suggesting that a lysine at position 150 may not be essential for the function. Taken together, these results suggest that ubiquitination of Lys150 may neither be a prerequisite for dimerization of STING nor necessary for association with TBK1.
Previously, STING has been shown to function as a dimer. Using the structure of dimeric STING CTD we selected residues for mutation to disrupt the dimer interface expecting a loss-of-function of IFN induction. V155R, W161A, and Y164A, mutants resulted in a complete loss-of-function of STING. Dimerization deficient G158L mutant showed partial activity. Thus, our structure-based mutagenesis data support the inference that dimerization of STING is essential for induction of IFN.
c-di-GMP was first reported by Benziman and co-workers as an allosterical activator in the membrane-bound cellulose synthase of Gluconacetobacter xylinus in 1987 (Ross et al., 1987). C-di-GMP is a ubiquitous second messenger that orchestrates the motile planktonic and sedentary biofilm-associated bacterial ‘lifestyles’ (Hengge, 2009). STING CTD has been shown to specifically recognize c-di-GMP (Burdette et al., 2011), but not GTP or ATP. Inspection of the binary complex of STING CTD with c-di-GMP revealed that there was no space to accommodate the second or third phosphate group of ADP/GDP or ATP/GTP. Therefore, STING has the ability to discriminate between c-di-GMP and host GTP or ATP molecules. Bacteria are also known to use c-di-AMP as a secondary messenger molecule for signaling (Romling, 2008). Such signaling molecules are not present in humans and therefore the human body can sense intrusion by the immunosurveillance pathway. Our structural analysis showed that c-di-AMP could be docked at the same site as c-di-GMP without any steric hindrance due to their structural similarity. C-di-AMP does not have the amine group of c-di-GMP which is involved in hydrogen bonding with a “conserved” (present in unliganded structure) water molecule. This could explain why STING has stronger binding with c-di-GMP over c-di-AMP. In addition, c-di-AMP has an amino group at C6 position that is opposite in charge to the keto group seen in c-di-GMP, which could further influence the binding of c-di-AMP to STING. In fact ITC and thermal shift assays did not detect any binding of c-di-AMP with STING. However, mutating Thr263 to arginine resulted in an affinity for c-di-AMP similar to that seen for c-di-GMP with STING CTD WT. Clearly, further studies are needed to ascertain whether STING signaling is c-di-GMP specific or can be triggered by c-di-AMP too. To find out how binding of c-di-GMP to STING resulted in initiation of downstream events leading to IFN-I production, we compared the structure of the c-di-GMP bound STING CTD with the structure of the unliganded STING CTD. Surprisingly, comparison of the structures revealed no large scale conformational changes in STING CTD upon c-di-GMP binding. So far, two different mechanisms of activation via c-di-GMP are known (Schirmer and Jenal, 2009). One mechanism involves cross-linking domains. For example, c-di-GMP has been shown to exert its effect by cross linking domains in PleD (Chan et al., 2004), DgcA (Christen et al., 2006) and WspR (De et al., 2008). The second mechanism involves signal-dependent ordering of loops. For example, an NMR study on the PilZ domain containing protein PA4608 revealed that upon binding c-di-GMP, an N-terminal region became ordered (Habazettl et al., 2011). Since dimerization of STING is not signal-dependent, we looked at disordered loops, especially the loop connecting strand β2 with β3. This loop was disordered in all the structures of STING CTD we solved. C-di-GMP binds in proximity; in fact, this loop became partially ordered and moved closer towards c-di-GMP upon its binding. It is tempting to speculate that binding of c-di-GMP to STING CTD makes these this loop more accessible for TBK1 binding. This is partly supported by the fact that recruitment of TBK1 was enhanced in presence of c-di-GMP. Further studies are warranted to explore this exciting possibility, which would also shed light on whether STING plays distinct roles in c-di-GMP and viral DNA mediated signaling.
In summary, our structural, functional and mutagenesis data unravel several hitherto unknown aspects of STING. STING forms homotypic interactions that are essential for stimulation of IFN production. The structures of STING CTD provide detailed molecular insights into the nature of this homotypic architecture. Notably, the structure of STING CTD is very different from any known structures of adaptors or proteins functioning in innate immune responses and therefore STING may represent a novel class of sensors involved in detection of bacterial intrusion. Our protein expression, analytical and structural evidences revealed that the AA153-173 region was not a transmembrane region as predicted previously, but is a hydrophobic dimer interface. The structure of the binary complex maps the exact location of the c-di-GMP binding site on STING and unveils a unique mode of binding of c-di-GMP to proteins. Structure guided mutagenesis studies showed that STING exists and functions as a dimer. Further, ubiquitination of Lys150 may not be a pre-requisite for dimerization. The c-di-GMP binding pocket is formed via dimerization of STING. We show that binding of c-di-GMP enhances the association of TBK1 with STING. An additional TBK1 binding site, other than the previously reported Ser358, is probably responsible for c-di-GMP mediated TBK1 signaling. Although the results further our understanding of the nature of the homotypic interactions of STING essential for stimulation of IFN production, questions on how the signal of intrusion sensed by STING is relayed, leading to recruitment of downstream effector molecules remain to be investigated.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human STING fragments were amplified from a human transcription library (Stratagene, USA) and cloned into pMCSG7 vector (Stols et al., 2002) for expression in E. coli BL21 (DE3). The recombinant protein was purified by Ni-affinity chromatography (Qiagen, USA) and gel filtration chromatography as described (Liang et al., 2011).
Crystallization and Data Collection
The initial crystallization conditions were examined using commercially available sparse-matrix screening kits. Hits were optimized by hand. 2 μl hanging drops containing 1 μl protein mixed with 1 μl mother liquor were equilibrated over 300 μl reservoir solution and incubated at 16 °C. The STING CTD and c-di-GMP complex was formed by mixing equal molar amounts of STING CTD with c-di-GMP (Biolog, USA) using the same crystallization condition as unliganded STING CTD.
Structure Determination and Analysis
Diffraction data for Se-Met, native and the binary complex of STING CTD with c-di-GMP were collected at wavelength of 0.9793 Å. Data were indexed and scaled using HKL2000 (Otwinowski Z., 1997). The initial phases were determined by Se-SAD (single-wavelength anomalous dispersion) method (Hendrickson, 1991). PHENIX AutoBuild was used to rebuild the model with the initial phase (Terwilliger et al., 2008). Other structures were solved by molecular replacement method with program Phaser (McCoy et al., 2007). The models were manually improved in Coot (Emsley and Cowtan, 2004). Refinement was carried out using REFMAC (Murshudov et al., 1997) and PHENIX (Adams et al., 2010) alternately. The quality of the final model was validated with MolProbity (Chen et al., 2010). Structures were analyzed using PDBePISA (Protein Interfaces, Surfaces and Assemblies) (Krissinel and Henrick, 2007), Dali (Holm and Rosenstrom, 2010), ProFunc (Laskowski et al., 2005).
Immunoblotting and Immunoprecipitation
HEK 293T cells were transfected with indicated plasmids using Lipofectamine 2000. Forty-eight hours post transfection, cells were homogenized. Cell lysates were then incubated with anti-Flag M2 affinity gel or anti-HA affinity gel (Sigma) for 2 hours at 4 ° and the immunoprecipitated complexes were separated by SDS-PAGE and blotted with indicated antibodies.
IFN-β Luciferase Reporter Assay
HEK 293T cells were transfected with IFN-β firefly luciferase and renilla luciferase reporter plasmids together with STING WT or STING mutants. Forty-eight hours post transfection, firefly and renilla luciferase activities were determined using a Dual Luciferase Assay System (Promega) and a SIRIUS Luminometer (Berthold Detection Systems) according to the manufacturer’s protocol.
Additional Methods
More detailed descriptions of methods for protein expression and purification, protein crystallization, structure refinement and analysis, cells and reagents, real-time Quantitative PCR, Isothermal Titration Calorimetry (ITC), GST pull-down, thermal shift, analytical ultracentrifugation and statistical analysis can be found in the Supplemental Information.
Supplementary Material
HIGHLIGHTS.
The STING structure and its complex with c-di-GMP revealed a unique architecture.
This structure showed c-di-GMP bound to a mammalian protein.
STING AA153-173 is not a transmembrane helix but a hydrophobic dimeric interface.
Binding of c-di-GMP enhanced the recruitment of TBK1 by STING.
Acknowledgments
The authors are grateful to G. Barber (University of Miami, USA) for providing the Tmem173-/- MEF cells, L.-W. Hung (Los Alamos National Laboratory) for the help on data collection; the staff at beamline 5.0.1 (ALS, Lawrence Berkeley National Laboratory, USA) and beamline BL17U1 (Shanghai Synchrotron Radiation Facility (SSRF), China) for help in the X-ray data collection; Y.Y. Chen at the Protein Science Core Facility of IBP for technical help with ITC experiments. This work was supported by the Ministry of Science and Technology of China (grants 2009DFB30310, 2009CB918803, 2011CB911103), the National Natural Science Foundation of China (grants 31070660, 30870483, 31021062), the Chinese Academy of Sciences (grantsKSCX2-EW-J-3, 2010Y1SA1) and NIH grants (AI069120, AI056154 and AI047868).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, 6 figures, and 6 tables and can be found with this article online at doi:xxxxxxxx.
ACCESSION NUMBERS
Coordinates and structure factors for the unliganded and liganded STING CTD have been deposited in the Protein Data Bank under the accession codes 4EF5 and 4EF4, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production rocess errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol. 2011;23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowzard JB, Ranjan P, Sambhara S, Fujita T. Antiviral defense: RIG-Ing the immune system to STING. Cytokine Growth Factor Rev. 2009;20:1–5. doi: 10.1016/j.cytogfr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Paul R, Samoray D, Amiot NC, Giese B, Jenal U, Schirmer T. Structural basis of activity and allosteric control of diguanylate cyclase. Proc Natl Acad Sci U S A. 2004;101:17084–17089. doi: 10.1073/pnas.0406134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y, et al. Activation of STAT6 by STING Is Critical for Antiviral Innate Immunity. Cell. 2011;147:436–446. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen B, Christen M, Paul R, Schmid F, Folcher M, Jenoe P, Meuwly M, Jenal U. Allosteric control of cyclic di-GMP signaling. J Biol Chem. 2006;281:32015–32024. doi: 10.1074/jbc.M603589200. [DOI] [PubMed] [Google Scholar]
- Christen M, Christen B, Folcher M, Schauerte A, Jenal U. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J Biol Chem. 2005;280:30829–30837. doi: 10.1074/jbc.M504429200. [DOI] [PubMed] [Google Scholar]
- De N, Pirruccello M, Krasteva PV, Bae N, Raghavan RV, Sondermann H. Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 2008;6:e67. doi: 10.1371/journal.pbio.0060067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–863. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Habazettl J, Allan MG, Jenal U, Grzesiek S. Solution structure of the PilZ domain protein PA4608 complex with cyclic di-GMP identifies charge clustering as molecular readout. J Biol Chem. 2011;286:14304–14314. doi: 10.1074/jbc.M110.209007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson WA. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science. 1991;254:51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell Mol Life Sci. 2011;68:1157–1165. doi: 10.1007/s00018-010-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud AL, Cambier JC, Lenz LL. MPYS Is Required for IFN Response Factor 3 Activation and Type I IFN Production in the Response of Cultured Phagocytes to Bacterial Second Messengers Cyclic-di-AMP and Cyclic-di-GMP. J Immunol. 2011a;187:2595–601. doi: 10.4049/jimmunol.1100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Lenz LL, Cambier JC. Cellular reactive oxygen species inhibit MPYS induction of IFNbeta. PLoS One. 2010;5:e15142. doi: 10.1371/journal.pone.0015142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28:5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Xu LG, Yang IV, Davidson EJ, Schwartz DA, Wurfel MM, Cambier JC. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun. 2011b;12:263–269. doi: 10.1038/gene.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33:W89–93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Ouyang S, Shaw N, Joachimiak A, Zhang R, Liu ZJ. Conversion of D-ribulose 5-phosphate to D-xylulose 5-phosphate: new insights from structural and biochemical studies on human RPE. FASEB J. 2011;25:497–504. doi: 10.1096/fj.10-171207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minasov G, Padavattan S, Shuvalova L, Brunzelle JS, Miller DJ, Basle A, Massa C, Collart FR, Schirmer T, Anderson WF. Crystal structures of YkuI and its complex with second messenger cyclic Di-GMP suggest catalytic mechanism of phosphodiester bond cleavage by EAL domains. J Biol Chem. 2009;284:13174–13184. doi: 10.1074/jbc.M808221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monie TP, Moncrieffe MC, Gay NJ. Structure and regulation of cytoplasmic adapter proteins involved in innate immune signaling. Immunol Rev. 2009;227:161–175. doi: 10.1111/j.1600-065X.2008.00735.x. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakhaei P, Hiscott J, Lin R. STING-ing the antiviral pathway. J Mol Cell Biol. 2010;2:110–112. doi: 10.1093/jmcb/mjp048. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, W M. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Vol. 276. New York: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- Romling U. Great Times for Small Molecules: c-di-AMP, a Second Messenger Candidate in Bacteria and Archaea. Sci Signal. 2008;1:pe39. doi: 10.1126/scisignal.133pe39. [DOI] [PubMed] [Google Scholar]
- Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinberger-Ohana P, Mayer R, Braun S, de Vroom E, van der Marel GA, van Boom JH, Benziman M. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature. 1987;325:279–281. doi: 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Yoshimori T, Akira S. Regulation of dsDNA-induced innate immune responses by membrane trafficking. Autophagy. 2010;6:430–432. doi: 10.4161/auto.6.3.11611. [DOI] [PubMed] [Google Scholar]
- Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer T, Jenal U. Structural and mechanistic determinants of c-di-GMP signalling. Nat Rev Microbiol. 2009;7:724–735. doi: 10.1038/nrmicro2203. [DOI] [PubMed] [Google Scholar]
- Smith KD, Lipchock SV, Ames TD, Wang J, Breaker RR, Strobel SA. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat Struct Mol Biol. 2009;16:1218–1223. doi: 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- Su J, Li Y, Shaw N, Zhou W, Zhang M, Xu H, Wang BC, Liu ZJ. Crystal structure of a novel non-Pfam protein PF2046 solved using low resolution B-factor sharpening and multi-crystal averaging methods. Protein Cell. 2010;1:453–458. doi: 10.1007/s13238-010-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. 2010;33:765–776. doi: 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth CL, Preissner R, Blundell TL. SDM--a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–222. doi: 10.1093/nar/gkr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, Wang YY, Zhang XL, Shu HB. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407. doi: 10.1016/j.immuni.2009.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.