

Abstract
Infertility is defined as the inability of a couple to conceive despite trying for a year, and it affects approximately 15% of the reproductive-age population. It is considered a genetically lethal factor, as the family lineage stops at that individual with no progeny produced. A genetic defect associated with an infertile individual cannot be transmitted to the offspring, ensuring the maintenance of reproductive fitness of the species. However, with the advent of assisted reproductive techniques (ART), we are now able to overcome sterility and bypass nature’s protective mechanisms that developed through evolution to prevent fertilization by defective or deficient sperm.
Keywords: mendelian genetics, male infertility, asthenozoospermia, oligospermia
Introduction
The causes of male factor infertility are multifactorial, with estimates reaching 50% due to, or contributed by, genetic abnormalities (Table 1, Refs. 2–11).1 Although the majority of the genetic causes of male infertility are still unknown, genetic defects, such as cystic fibrosis, have important implications for offspring conceived through intracytoplasmic sperm injection (ICSI). However, in the absence of a comprehensive clinical evaluation, many of these genetic etiologies may go undiagnosed.
Table 1.
Frequency and associated phenotypes of the most common genetic abnormalities in male infertility
| Reference | Genetic abnormality | Phenotype | Prevalence (%) |
|---|---|---|---|
| Foresta et al.11 | Chromosomal aberrations | Azo → normospermia | 2–10 |
| Foresta et al.4 | Klinefelter’s syndrome | Azo → severe oligospermia | 5–10 azoospermia 2–5 severe oligospermia |
| Mau-Holzmann et al.6 | Other sex chromosome | Azo → normospermia | 0.1–0.2 |
| De Braekeleer et al.2 | Robertsonian translocations | Azo → severe oligospermia | 0.5–1.0 |
| De Braekeleer et al.2 | Reciprocal translocations | Azo → severe oligospermia | 0.5–1.0 |
| Kuroda-Kawaguchi et al. 5 | Y chromosome microdeletions | Azo → severe oligospermia | 5–10 |
| Vogt et al. 8 | AZFa | Azo-SCOS | 0.5–1.0 |
| Vogt et al.8 | AZFb | Azo-spermatogenic arrest | 0.5–1.0 |
| Ferlin et al.10 | AZFc | Azo → severe oligospermia | 3–7 |
| Vogt et al.8 | AZFb+c | SCOS/spermatogenic arrest | 0.5–1.0 |
| Vogt et al.7 | Partial AZFc deletion Gene mutations |
Azo → normospermia | 3–5 |
| Foresta et al.4 | CFTR | Obstructive azo | 60–70 5% in infertile men |
| Ferlin et al.9 | AR | Azo-oligospermia | 2–3 |
| Bogatcheva et al.3 | INSL3-LGR8 | Cryptorchidism | 4–5 |
Azo = Azoospermia
The foundation of diagnosis for males is the routine semen analysis (SA), where sperm concentration, motility, morphology, and the presence of other cells are evaluated. Indicators of patency and function of the male genital tract are assessed including semen volume, liquefaction time, pH, and the presence or absence of fructose. While the routine SA evaluates the ejaculate for abnormalities in the sperm number, morphology, and motility, it only provides clues and suggestions to indicate the need to pursue further testing. Current genetic testing performed for male infertility includes karyotype, CFTR gene analysis, PCR testing for Y-chromosome microdeletions, and sperm FISH analysis for specific chromosome aberrations.
The primary goals of the evaluation of the male presenting with infertility are to identify etiological conditions that can be reversed with resulting improvement of fertility status, medically significant and potentially dangerous diagnoses underlying the male’s infertility, genetic etiologies that may have implications for the patient and/or his offspring; and irreversible conditions that may be best managed with the use of assisted reproductive techniques (ART) or the recommendation of donor insemination or adoption. The genetic basis of male infertility, although relatively poorly understood, may ultimately represent one of the most clinically important aspects of male infertility (Table 2, Ref. 12). However, with the advent of IVF/ICSI, while these hurdles of infertility can be overcome, the underlying causes of infertility–defective genes–have the potential to be transmitted to offspring.
Table 2.
Genetic basis of human male infertility defects: spermatogenesis and sperm function
| Abnormal spermatogenesis | |
| ATM; ATMAC; DAZL; ERCC2; GTF2A1L; JUN; NLRP14; NRB0B1; POLG; PRM1; PRM2; SDHA; SOX8; XRCC1; YBX2 | |
| Azoospermia | |
| APOB; ACSBG2; ART3; ATM; BOULE; BPY2; BRCA2; CDY1; CFTR; CREM; DAZ; DDX25; DDX3Y; DRFFY; ERCC1; ERCC2; FASLG; FHL5; FKBP6; HNRNPC; HSFY1; KLHL10; LAP3; MBOAT1; MEI1; MLH1; MLH3; MTR; NLRP14; PRDM16; RBMX; RBMY1A1; RBMY1F; SPATA16; SYCP1; SYCP3; TAF7L; TGIF2LX; TSPY; TSSK4; UBE2B; USP26; UTP14C; USP9Y; UTY; XPC; XPD; XRCC1; YBX2; ZNF230 | |
| Oligozoospermia | |
| MT-ATP6; EGF; FASL; H19 and MEST; KLHL10; PIGA; PRM1; PRM2; SHBG; SDHA; TSSK4; UBE2B; VASA | |
| Asthenozoospermia | |
| AKAP3; AKAP4C; CATSPER2; DNMT3B; DHAH5; DNAH11; DNAL1; PDYN; GNA12; Mitochondrial DNA; MTHFR; MT-ND4; PIGA; POLG; PPM1G; PRKAR1A; SHBG; SPAG16; TEKT1; TEKT2; TPN1; TPN2; TXNDC3; T | |
| mt DNA haplotypes | |
| Teratozoospermia | |
| AURKC; PRM1; PVRL2; SPATA16; SP1 | |
| Oligoasthenozoospermia | |
| JUND; MT-ND4; NALP14 | |
| Oligoasthenoteratozoospermia | |
| MTRR; IL1B; SABP | |
| Acrosome or Fertilization | |
| POIA3 | |
| Varicocele effect | |
| MT-ATP6; MT-ATP8; CACNA1C; MT-CO1; MT-CO2; MT-ND3 | |
| Chromosome defect | |
| Numerical sex chromosome (Klinefelter’s; XXY–XXXXY) | |
| Structural chromosome (translocations, inversions or deletions) | |
| Y chromosome microdeletions, XX male or XY female | |
| Systemic Disorders Affecting Fertility | |
| Kartagener’s syndrome | Sickle cell anemia (HBB) |
| Fanconi anemia (FANCA) | ®-thalassemia |
| Myotonic dystrophy (DMPK) | Noonan (PTPN11) |
| DNA Damage | Infertility |
| GSTM1 | AR; GSTM1 KIT; KITLG; IL1A; OAZ3; PRM1; TSPY; TSSK4; USP26; YBX2 |
Note: SNPs of unknown significance shown in bold. Adapted by permission from Macmillan Publishers Ltd: Matzuk & Lamb Nature Medicine 2008.12
When faced with the evaluation of a man with azoospermia, the focus should be placed on deciphering between whether the azoospermia is a result of abnormal spermatogenesis or obstruction (Fig. 1).
Figure 1.
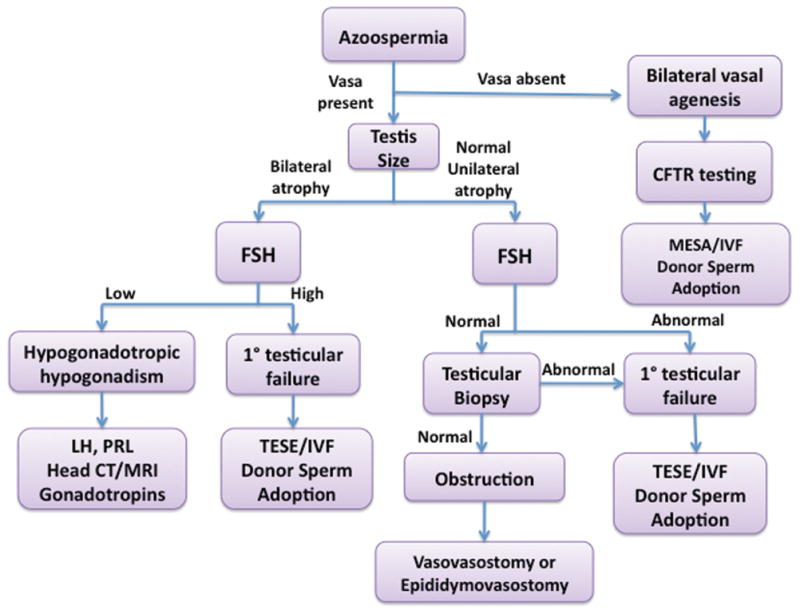
Evaluation of patient with azoospermia. CFTR, cystic fibrosis transmembrane conductance regulator gene; FSH, follicle stimulating hormone; IVF, in vitro fertilization; MESA, microsurgical epididymal sperm aspiration; LH, luteinizing hormone; TESSE, testicular sperm extraction; PRL, prolactin.
In Mendelian recessive inheritance, each parent contributes one of two mutant alleles for a disease trait. However, if a mutant allele has a dominant effect, it requires inheritance of only one parental allele. If the genotypes of both parents in a genetic cross are known, Mendel’s principles of segregation and independent assortment can be used to determine the distribution of phenotypes expected for the population of offspring.
Endocrinopathies and male infertility
There are relatively few diagnosed genetic causes of male infertility. Diagnoses are largely descriptive and reflect the lack of understanding of the factors that regulate sperm production, maturation, and function. Endocrine factors are one of the components that are relatively well-established and have become an essential component of the male evaluation, with either an abnormal physical examination suggestive of a disorder in testosterone production and action, an abnormal semen examination, or evidence of impaired sexual dysfunction (Table 3). However, true endocrinologic causes of male infertility are relatively uncommon, being present in less than 3% of cases13 (Table 4).
Table 3.
Endocrinopathies in male infertility
| Hypergonadotropic hypogonadism |
| Classic (Klinefelter syndrome) |
| Isolated spermatogenic compartment failure/primary germ cell failure: postpubertal viral or bacterial orchitis, chemotherapeutic agents, idiopathic, secondary to exposure to environmental toxicants |
| Hypogonadotropic hypogonadism |
| Congenital (Kallman syndrome) |
| Acquired: tumor, infection, autoimmune, infiltrative diseases, pituitary infarction, and drug use |
| Defective androgen synthesis or response |
| 5 α-reductase deficiency |
| Complete androgen insensitivity |
| Partial androgen resistance |
Table 4.
Distribution of final diagnostic categories found in a male infertility clinic
| Category | Number | % |
|---|---|---|
| Idiopathic | 1535 | 32.6% |
| Varicocele | 1253 | 26.6% |
| Obstruction | 720 | 15.3% |
| Normal female factorb | 503 | 10.7% |
| Cryptorchidism | 129 | 2.7% |
| Ejaculatory failure | 95 | 2.0% |
| Endocrinopathies | 70 | 1.5% |
| Drug/radiation | 64 | 1.4% |
| Genetica | 56 | 1.2% |
| Testicular failure | 52 | 1.1% |
| Sexual dysfunction | 32 | 0.7% |
| Other | 80 | 1.6% |
| Total | 4589 | 100% |
Adapted from Sigman M. 2009. Office evaluation of the subfertile male. In Infertility in the Male. L. I. Lipshultz, S. S. Howards, & C. S. Neiderberger, Eds.: 153–176. Cambridge University Press.
Underestimation as most patients are not evaluated.
Normal female factor indicates no clinically detected abnormalities in female partner.
The hypothalamic–pituitary–adrenal (HPA) axis, and the hormonal reproductive system, that is, the hypothalamic–pituitary–gonadal (HPG) axis, are intimately interlinked. The HPG axis is an important mediator of infertility. Stress can inhibit the HPG axis and negatively impact fertility. Abnormal development of the HPG axis can result in acquired hypogonadotropic hypogonadism (IHH) and Kallman syndrome (KS). Finally, spontaneously occurring mutations in the genes involved in the HPG axis, can also contribute to infertility.14,15
Kallmann syndrome (MIM308700) is a rare condition characterized by isolated hypogonadotropic hypogonadism (HH) and anosmia due to agenesis of the olfactory bulb; it occurs in approximately 1 in 10,000–60,000 live births.16 A subset of Kallmann syndrome, inherited in an X-linked fashion, is caused by a mutation of the KAL1 gene, which produces a cell adhesion molecule.17–19 Autosomal dominant inheritance is caused by mutations in FGFR1, FGFR8, PROKR2, and PROK2 genes.20–22 In about 75–80% of KS patients, the causative gene is unknown.
Androgen insensitivity syndrome (AIS, MIM300068) is an X-linked disorder caused by mutations in the androgen receptor (AR) gene (MIM313700), resulting in end-organ resistance to androgens. More than 800 different AR gene mutations, resulting in AIS have been reported. However, the mutation–receptor structure–function relationship is not yet fully understood. Thus, the phenotypes resulting from these mutations are not always predictable. The vast majority of androgen receptor mutations are single base substitutions, while deletions or insertions are rare. Most of these mutations are located in a few “hot spots” in the ligand-binding domain of the AR protein. About two thirds of these androgen receptor mutations are inherited in an X-linked fashion. The remainders of these mutations are either germ line or somatic de novo mutations. Somatic mutations can result in somatic mosaicism, where both mutant and wild-type receptors are expressed in different proportions.23
AIS is a disorder affecting sexual differentiation in variable severity, ranging from phenotypic females (complete AIS, CAIS) to defective spermatogenesis in otherwise normal males (partial AIS, PAIS, or minimal AIS, MAIS) depending on the type and localization of these mutations.24,25 Haploinsufficiency of the AR gene due to complete or partial gene deletions or nonsense and frameshift mutations usually lead to complete AIS. Splicing mutations or missense mutations, on the other hand, can result in diverse phenotypes that are impossible to predict. Alternative factors that can influence the phenotype include defects in the protein coactivators and corepressors, somatic mosaicism, and the length of polyglutamine (PolyGln) repeats in exon 1. There are at least 864 mutations known, of which 724 associated with AIS have been reported in the AR mutation database (www.mcgill.ca/androgendb). Until now, only 91 mutations have been reported in exon 1 of the AR gene in patients suffering from any form of AIS, despite the fact that it encodes for more than half of the AR protein. Interestingly, many patients presenting with clinical features of AIS do not have documented mutations in the AR gene and may have alternative defects in androgen signaling pathway. For example, defects in coregulators such as transcriptional intermediary factor 2 (TIF2)26–28 and AR coactivators29 are also implicated in the pathogenesis of AIS. Reduced AR transcription was also found in patients with AIS.30 Several mutations have been identified in the AR gene linked to endocrine dysfunction. Most of these mutations are due to loss-of-function of AR activity.
Systemic genetic disorders/syndromes
The expansion of the CAG trinucleotide repeats encoding the polyglutamine (PolyGln) tract, including the amino terminal transactivation domain of the AR protein, is involved in various neurodegenerative disorders. Spinal and bulbar muscular atrophy (SBMA, MIM313200) or Kennedy’s disease (Kennedy-Alter-Sung disease) is a rare inherited X-linked neurodegenerative disorder of motor neurons.31,32 SBMA usually affects males in the third decade. It is characterized by muscle cramps, fasciculations followed by weakness, and atrophy of the proximal limb and bulbar muscles.33–35 The severity of the disease correlates with the increased number of CAG repeats of the AR gene. Expansion over 38–62 repeats leads to degeneration of facial, hypoglossal, and spinal motor neurons with neurogenic wasting of the corresponding skeletal muscle. Clinically, patients with SBMA present with progressive AIS, severe oligozoospermia, infertility, testicular atrophy, and gynecomastia.32,36,37
SBMA is a member of a new class of trinucleotide repeat disorders (or PolyGln-related) and inherited neurodegenerative diseases38,39 that include Huntington’s disease (HD), several spinocerebellar ataxias (SCAs),40 and dentatorubral-pallidoluysian atrophy (DRPLA).41,42 These conditions are caused by gain-of-function mutations leading to accumulation of abnormal proteins with the PolyGln tract that produce neurotoxic effects and cause cell death of motor neurons,38 mostly in the anterior horns of the spinal cord and in the bulbar region of the brain stem.
Frequently, A43 and myotonic dystrophy (DM) (MIM160900) are associated with idiopathic azoospermia.44 DM is caused by a CTG trinucleotide repeat expansion in the 3′-untranslated region of the DM protein kinase gene (DMPK, MIM605377, 19q13.3). However, the results are controversial since one report found more than 18 repeats at the DM locus in azoospermia patients, and not in controls,45 while others have failed to find any differences.44 A less frequent cause of DM is an expansion of the CCTG repeat in intron 1 of zinc finger protein 9 (ZNF9, MIM116955, 3q13.3).46
Rare instances of isolated HH can also result from rare dominant or recessive mutations in GNRHR, KISS1R, NROB1, LEPR, TAC3, and TACR3 affecting the various components of the HPG axis.15 The effect of inactivating mutations of GNRHR has been associated with impaired LH/FSH secretion, HH, and delayed puberty in both males and females. Male patients present with microphallus and undescended testes in childhood. Inactivating mutations of LH receptors have also been described in both sexes. In men, the phenotype ranges from female genitalia to micropenis. Testicular biopsies show various degrees of Leydig cell hypoplasia with resultant oligozoospermia. Isolated selective LH hormone deficiency has been described in consanguineous families due to mutations in the luteinizing hormone β (LHB) gene, resulting in hypogonadism in both sexes.47 Recently, a polymorphism in the promoter of the follicle stimulating hormone β (FSHB) gene, which is associated with hypogonadism has been identified in a young Estonian cohort.48
Prader-Willi syndrome (PWS) (MIM176270) is a complex genetic disorder caused by deficiency of one or more paternally expressed imprinted transcripts within chromosome 15q11-q13, affecting 1:25,000 live births. Thus, PWS is not usually inherited in a Mendelian fashion. PWS is characterized by hypothalamic dysfunction resulting in hypogonadism associated with delayed puberty, obesity, hypotonia, developmental delay, behavioral abnormalities, short stature, and genital abnormalities that include cryptorchidism, scrotal hypoplasia, and microphallus.49,50 PWS is a phenotypically variable disorder with regards to sexual maturation. Pubertal variability is common in most of the individuals who are unable to complete puberty, likely due to degeneration of GnRH secreting neurons.50,51 Fertility has not been documented in any male PWS patients to date. PWS infertility could be a result of multiple factors, including low pituitary-testicular axis hormones including FSH, LH, estradiol, testosterone, and inhibin B.52,53 PWS patients have low sexual libido with testicular histology varying from normal to Sertoli cell only.54
Chromosome abnormalities
Klinefelter’s syndrome is the most common cause of hypogonadism and infertility in males (1 in 500). The classic Klinefelter’s syndrome is associated with a 47, XXY karyotype due to maternal or paternal meiotic nondisjunction. Maternal nondisjunction is associated with advanced maternal age.55 A variety of mosaic patterns constitute nearly 15% of the cases, most common being 46,XY/47,XXY and the remainder of cases are due to polysomy of X chromosome (48, XXXY or 49,XXXXY). Rare cases of Klinefelter syndrome are caused by isochromosome Xq i (Xq), or X-Y translocations in 0.3–0.9% of males with X chromosome polysomies.56,57 Clinically, patients with this syndrome present with azoospermia or severe oligozoospermia, gynecomastia in late puberty, atrophy and hyalinization of the seminiferous tubules, and elevated urinary gonadotropin levels.
CFTR and reproductive tract obstruction
Cystic fibrosis transmembrane conductance regulator (CFTR) gene-related disorders encompass a disease spectrum from focal male reproductive tract involvement in congenital bilateral absence of the vas deferens (CBAVD) to multiorgan involvement in classic cystic fibrosis. CFTR-related disorders are inherited in an autosomal recessive manner. Severe dysfunction of the CFTR gene causes cystic fibrosis (CF), a life-threatening disease manifesting with progressive lung disease. CBAVD, resulting in primary obstructive azoospermia is a well-recognized cause of male infertility.
The CFTR gene (MIM602421) is located on the long arm of chromosome 7q31.2 and contains 27 coding exons that spread over 230 kb.58 Its normal allele produces a 6.5-kb mRNA that encodes a 1480-amino acid integral membrane protein that functions as a regulated chloride channel in a variety of epithelial cells. The most common gene mutation is a 3-bp deletion, resulting in loss of a phenylalanine at amino acid position 508 of the CFTR polypeptide (ΔF508); this mutation accounts for about 30–80% of all mutant alleles depending on the ethnic group.59 CBAVD commonly results from the combination of two mutant alleles, one allele with severe CFTR mutation and a second allele with either a mild CFTR mutation or the common splicing mutant allele 5T.60
Young’s syndrome (MIM279000) is a condition characterized by respiratory infections, ranging from simple bronchitis to bronchiectasis, and obstructive azoospermia secondary to inspissated secretions, although spermatogenesis is unaffected. Due to the similarities, parallels to CBAVD secondary to mutations in the CFTR gene were noted; however, further investigations concluded that classical Young’s syndrome was unlikely associated with defects in CFTR.61,62
Men without clinically apparent pulmonary manifestations of CF may have CBAVD. Hypoplasia or aplasia of the vas deferens and seminal vesicles may occur either bilaterally or unilaterally. CBAVD does not pose a health risk per se to the affected men, and testicular development, function, and spermatogenesis are usually normal. CBAVD is commonly identified during evaluation of infertility or as an incidental finding at the time of a surgical procedure, such as orchidopexy. However, the genetic fundamental basis of CBAVD has a significant impact on ART. For fathers with CBAVD, spermatogenesis may be normal, though it is imperative to determine whether the female partner is a carrier of the CFTR mutation as their offspring will be at increased risk of transmitting infertility to their male offspring or cystic fibrosis to both male and female offspring.
The treatment options include both microsurgical epididymal sperm aspiration (MESA) and testicular sperm extraction (TESE), or donor sperm in conjunction with ICSI to treat men with obstructive azoospermia (Fig. 1). Genetic counseling is standard in family planning for male factor couples with CBAVD.
Sperm motility defects
Asthenozoospermia (AZS) or reduced sperm motility is defined by the proportion of motile spermatozoa in semen (usually <40%). It is one of the four major semen defects found in at least half of infertile men. AZS is one of the phenotypes present in primary ciliary dyskinesia (PCD) (MIM242650), a predominantly autosomal recessive condition characterized by ciliary dysfunction and impaired mucociliary clearance that causes respiratory tract infections and infertility. The most severe form, Kartagener’s syndrome (KS), presents in combination with situs inversus (MIM244400). The incidence of PCD is estimated at 1:16,000 births with KS accounting for 50% of the cases. A high percentage of PCD males are infertile, and the majority of cases of KS are due to immotility or dysmotility of the spermatozoa; however, the penetrance of the defect varies among individuals. PCD is caused by different structural abnormalities in dynein arms of axoneme, the internal structure of cilia and flagella responsible for their motility. The axonemal structure is highly conserved through evolution and consists of a complex made of a central pair of two microtubules and nine outer-doublet microtubules with attached inner and outer dynein arms (9 + 2 structure).63 There are more than 250 genes involved in the structure, assembly, and regulation of a cilium; however, only three genes have been associated with PCD: DNAI1 (MIM603332; 9p21-p13), DNAH5 (MIM603335; 5p15.2), and DNAH11 (MIM603339; 7p21). Recent screening in 90 non-syndromic AZS patients and 200 controls revealed one mutation in each of the three dynein genes in seven patients, all inherited from their mothers.64
Sperm motility requires an increase in the concentration of intracellular calcium ions.65 Several calcium channel genes residing in the sperm may be responsible for infertility. The CATSPER gene family (CATSPER1–4) is a unique cation channel expressed exclusively in sperm. CATSPER2 (MIM607249) (expressed in the sperm and inner ear) together with STRC, KIAA0377, and CKMT1B are associated with deafness-infertility syndrome (DIS) (MIM61102) due to a homozygous contiguous gene deletion at 15q15.3. Four families have been identified with homozygous deletions in this area that cause deafness and sperm dysmotility.66,67 CATSPER1 (MIM606389; 11q12.1), a gene responsible for infertility in mice due to lack of sperm motility, has been mutated in infertile men of two consanguineous families.68,69 Two separate insertion mutations that lead to frameshifts and a premature stop codon in CATSPER1 were found in three infertile Iranian men, and absent in 576 Iranian controls.68
SPAG16 (MIM612173; 2q34), which encodes a protein from the axoneme central apparatus, is associated with ciliary dyskinesia and mouse infertility due to impaired sperm motility.70 In humans, two fertile males heterozygous for the SPAG16 mutation were reported with only one having sperm count and motility levels significantly below the normal range. SPAG16 likely influences the stability of protein interactions in the sperm axoneme; however, it may be not significant enough to cause infertility.71
Another family of proteins important for axoneme stability and structure are the tektins, playing a fundamental role in ciliary movement. A screening of 90 nonsyndromic AZS patients for TEKT2 (MIM608953; 1p35.3-p34.1) revealed a heterozygous mutation in one patient that was maternally inherited.72 Dysplasia of the fibrous sheath (DFS) is a genetic sperm defect with hypertrophy and hyperplasia of random fibrous sheath associated with classical dynein arm deficiency in spermatozoa. Mutations in the cyclic-dependent protein kinases AKAP3 (MIM604689; 12p13.3) and AKAP4 (MIM300185; Xp11.2), the most abundant structural proteins of the sheath, were found in an AZS patient indicating the importance of motility of the sperm for fertility.73 These are examples of Mendelian inheritance of nonsyndromic male infertility. Most of the mutations are heterozygous displayed by the variable penetrance.
Currently, there are numerous mouse models described that manifest asthenozoospermia (Table 5). These models provide growing evidence that a myriad of the gene products influence spermatozoal motility in mammals, and defects in many human genes could result in spermatozoal immotility. Moreover, recent proteomic studies estimate that several hundred genes encode structural proteins of the flagellum, the axoneme fibrous sheath, outer dense fibers, and enzymes of the glycolytic machinery in spermatozoa and could therefore be considered as additional gene candidates for AZS in human males.74,75
Table 5.
Mouse male infertility and motility models.
| Mutant gene (gene symbol; alternate protein symbol) | Chr. | Reproductive phenotype | Fertility status | Ref. |
|---|---|---|---|---|
| Apolipoprotein B (Apob) | 12 | Oligoasthenozoospermia, decreased spermatozoan survival time and ability to fertilize ova | Infertile | 76 |
| Adenylate cyclase 3 (Adcy3) | 12 | Reduced sperm motility and defects in acrosome reaction | Subfertile | 77 |
| Adenylate cyclase 10 (Adcy10; soluble adenylate cyclase) | 1 | Abnormal sperm motility defect | Infertile | 78 |
| A kinase (PRKA) anchor protein 4 (Akap4) | X | Lack of progressive motility in spermatozoa | Infertile | 79 |
| ATP/GTP binding protein 1 (Agtpbp1; Nna1; pcd) | 13 | Spontaneous mutant; oligoasthenoteratozoospermia | Infertile | 80,81 |
| ATPase, Ca++ transporting, plasma membrane 4 (Atp2b4; PMCA4) | 1 | Defect in hyperactivated motility | Infertile | 82,83 |
| Bardet-Biedl syndrome 1 (Bbs1) | 19 | M390R knock in allele; absence of sperm flagella, motility defect | Infertile; Multiple defects | 84 |
| Bardet-Biedl syndrome 4 (Bbs4) | 9 | Absence of sperm flagella | Infertile, lethality variable; | 85 |
| Cation channel, sperm associated 1 (CatSper1) | 19 | Defects in hyperactivated motility and fertilization | Infertile | 86,87 |
| Cation channel, sperm associated 2 (CatSper2) | 2 | Defects in hyperactivated motility and fertilization | Infertile | 86 |
| Cation channel, sperm associated 3 (CatSper3) | 13 | Defects in hyperactivated motility and fertilization | Infertile | 88,89 |
| Cation channel, sperm associated 4 (CatSper4) | 4 | Defects in hyperactivated motility and fertilization | Infertile | 88,89 |
| CD59b antigen (Cd59b) | 2 | Teratozoospermia | Progressive infertility | 90 |
| Glycoprotein hormone α-subunit (Cga) | 4 | Hypogonadal due to FSH and LH deficiency | Infertile | 91 |
| F11 receptor (F11r; JAM-A) | 1 | Gene trap; decreased motility | Subfertile | 92 |
| Glyceraldehyde-3-phosphate dehy drogenase, spermatogenic (Gapdhs) | 7 | Motility defect | Infertile | 93 |
| Gene model 101 (Gm101; PCDP1) | 1 | Nm1054 mutant; lack of mature flagella, motility defect | Infertile, lethality; | 94 |
| Inositol polyphosphate-5-phosphatase (Inpp5b) | 4 | Asthenozoospermia, reduced ability of sperm to fertilize eggs, defects in fertilin β processing | Infertile | 95 |
| Lactate dehydrogenase C (Ldhc) | 7 | Rapid loss of motility, absence of hyperactivated motility, and capacitation defects | Variable infertility/subfertility | 96 |
| Low density lipoprotein receptor-related protein 8, apolipoprotein e receptor (Lrp8) | 4 | Defective osmotic regulation and motility in epididymal spermatozoa | Infertile | 97 |
| 5, 10-Methylenetetrahydrofolate reductase (Mthfr) | 4 | Reduced sperm numbers and abnormal spermatogenesis | Infertile | 98 |
| NOL1/NOP2/Sun domain family, member 7 (Nsun7) | 5 | ENU mutant; decreased motility | Infertile | 99 |
| Proprotein convertase subtilisin/kexin type 4 (Pcsk4; PC4) | 10 | Sperm have impaired fertilization | Infertile | 100 |
| Phospholipase A2, group IVC (Pla2g4c) | 7 | Asthenozoospermia, decreased fertilization capacity | Subfertile | 101 |
| Phosphatidylglycero-phosphate synthase 1 (Pgs1; ROSA22) | 11 | Spermatid flagella defect | Infertile | 102 |
| Phospholipid transfer protein (Pltp) | 2 | Asthenozoospermia | Subfertile | 103 |
| Polymerase (DNA-directed), delta 4 (Pold4) | 19 | Immotile spermatozoa | Infertile lethality; | 104 |
| Protein kinase, cAMP dependent, catalytic, α (Prkaca) | 8 | Ranting, sperm motility defects | Lethality; Fertility not assessed | 105 |
| Protein kinase, cAMP dependent regulatory, type I, α (Prkar1a) | 11 | Abnormal spermatozoa, oligozoospermia, decreased fertilization | Subfertile | 106 |
| Ros1 proto-oncogene (Ros1; c-ros) | 10 | Sperm motility defects | Infertile | 107,108 |
| Sirtuin 1 (Sirt1; SIR2α) | 10 | Failure of ovulation (F); azoospermia and asthenozoospermia (M) | Variable lethality; Infertile | 109 |
| Solute carrier family 9, member 10 (Slc9a10; sNHE) | 16 | Asthenozoospermia | Infertile | 110 |
| Sperm mitochondrion-associated cysteine-rich protein (Smcp) | 3 | 129 background; defects in sperm motility and migration into the oviduct, defects in fertilization | Infertile | 111 |
| Sperm associated antigen 6 (Spag6) | 16 | Dysmorphic and immotile sperm | Infertile postnatal lethal; | 112 |
| Sulfotransferase family 1E, member 1 (Sult1e1) | 5 | Progressive defects in sperm motility (M); abnormal ovulation and cumulus expansion (F) | Subfertile | 113,114 |
| Transaldolase 1 (Taldo1) | 7 | Motility defect, defective sperm mitochondrial potential | Infertile | 115 |
| Transcription factor 21 (Tcf21; Pod1) | 10 | Male to female sex reversal (M); both sex gonadal agenesis, germ cell loss | Postnatal lethal | 116 |
| Tektin 2 (Tekt2) | 4 | Spermatozoa have defective motility and ultrastructure | Infertile | 117 |
| Tektin 3 (Tekt3) | 11 | Sperm motility defects | Fertile | 118 |
| Tektin 4 (Tekt4) | 17 | Sperm motility defects and ultrastructural defects in flagellum | Subfertile | 119 |
| Testis expressed gene 18 (Tex18) | 10 | Asthenoteratozoospermia | Subfertile | 120 |
| Transforming growth factor β1 (Tgfb1) | 7 | Decreased testosterone production due to decreased LH, infrequent intromission, absence of ejaculation (M), SCID background; decreased ovulation frequency due to impaired LH surge, decreased progesterone synthesis, preimplantation embryo defects (F) | Subfertile (F) Infertile (M) | 121,122 |
| Testicular haploid expressed gene (Theg; kisimo) | 10 | Transgene insertion; abnormal elongated spermatids, asthenozoospermia | Infertile | 123 |
| Voltage-dependent Anion Channel 3 (Vdac3) | 8 | Immotile sperm, axonemal defects as sperm mature | Infertile | 124 |
Chr. abbreviates chromosome locations, and Ref. denotes corresponding references
Sperm count and morphology defects
Oligozoospermia (OZS), or reduced sperm count, is one of the most common categories of semen defects and is found in nearly half of infertile men. According to the World Health Organization, OZS is defined as a sperm concentration with less than 20 × 106 sperm/mL.125 However, several prominent studies indicate that the diagnosis of OZS must reflect the inherent intra- and inter-individual variability of sperm concentration, and therefore OZS should be divided into subcategories that are more specifically defined, that is, severe OZS with concentration < 1 × 106 sperm/mL, moderate OZS with 1–10 × 106 sperm/mL, and mild OZS >10 × 106 to 20 × 106 sperm/mL.126–128 Other major semen categories are absence of sperm (azoospermia) and morphology defects (teratozoospermia). Clinically, azoospermia overlaps with severe oligozoospermia. To date, only a limited number of genes are associated with low sperm count and male infertility.
One of the first successful studies reported is an association of nonsense mutations in SYCP3 with azoospermia and severe OZS in infertile patients.129 SYCP3 encodes a DNA-binding protein and is an important structural component of the synaptonemal complex, which mediates the synapsis of homologous chromosomes pairing during meiosis in male germ cells. The Sycp3 knockout mouse model exhibits meiosis arrest during spermatogenesis and resultant azoospermia and male infertility.130 The patients selected for this analysis represented a highly selected group of men with a meiotic arrest phenotypically similar to that observed in the mouse model.
Likewise, mutations in two related genes, CREM and FHL5 (formerly ACT, activator of CREM in testis), were shown to be responsible for azoospermia and OZS in men.131,132 CREM encodes a testis-specific transcription factor, cAMP responsive element modulator protein, and a CREM recognition site was found in the promoters of many testis-specific genes. Crem knockout mice show round spermatid arrest leading to male infertility.133,134 FHL5 protein contains a four-and-a-half LIM (FHL) domain that binds to CREM as a cofactor and modulates its activity. The Flh5 knockout mouse model demonstrates severe OZS and abnormal cell morphology, resulting in subfertility.135
Similarly, investigation of PRM1 gene mutations in infertile patients with severe OZS, abnormal morphology, and DNA damage revealed nonsense and missense alterations.136–138 PRM1 encodes protamine 1, which functions to compact, stabilize, and protect the DNA-protein complexes in the nucleus during spermatid development. The study was prompted by the Prm1 haploinsufficiency effect in mice, which causes abnormal sperm compaction, DNA damage, OZS, and male infertility.139 Recently, NALP14 mutations were identified in patients with AS and severe OZS.140 The study was initiated by delineation of gene-candidate disrupted by chromosome aberrations identified in patient with OZS.141 The gene was mapped to the chromosome 11p15 region. NALP14 displays testis-specific expression and encodes protein that plays important roles in apoptosis.
One noteworthy alternative approach for the OZS research is the RNA-based study of human kelch-like 10 (KLHL10) in infertile OZS patients.142 It employs the presence of stable mRNAs in human ejaculate spermatozoa. Our investigations focused on the detection of missense and splicing KLHL10 mutations in male germline mRNAs obtained from semen samples of OZS infertile men. We identified seven KLHL10 missense and splicing mutations out of 550 (1.2%) OZS patients examined. A concurrent study of 400 normozoospermic controls did not reveal these described KLHL10 mutations. Using reverse transcription-polymerase chain reaction (RT-PCR), we demonstrated that the splicing mutation caused a frameshift of the KLHL10 open reading frame. Pathogenic effects of the identified KLHL10 missense mutations were confirmed using a functional assay, an in vitro yeast 2-hybrid screen, which showed abnormal protein dimerization of mutant proteins. These findings were consistent with earlier mouse studies. KLHL10 is germ cell-specific protein involved in protein ubiquitination and is critical for maturation of mouse spermatozoa. Male mice heterozygous for a deletion in the Klhl10 gene demonstrate a failure of spermatozoal maturation, resulting in severe oligozoospermia.143,144
Despite a low frequency of described gene defects in OZS, significance of mutations in SYCP3, CREM1, FLH5, NALP14, PRM1, and KLHL10 were supported by statistical and/or functional evidence. However, many other investigations report limited statistical or functional evidence to support the pathological roles of the gene mutations in oligozoospermia; these include DDX25, PRM2, PRM3, TNP1, TNP2, UBE2B, USP26, FKBP6, mitochondrial ATPase8, and ATPase6.136,138,145–151
Teratozoospermia (TZS) is a condition characterized by the presence of sperm with abnormal morphology that affects fertility in males. Despite its relatively high occurrence, the progress toward understanding TZS has been slow. To date, there are only a few dozen genes that exhibit abnormal spermatozoal morphology in mouse knockout models,12 and even fewer genes show an association with TZS in infertile men.
One of the encouraging discoveries in the genetics of TZS and male infertility has been reported recently. Using a conventional identical-by-descent genetic approach, researchers reported that a testis-expressed gene, aurora kinase C (AURKC), is responsible for TZS in 10 patients with male infertility.152 The study demonstrated that all male patients carried same founder mutation, homozygous single-nucleotide deletion in the AURKC gene. These patients presented with nearly 100% morphologically abnormal spermatozoa, including oversized irregular heads, abnormal midpiece and acrosome, up to six flagella, and large-headed multiflagellar polyploid spermatozoa. In addition, AURKC protein plays an important role in meiotic division during spermatogenesis in mice.153
In another report, investigation focused on globozoospermia, rare form of TZS with characteristic round-headed spermatozoa that lack an acrosome and showed that the gene SPATA16 is responsible for globozoospermia in three related male infertile patients.154 The study described three affected brothers from a consanguineous family, in which each brother is homozygous for mutation in the SPATA16. The gene is testis-specific and SPATA16 specifically located in cytoplasm and involved in acrosome formation.155 However, a follow-up study of unrelated patients with complete or partial globozoospermia failed to identify SPATA16 mutation, highlighting the common difficulty in identifying genetic mutations in such highly heterogeneous etiology as TZS.
Y chromosome microdeletion
The Y chromosome comprises 60 million base pairs with a short arm (Yp) and a long arm (Yq). The sex determining region (SRY) is located on Yp and is an essential member of the group of genes that ultimately determines the fate of the bipotential gonad.156 The Y chromosome contains vital components needed for male differentiation and sperm function. The azoospermia factor region (AZF) on the long arm of the Y chromosome (Yq) is responsible for sperm development. The male-specific Y is the chromosomal material that bridges the two polar pseudoautosomal regions and is unique in the human genome. It comprises approximately 95% of the Y chromosome and houses multiple genes that help drive spermatogenesis such as DAZ, USP9Y, RBMY1, and BPY2.157 The AZF region is subdivided by location into AZFa, AZFb, and AZFc, which correspond to proximal, middle, and distal portions of the chromosome.5 Deletions in these locations are responsible for varying degrees of spermatogenic dysfunction. Entire microdeletions of AZFa or AZFb regions of the Y chromosome portends an exceptionally poor prognosis in sperm retrieval, such that microscopic sperm extraction is predictably negative.158
Depending on the severity of the deletion, a microdeletion in AZFc can result in a spectrum of spermatogenic deficiency including oligospermia and azoospermia.158 Deletions in the Y(q) are too small to be detected with a karyotype and thus are termed microdeletions. These deletions are identified using polymerase chain reaction techniques to analyze sequence tagged sites. Indications for testing AZF microdeletions are sperm concentrations less then 5 million/mL. Importantly, male offspring of patients with Y microdeletions will inherit the abnormal gene, rendering them likely to be infertile. Thus, ICSI with preimplantation genetic diagnosis (PGD) should be discussed.
Conclusions
As additional mouse models are created and high throughput screening approaches are used, we are beginning to understand more about the Mendelian genetics of male infertility. While it is difficult to fully understand the impact of Mendelian genetics on male infertility, the quick evolution of advanced reproductive therapies has allowed the achievement of biological paternity by men, who, by nature’s standard, would have never been permitted to procreate. IVF/ICSI has allowed the technical potential for these men with severe male factor infertility the ability to father children. However, finding a genetic basis and understanding this mishap before proceeding with any surgical or in vitro technology allows appropriate counseling of the couple with regard to immediate and long-term issues not only for the parents, but also for the future offspring that they should be aware of before moving forward.
Acknowledgments
This study was supported in part by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (P01HD36289), D.J.L. and M.M.M.; 1R01DK078121 to D.J.L.; and 1K12DK083014 Multidisciplinary K12 Urology Research Career Development Program at Baylor to D.J.L., K.H., and C.J.J., from the National Institute of Kidney and Digestive Diseases., and Ko8HD58073 to A.N.Y. R.L.N. is supported by the Edward J. and Josephine G. Hudson Scholar Fund and the Baylor College of Medicine Medical Scientist Training Program.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Lipshultz LI, Lamb DJ. Risk of transmission of genetic diseases by assisted reproduction. Nat Clin Pract Urol. 2007;4:460–461. doi: 10.1038/ncpuro0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Braekeleer M, Dao TN. Cytogenetic studies in male infertility: a review. Hum Reprod. 1991;6:245–250. [PubMed] [Google Scholar]
- 3.Bogatcheva NV, Truong A, Feng S, et al. GREAT/LGR8 is the only receptor for insulin-like 3 peptide. Mol Endocrinol. 2003;17:2639–2646. doi: 10.1210/me.2003-0096. [DOI] [PubMed] [Google Scholar]
- 4.Foresta C, Ferlin A, Gianaroli L, Dallapiccola B. Guidelines for the appropriate use of genetic tests in infertile couples. Eur J Hum Genet. 2002;10:303–312. doi: 10.1038/sj.ejhg.5200805. [DOI] [PubMed] [Google Scholar]
- 5.Kuroda-Kawaguchi T, Skaletsky H, Brown LG, et al. The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat Genet. 2001;29:279–286. doi: 10.1038/ng757. [DOI] [PubMed] [Google Scholar]
- 6.Mau-Holzmann UA. Somatic chromosomal abnormalities in infertile men and women. Cytogenet Genome Res. 2005;111:317–336. doi: 10.1159/000086906. [DOI] [PubMed] [Google Scholar]
- 7.Vogt PH. Genomic heterogeneity and instability of the AZF locus on the human Y chromosome. Mol Cell Endocrinol. 2004;224:1–9. doi: 10.1016/j.mce.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Vogt PH, Edelmann A, Kirsch S, et al. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet. 1996;5:933–943. doi: 10.1093/hmg/5.7.933. [DOI] [PubMed] [Google Scholar]
- 9.Ferlin A, Vinanzi C, Garolla A, et al. Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations. Clin Endocrinol (Oxf) 2006;65:606–610. doi: 10.1111/j.1365-2265.2006.02635.x. [DOI] [PubMed] [Google Scholar]
- 10.Ferlin A, Arredi B, Speltra E, et al. Molecular and clinical characterization of Y chromosome microdeletions in infertile men: a 10-year experience in Italy. J Clin Endocrinol Metab. 2007;92:762–770. doi: 10.1210/jc.2006-1981. [DOI] [PubMed] [Google Scholar]
- 11.Foresta C, Garolla A, Bartolon L, et al. Genetic abnormalities among severely oligospermic men who are candidates for intracytoplasmic sperm injection. J Clin Endocrinol Metab. 2005;90:152–156. doi: 10.1210/jc.2004-1469. [DOI] [PubMed] [Google Scholar]
- 12.Matzuk MM, Lamb DJ. The biology of infertility: research advances and clinical challenges. Nat Med. 2008;14:1197–1213. doi: 10.1038/nm.f.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigman M, Jarow JP. Endocrine evaluation of infertile men. Urology. 1997;50:659–664. doi: 10.1016/S0090-4295(97)00340-3. [DOI] [PubMed] [Google Scholar]
- 14.Poongothai J, Gopenath TS, Manonayaki S. Genetics of human male infertility. Singapore Med J. 2009;50:336–347. [PubMed] [Google Scholar]
- 15.Latronico AC. The neurokinin B pathway in human reproduction. Nat Genet. 2009;41:269–270. doi: 10.1038/ng0309-269. [DOI] [PubMed] [Google Scholar]
- 16.Tsai PS, Gill JC. Mechanisms of disease: insights into X-linked and autosomal-dominant Kallmann syndrome. Nat Clin Pract Endocrinol Metab. 2006;2:160–171. doi: 10.1038/ncpendmet0119. [DOI] [PubMed] [Google Scholar]
- 17.Hou JW, Tsai WY, Wang TR. Detection of KAL-1 gene deletion with fluorescence in situ hybridization. J Formos Med Assoc. 1999;98:448–451. [PubMed] [Google Scholar]
- 18.Waldstreicher J, Seminara SB, Jameson JL, et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81:4388–4395. doi: 10.1210/jcem.81.12.8954047. [DOI] [PubMed] [Google Scholar]
- 19.Oliveira LM, Seminara SB, Beranova M, et al. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86:1532–1538. doi: 10.1210/jcem.86.4.7420. [DOI] [PubMed] [Google Scholar]
- 20.Dode C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 21.Sato N, Katsumata N, Kagami M, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab. 2004;89:1079–1088. doi: 10.1210/jc.2003-030476. [DOI] [PubMed] [Google Scholar]
- 22.Dode C, Teixeira L, Levilliers J, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. doi: 10.1371/journal.pgen.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galani A, Kitsiou-Tzeli S, Sofokleous C, et al. Androgen insensitivity syndrome: clinical features and molecular defects. Hormones (Athens) 2008;7:217–229. doi: 10.14310/horm.2002.1201. [DOI] [PubMed] [Google Scholar]
- 24.Hiort O, Naber SP, Lehners A, et al. The role of androgen receptor gene mutations in male breast carcinoma. J Clin Endocrinol Metab. 1996;81:3404–3407. doi: 10.1210/jcem.81.9.8784104. [DOI] [PubMed] [Google Scholar]
- 25.Hiort O, Holterhus PM, Horter T, et al. Significance of mutations in the androgen receptor gene in males with idiopathic infertility. J Clin Endocrinol Metab. 2000;85:2810–2815. doi: 10.1210/jcem.85.8.6713. [DOI] [PubMed] [Google Scholar]
- 26.Ghali SA, Gottlieb B, Lumbroso R, et al. The use of androgen receptor amino/carboxyl-terminal interaction assays to investigate androgen receptor gene mutations in subjects with varying degrees of androgen insensitivity. J Clin Endocrinol Metab. 2003;88:2185–2193. doi: 10.1210/jc.2002-021324. [DOI] [PubMed] [Google Scholar]
- 27.Quigley CA, Tan JA, He B, et al. Partial androgen insensitivity with phenotypic variation caused by androgen receptor mutations that disrupt activation function 2 and the NH(2)- and carboxyl-terminal interaction. Mech Ageing Dev. 2004;125:683–695. doi: 10.1016/j.mad.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Umar A, Berrevoets CA, Van NM, et al. Functional analysis of a novel androgen receptor mutation, Q902K, in an individual with partial androgen insensitivity. J Clin Endocrinol Metab. 2005;90:507–515. doi: 10.1210/jc.2004-0057. [DOI] [PubMed] [Google Scholar]
- 29.Adachi M, Takayanagi R, Tomura A, et al. Androgen-insensitivity syndrome as a possible coactivator disease. N Engl J Med. 2000;343:856–862. doi: 10.1056/NEJM200009213431205. [DOI] [PubMed] [Google Scholar]
- 30.Holterhus PM, Werner R, Hoppe U, et al. Molecular features and clinical phenotypes in androgen insensitivity syndrome in the absence and presence of androgen receptor gene mutations. J Mol Med. 2005;83:1005–1013. doi: 10.1007/s00109-005-0704-y. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 32.La Spada AR, Wilson EM, Lubahn DB, et al. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 33.Thomas M, Dadgar N, Aphale A, et al. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem. 2004;279:8389–8395. doi: 10.1074/jbc.M311761200. [DOI] [PubMed] [Google Scholar]
- 34.Thomas M, Harrell JM, Morishima Y, et al. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum Mol Genet. 2006;15:1876–1883. doi: 10.1093/hmg/ddl110. [DOI] [PubMed] [Google Scholar]
- 35.Lieberman AP, Fischbeck KH. Triplet repeat expansion in neuromuscular disease. Muscle Nerve. 2000;23:843–850. doi: 10.1002/(sici)1097-4598(200006)23:6<843::aid-mus2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 36.Dowsing AT, Yong EL, Clark M, et al. Linkage between male infertility and trinucleotide repeat expansion in the androgen-receptor gene. Lancet. 1999;354:640–643. doi: 10.1016/s0140-6736(98)08413-x. [DOI] [PubMed] [Google Scholar]
- 37.Dadze S, Wieland C, Jakubiczka S, et al. The size of the CAG repeat in exon 1 of the androgen receptor gene shows no significant relationship to impaired spermatogenesis in an infertile Caucasoid sample of German origin. Mol Hum Reprod. 2000;6:207–214. doi: 10.1093/molehr/6.3.207. [DOI] [PubMed] [Google Scholar]
- 38.Palazzolo I, Gliozzi A, Rusmini P, et al. The role of the polyglutamine tract in androgen receptor. J Steroid Biochem Mol Biol. 2008;108:245–253. doi: 10.1016/j.jsbmb.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 39.Fischbeck KH. Polyglutamine expansion neurodegenerative disease. Brain Res Bull. 2001;56:161–163. doi: 10.1016/s0361-9230(01)00577-9. [DOI] [PubMed] [Google Scholar]
- 40.Lieberman AP, Trojanowski JQ, Leonard DG, et al. Ataxin 1 and ataxin 3 in neuronal intranuclear inclusion disease. Ann Neurol. 1999;46:271–273. doi: 10.1002/1531-8249(199908)46:2<271::aid-ana21>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 41.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 42.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Ann Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 43.Cram DS, Song B, McLachlan RI, Trounson AO. CAG trinucleotide repeats in the androgen receptor gene of infertile men exhibit stable inheritance in female offspring conceived after ICSI. Mol Hum Reprod. 2000;6:861–866. doi: 10.1093/molehr/6.9.861. [DOI] [PubMed] [Google Scholar]
- 44.Kunej T, Teran N, Zorn B, Peterlin B. CTG amplification in the DM1PK gene is not associated with idiopathic male subfertility. Hum Reprod. 2004;19:2084–2087. doi: 10.1093/humrep/deh382. [DOI] [PubMed] [Google Scholar]
- 45.Pan H, Li YY, Li TC, et al. Increased (CTG/CAG)(n) lengths in myotonic dystrophy type 1 and Machado-Joseph disease genes in idiopathic azoospermia patients. Hum Reprod. 2002;17:1578–1583. doi: 10.1093/humrep/17.6.1578. [DOI] [PubMed] [Google Scholar]
- 46.Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science (New York, NY) 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 47.Lofrano-Porto A, Barra GB, Giacomini LA, et al. Luteinizing hormone beta mutation and hypogonadism in men and women. N Engl J Med. 2007;357:897–904. doi: 10.1056/NEJMoa071999. [DOI] [PubMed] [Google Scholar]
- 48.Grigorova M, Punab M, Ausmees K, Laan M. FSHB promoter polymorphism within evolutionary conserved element is associated with serum FSH level in men. Hum Reprod. 2008;23:2160–2166. doi: 10.1093/humrep/den216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whittington JE, Holland AJ, Webb T, et al. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet. 2001;38:792–798. doi: 10.1136/jmg.38.11.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crino A, Schiaffini R, Ciampalini P, et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. 2003;162:327–333. doi: 10.1007/s00431-002-1132-4. [DOI] [PubMed] [Google Scholar]
- 51.Fillion M, Deal CL, Van Vliet G. Normal minipuberty of infancy in boys with Prader-Willi syndrome. J Pediatr. 2006;149:874–876. doi: 10.1016/j.jpeds.2006.08.077. [DOI] [PubMed] [Google Scholar]
- 52.Eiholzer U, l’;Allemand D, Rousson V, et al. Hypothalamic and gonadal components of hypogonadism in boys with Prader-Labhart-Willi syndrome. J Clin Endocrinol Metab. 2006;91:892–898. doi: 10.1210/jc.2005-0902. [DOI] [PubMed] [Google Scholar]
- 53.Vogels A, Moerman P, Frijns JP, Bogaert GA. Testicular histology in boys with Prader-Willi syndrome: fertile or infertile? J Urol. 2008;180(4 Suppl):1800–1804. doi: 10.1016/j.juro.2008.03.113. [DOI] [PubMed] [Google Scholar]
- 54.Katcher ML, Bargman GJ, Gilbert EF, Opitz JM. Absence of spermatogonia in the Prader-Willi syndrome. Eur J Pediatr. 1977;124:257–260. doi: 10.1007/BF00441933. [DOI] [PubMed] [Google Scholar]
- 55.Donlan MA, Dolan CR, Metcalf MJ, et al. Trisomy Xq in a male: the isochromosome X Klinefelter syndrome. Am J Med Genet. 1987;27:189–194. doi: 10.1002/ajmg.1320270120. [DOI] [PubMed] [Google Scholar]
- 56.Arps S, Koske-Westphal T, Meinecke P, et al. Isochromosome Xq in Klinefelter syndrome: report of 7 new cases. Am J Med Genet. 1996;64:580–582. doi: 10.1002/(SICI)1096-8628(19960906)64:4<580::AID-AJMG10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 57.Kalousek D, Biddle CJ, Rudner M, et al. 47,X,i(Xq),Y karyotype in Klinefelter’s syndrome. Hum Genet. 1978;43:107–110. doi: 10.1007/BF00396486. [DOI] [PubMed] [Google Scholar]
- 58.Moskowitz SM, Gibson RL, Effmann EL. Cystic fibrosis lung disease: genetic influences, microbial interactions, and radiological assessment. Pediatr Radiol. 2005;35:739–757. doi: 10.1007/s00247-005-1445-3. [DOI] [PubMed] [Google Scholar]
- 59.McKone EF, Emerson SS, Edwards KL, Aitken ML. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet. 2003;361:1671–1676. doi: 10.1016/S0140-6736(03)13368-5. [DOI] [PubMed] [Google Scholar]
- 60.Chillon M, Casals T, Mercier B, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475–1480. doi: 10.1056/NEJM199506013322204. [DOI] [PubMed] [Google Scholar]
- 61.Friedman KJ, Teichtahl H, De Kretser DM, et al. Screening Young syndrome patients for CFTR mutations. Am J Respir Crit Care Med. 1995;152(4 Pt 1):1353–1357. doi: 10.1164/ajrccm.152.4.7551394. [DOI] [PubMed] [Google Scholar]
- 62.Le Lannou D, Jezequel P, Blayau M, et al. Obstructive azoospermia with agenesis of vas deferens or with bronchiectasia (Young’s syndrome): a genetic approach. Hum Reprod. 1995;10:338–341. doi: 10.1093/oxfordjournals.humrep.a135939. [DOI] [PubMed] [Google Scholar]
- 63.Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 64.Zuccarello D, Ferlin A, Cazzadore C, et al. Mutations in dynein genes in patients affected by isolated non-syndromic asthenozoospermia. Hum Reprod. 2008;23:1957–1962. doi: 10.1093/humrep/den193. [DOI] [PubMed] [Google Scholar]
- 65.Cook SP, Brokaw CJ, Muller CH, Babcock DF. Sperm chemotaxis: egg peptides control cytosolic calcium to regulate flagellar responses. Dev Biol. 1994;165:10–19. doi: 10.1006/dbio.1994.1229. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, Malekpour M, Al-Madani N, et al. Sensorineural deafness and male infertility: a contiguous gene deletion syndrome. J Med Genet. 2007;44:233–240. doi: 10.1136/jmg.2006.045765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avidan N, Tamary H, Dgany O, et al. CATSPER2, a human autosomal nonsyndromic male infertility gene. Eur J Hum Genet. 2003;11:497–502. doi: 10.1038/sj.ejhg.5200991. [DOI] [PubMed] [Google Scholar]
- 68.Avenarius MR, Hildebrand MS, Zhang Y, et al. Human male infertility caused by mutations in the CATSPER1 channel protein. Am J Hum Genet. 2009;84:505–510. doi: 10.1016/j.ajhg.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ren D, Navarro B, Perez G, et al. A sperm ion channel required for sperm motility and male fertility. Nature. 2001;413:603–609. doi: 10.1038/35098027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Z, Kostetskii I, Tang W, et al. Deficiency of SPAG16L causes male infertility associated with impaired sperm motility. Biol Reprod. 2006;74:751–759. doi: 10.1095/biolreprod.105.049254. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Z, Zariwala MA, Mahadevan MM, et al. A heterozygous mutation disrupting the SPAG16 gene results in biochemical instability of central apparatus components of the human sperm axoneme. Biol Reprod. 2007;77:864–871. doi: 10.1095/biolreprod.107.063206. [DOI] [PubMed] [Google Scholar]
- 72.Zuccarello D, Ferlin A, Garolla A, et al. A possible association of a human tektin-t gene mutation (A229V) with isolated non-syndromic asthenozoospermia: case report. Hum Reprod. 2008;23:996–1001. doi: 10.1093/humrep/dem400. [DOI] [PubMed] [Google Scholar]
- 73.Baccetti B, Collodel G, Estenoz M, et al. Gene deletions in an infertile man with sperm fibrous sheath dysplasia. Hum Reprod. 2005;20:2790–2794. doi: 10.1093/humrep/dei126. [DOI] [PubMed] [Google Scholar]
- 74.Cao W, Gerton GL, Moss SB. Proteomic profiling of accessory structures from the mouse sperm flagellum. Mol Cell Proteomics. 2006;5:801–810. doi: 10.1074/mcp.M500322-MCP200. [DOI] [PubMed] [Google Scholar]
- 75.Martinez-Heredia J, de Mateo S, Vidal-Taboada JM, et al. Identification of proteomic differences in asthenozoospermic sperm samples. Hum Reprod. 2008;23:783–791. doi: 10.1093/humrep/den024. [DOI] [PubMed] [Google Scholar]
- 76.Huang LS, Voyiaziakis E, Chen HL, et al. A novel functional role for apolipoprotein B in male infertility in heterozygous apolipoprotein B knockout mice. Proc Natl Acad Sci USA. 1996;93:10903–10907. doi: 10.1073/pnas.93.20.10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Livera G, Xie F, Garcia MA, et al. Inactivation of the mouse adenylyl cyclase 3 gene disrupts male fertility and spermatozoon function. Mol Endocrinol. 2005;19:1277–1290. doi: 10.1210/me.2004-0318. [DOI] [PubMed] [Google Scholar]
- 78.Esposito G, Jaiswal BS, Xie F, et al. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc Natl Acad Sci USA. 2004;101:2993–2998. doi: 10.1073/pnas.0400050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miki K, Willis WD, Brown PR, et al. Targeted disruption of the Akap4 gene causes defects in sperm flagellum and motility. Dev Biol. 2002;248:331–342. doi: 10.1006/dbio.2002.0728. [DOI] [PubMed] [Google Scholar]
- 80.Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci USA. 1976;73:208–212. doi: 10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fernandez-Gonzalez A, La Spada AR, Treadaway J, et al. Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science (New York, NY) 2002;295:1904–1906. doi: 10.1126/science.1068912. [DOI] [PubMed] [Google Scholar]
- 82.Okunade GW, Miller ML, Pyne GJ, et al. Targeted ablation of plasma membrane Ca2+-ATPase (PMCA) 1 and 4 indicates a major housekeeping function for PMCA1 and a critical role in hyperactivated sperm motility and male fertility for PMCA4. J Biol Chem. 2004;279:33742–33750. doi: 10.1074/jbc.M404628200. [DOI] [PubMed] [Google Scholar]
- 83.Schuh K, Cartwright EJ, Jankevics E, et al. Plasma membrane Ca2+ ATPase 4 is required for sperm motility and male fertility. J Biol Chem. 2004;279:28220–28226. doi: 10.1074/jbc.M312599200. [DOI] [PubMed] [Google Scholar]
- 84.Davis RE, Swiderski RE, Rahmouni K, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci USA. 2007;104:19422–19427. doi: 10.1073/pnas.0708571104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mykytyn K, Mullins RF, Andrews M, et al. Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA. 2004;101:8664–8669. doi: 10.1073/pnas.0402354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carlson AE, Quill TA, Westenbroek RE, et al. Identical phenotypes of CatSper1 and CatSper2 null sperm. J Biol Chem. 2005;280:32238–32244. doi: 10.1074/jbc.M501430200. [DOI] [PubMed] [Google Scholar]
- 87.Carlson AE, Westenbroek RE, Quill T, et al. CatSper1 required for evoked Ca2+ entry and control of flagellar function in sperm. Proc Natl Acad Sci USA. 2003;100:14864–14868. doi: 10.1073/pnas.2536658100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jin J, Jin N, Zheng H, et al. Catsper3 and catsper4 are essential for sperm hyperactivated motility and male fertility in the mouse. Biol Reprod. 2007;77:37–44. doi: 10.1095/biolreprod.107.060186. [DOI] [PubMed] [Google Scholar]
- 89.Qi H, Moran MM, Navarro B, et al. All four CatSper ion channel proteins are required for male fertility and sperm cell hyperactivated motility. Proc Natl Acad Sci USA. 2007;104:1219–1223. doi: 10.1073/pnas.0610286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qin X, Krumrei N, Grubissich L, et al. Deficiency of the mouse complement regulatory protein mCd59b results in spontaneous hemolytic anemia with platelet activation and progressive male infertility. Immunity. 2003;18:217–227. doi: 10.1016/s1074-7613(03)00022-0. [DOI] [PubMed] [Google Scholar]
- 91.Kendall SK, Samuelson LC, Saunders TL, et al. Targeted disruption of the pituitary glycoprotein hormone α-subunit produces hypogonadal and hypothyroid mice. Genes Dev. 1995;9:2007–2019. doi: 10.1101/gad.9.16.2007. [DOI] [PubMed] [Google Scholar]
- 92.Shao M, Ghosh A, Cooke VG, et al. JAM-A is present in mammalian spermatozoa where it is essential for normal motility. Dev Biol. 2008;313:246–255. doi: 10.1016/j.ydbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miki K, Qu W, Goulding EH, et al. Glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme, is required for sperm motility and male fertility. Proc Natl Acad Sci USA. 2004;101:16501–16506. doi: 10.1073/pnas.0407708101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee L, Campagna DR, Pinkus JL, et al. Primary ciliary dyskinesia in mice lacking the novel ciliary protein Pcdp1. Mol Cell Biol. 2008;28:949–957. doi: 10.1128/MCB.00354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hellsten E, Evans JP, Bernard DJ, et al. Disrupted sperm function and fertilin beta processing in mice deficient in the inositol polyphosphate 5-phosphatase Inpp5b. Dev Biol. 2001;240:641–653. doi: 10.1006/dbio.2001.0476. [DOI] [PubMed] [Google Scholar]
- 96.Odet F, Duan C, Willis WD, et al. Expression of the gene for mouse lactate dehydrogenase C (Ldhc) is required for male fertility. Biol Reprod. 2008;79:26–34. doi: 10.1095/biolreprod.108.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Andersen OM, Yeung CH, Vorum H, et al. Essential role of the apolipoprotein E receptor-2 in sperm development. J Biol Chem. 2003;278:23989–23995. doi: 10.1074/jbc.M302157200. [DOI] [PubMed] [Google Scholar]
- 98.Kelly TL, Neaga OR, Schwahn BC, et al. Infertility in 5,10-methylenetetrahydrofolate reductase (MTHFR)-deficient male mice is partially alleviated by lifetime dietary betaine supplementation. Biol Reprod. 2005;72:667–677. doi: 10.1095/biolreprod.104.035238. [DOI] [PubMed] [Google Scholar]
- 99.Harris T, Marquez B, Suarez S, Schimenti J. Sperm motility defects and infertility in male mice with a mutation in Nsun7, a member of the Sun domain-containing family of putative RNA methyltransferases. Biol Reprod. 2007;77:376–382. doi: 10.1095/biolreprod.106.058669. [DOI] [PubMed] [Google Scholar]
- 100.Mbikay M, Tadros H, Ishida N, et al. Impaired fertility in mice deficient for the testicular germ-cell protease PC4. Proc Natl Acad Sci USA. 1997;94:6842–6846. doi: 10.1073/pnas.94.13.6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bao S, Miller DJ, Ma Z, et al. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Campbell PK, Waymire KG, Heier RL, et al. Mutation of a novel gene results in abnormal development of spermatid flagella, loss of intermale aggression and reduced body fat in mice. Genetics. 2002;162:307–320. doi: 10.1093/genetics/162.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Drouineaud V, Lagrost L, Klein A, et al. Phospholipid transfer protein deficiency reduces sperm motility and impairs fertility of mouse males. FASEB J. 2006;20:794–796. doi: 10.1096/fj.05-5385fje. [DOI] [PubMed] [Google Scholar]
- 104.Kobayashi Y, Watanabe M, Okada Y, et al. Hydrocephalus, situs inversus, chronic sinusitis, and male infertility in DNA polymerase lambda-deficient mice: possible implication for the pathogenesis of immotile cilia syndrome. Mol Cell Biol. 2002;22:2769–2776. doi: 10.1128/MCB.22.8.2769-2776.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Skalhegg BS, Huang Y, Su T, et al. Mutation of the calpha subunit of PKA leads to growth retardation and sperm dysfunction. Mol Endocrinol. 2002;16:630–639. doi: 10.1210/mend.16.3.0793. [DOI] [PubMed] [Google Scholar]
- 106.Burton KA, McDermott DA, Wilkes D, et al. Haploinsufficiency at the protein kinase A RI alpha gene locus leads to fertility defects in male mice and men. Mol Endocrinol. 2006;20:2504–2513. doi: 10.1210/me.2006-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yeung CH, Sonnenberg-Riethmacher E, Cooper TG. Infertile spermatozoa of c-ros tyrosine kinase receptor knockout mice show flagellar angulation and maturational defects in cell volume regulatory mechanisms. Biol Reprod. 1999;61:1062–1069. doi: 10.1095/biolreprod61.4.1062. [DOI] [PubMed] [Google Scholar]
- 108.Yeung CH, Wagenfeld A, Nieschlag E, Cooper TG. The cause of infertility of male c-ros tyrosine kinase receptor knockout mice. Biol Reprod. 2000;63:612–618. doi: 10.1095/biolreprod63.2.612. [DOI] [PubMed] [Google Scholar]
- 109.McBurney MW, Yang X, Jardine K, et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang D, King SM, Quill TA, et al. A new sperm-specific Na+/H + exchanger required for sperm motility and fertility. Nat Cell Biol. 2003;5:1117–1122. doi: 10.1038/ncb1072. [DOI] [PubMed] [Google Scholar]
- 111.Nayernia K, I, Adham M, Burkhardt-Gottges E, et al. Asthenozoospermia in mice with targeted deletion of the sperm mitochondrion-associated cysteine-rich protein (Smcp) gene. Mol Cell Biol. 2002;22:3046–3052. doi: 10.1128/MCB.22.9.3046-3052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sapiro R, Kostetskii I, Olds-Clarke P, et al. Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm-associated antigen 6. Mol Cell Biol. 2002;22:6298–6305. doi: 10.1128/MCB.22.17.6298-6305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Qian YM, Sun XJ, Tong MH, et al. Targeted disruption of the mouse estrogen sulfotransferase gene reveals a role of estrogen metabolism in intracrine and paracrine estrogen regulation. Endocrinology. 2001;142:5342–5350. doi: 10.1210/endo.142.12.8540. [DOI] [PubMed] [Google Scholar]
- 114.Gershon E, Hourvitz A, Reikhav S, et al. Low expression of COX-2, reduced cumulus expansion, and impaired ovulation in SULT1E1-deficient mice. FASEB J. 2007;21:1893–1901. doi: 10.1096/fj.06-7688com. [DOI] [PubMed] [Google Scholar]
- 115.Perl A, Qian Y, Chohan KR, et al. Transaldolase is essential for maintenance of the mitochondrial transmembrane potential and fertility of spermatozoa. Proc Natl Acad Sci USA. 2006;103:14813–14818. doi: 10.1073/pnas.0602678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cui S, Ross A, Stallings N, et al. Disrupted gonadogenesis and male-to-female sex reversal in Pod1 knockout mice. Development. 2004;131:4095–4105. doi: 10.1242/dev.01266. [DOI] [PubMed] [Google Scholar]
- 117.Tanaka H, Iguchi N, Toyama Y, et al. Mice deficient in the axonemal protein Tektin-t exhibit male infertility and immotile-cilium syndrome due to impaired inner arm dynein function. Mol Cell Biol. 2004;24:7958–7964. doi: 10.1128/MCB.24.18.7958-7964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Roy A, Lin YN, Agno JE, et al. Tektin 3 is required for progressive sperm motility in mice. Mol Reprod Dev. 2009;76:453–459. doi: 10.1002/mrd.20957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Roy A, Lin YN, Agno JE, et al. Absence of tektin 4 causes asthenozoospermia and subfertility in male mice. FASEB J. 2007;21:1013–1025. doi: 10.1096/fj.06-7035com. [DOI] [PubMed] [Google Scholar]
- 120.Jaroszynski L, Dev A, Li M, et al. Asthenoteratozoospermia in mice lacking testis expressed gene 18 (Tex18) Mol Hum Reprod. 2007;13:155–163. doi: 10.1093/molehr/gal107. [DOI] [PubMed] [Google Scholar]
- 121.Ingman WV, Robker RL, Woittiez K, Robertson SA. Null mutation in transforming growth factor beta1 disrupts ovarian function and causes oocyte incompetence and early embryo arrest. Endocrinology. 2006;147:835–845. doi: 10.1210/en.2005-1189. [DOI] [PubMed] [Google Scholar]
- 122.Ingman WV, Robertson SA. Transforming growth factor-beta1 null mutation causes infertility in male mice associated with testosterone deficiency and sexual dysfunction. Endocrinology. 2007;148:4032–4043. doi: 10.1210/en.2006-1759. [DOI] [PubMed] [Google Scholar]
- 123.Yanaka N, Kobayashi K, Wakimoto K, et al. Insertional mutation of the murine kisimo locus caused a defect in spermatogenesis. J Biol Chem. 2000;275:14791–14794. doi: 10.1074/jbc.C901047199. [DOI] [PubMed] [Google Scholar]
- 124.Sampson MJ, Decker WK, Beaudet AL, et al. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J Biol Chem. 2001;276:39206–39212. doi: 10.1074/jbc.M104724200. [DOI] [PubMed] [Google Scholar]
- 125.WHO. Towards more objectivity in diagnosis and management of male infertility. Int J Androl. 1987;7(Suppl):1–53. [Google Scholar]
- 126.Nallella KP, Sharma RK, Aziz N, Agarwal A. Significance of sperm characteristics in the evaluation of male infertility. Fertil Steril. 2006;85:629–634. doi: 10.1016/j.fertnstert.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 127.Guzick DS, Overstreet JW, Factor-Litvak P, et al. Sperm morphology, motility, and concentration in fertile and infertile men. N Engl J Med. 2001;345:1388–1393. doi: 10.1056/NEJMoa003005. [DOI] [PubMed] [Google Scholar]
- 128.WHO. WHO (World Health Organization) Laboratory Manual for the Examination of Human Semen and Sperm-Cervical Mucus Interaction. 3. New York, NY, USA: Published on behalf of the World Health Organization by Cambridge University Press; Cambridge, England: 1992. Special Programme of Research Development and Research Training in Human Reproduction. [Google Scholar]
- 129.Miyamoto T, Hasuike S, Yogev L, et al. Azoospermia in patients heterozygous for a mutation in SYCP3. Lancet. 2003;362:1714–1719. doi: 10.1016/S0140-6736(03)14845-3. [DOI] [PubMed] [Google Scholar]
- 130.Yuan L, Liu JG, Zhao J, et al. The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol Cell. 2000;5:73–83. doi: 10.1016/s1097-2765(00)80404-9. [DOI] [PubMed] [Google Scholar]
- 131.Vouk K, Hudler P, Strmsnik L, et al. Combinations of genetic changes in the human cAMP-responsive element modulator gene: a clue towards understanding some forms of male infertility? Mol Hum Reprod. 2005;11:567–574. doi: 10.1093/molehr/gah209. [DOI] [PubMed] [Google Scholar]
- 132.Christensen GL, Wooding SP, Ivanov IP, et al. Sequencing and haplotype analysis of the activator of CREM in the testis (ACT) gene in populations of fertile and infertile males. Mol Hum Reprod. 2006;12:257–262. doi: 10.1093/molehr/gal006. [DOI] [PubMed] [Google Scholar]
- 133.Blendy JA, Kaestner KH, Weinbauer GF, et al. Severe impairment of spermatogenesis in mice lacking the CREM gene. Nature. 1996;380:162–165. doi: 10.1038/380162a0. [DOI] [PubMed] [Google Scholar]
- 134.Nantel F, Monaco L, Foulkes NS, et al. Spermiogenesis deficiency and germ-cell apoptosis in CREM-mutant mice. Nature. 1996;380:159–162. doi: 10.1038/380159a0. [DOI] [PubMed] [Google Scholar]
- 135.Kotaja N, De Cesare D, Macho B, et al. Abnormal sperm in mice with targeted deletion of the act (activator of cAMP-responsive element modulator in testis) gene. Proc Natl Acad Sci USA. 2004;101:10620–10625. doi: 10.1073/pnas.0401947101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ravel C, Chantot-Bastaraud S, El Houate B, et al. Mutations in the protamine 1 gene associated with male infertility. Mol Hum Reprod. 2007;13(7):461–464. doi: 10.1093/molehr/gam031. [DOI] [PubMed] [Google Scholar]
- 137.Iguchi N, Yang S, Lamb DJ, Hecht NB. A protamine SNP: one genetic cause of male infertility. J Med Genet. 2006;13:83–92. doi: 10.1136/jmg.2005.037168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Imken L, Rouba H, El Houate B, et al. Mutations in the protamine locus: association with spermatogenic failure? Mol Hum Reprod. 2009;15:733–738. doi: 10.1093/molehr/gap056. [DOI] [PubMed] [Google Scholar]
- 139.Cho C, Willis WD, Goulding EH, et al. Haploin-sufficiency of protamine-1 or -2 causes infertility in mice. Nat Genet. 2001;28:82–86. doi: 10.1038/ng0501-82. [DOI] [PubMed] [Google Scholar]
- 140.Westerveld GH, Korver CM, van Pelt AM, et al. Mutations in the testis-specific NALP14 gene in men suffering from spermatogenic failure. Hum Reprod (Oxford, England) 2006;21:3178–3184. doi: 10.1093/humrep/del293. [DOI] [PubMed] [Google Scholar]
- 141.Gianotten J, Van Der Veen F, Alders M, et al. Chromosomal region 11p15 is associated with male factor sub-fertility. Mol Hum Reprod. 2003;9:587–592. doi: 10.1093/molehr/gag081. [DOI] [PubMed] [Google Scholar]
- 142.Yatsenko AN, Roy A, Chen R, et al. Non-invasive genetic diagnosis of male infertility using spermatozoal RNA: KLHL10 mutations in oligozoospermic patients impair homodimerization. Hum Mol Genet. 2006;15:3411–3419. doi: 10.1093/hmg/ddl417. [DOI] [PubMed] [Google Scholar]
- 143.Yan W, Ma L, Burns KH, Matzuk MM. Haploin-sufficiency of kelch-like protein homolog 10 causes infertility in male mice. Proc Natl Acad Sci USA. 2004;101:7793–7798. doi: 10.1073/pnas.0308025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang S, Zheng H, Esaki Y, et al. Cullin3 is a KLHL10-interacting protein preferentially expressed during late spermiogenesis. Biol Reprod. 2006;74:102–108. doi: 10.1095/biolreprod.105.045484. [DOI] [PubMed] [Google Scholar]
- 145.Suryavathi V, Khattri A, Gopal K, et al. Novel variants in UBE2B gene and idiopathic male infertility. J Androl. 2008;29:564–571. doi: 10.2164/jandrol.107.004580. [DOI] [PubMed] [Google Scholar]
- 146.Zhang AZS, Yang Y, et al. Single nucleotide polymorphisms of the gonadotrophin-regulated testicular helicase (GRTH) gene may be associated with the human spermatogenesis impairment. Hum Reprod. 2006;21:755–759. doi: 10.1093/humrep/dei388. [DOI] [PubMed] [Google Scholar]
- 147.Huang I, Emery BR, Christensen GL, et al. Novel UBE2B-associated polymorphisms in an azoospermic/oligozoospermic population. Asian J Androl. 2008;10:461–466. doi: 10.1111/j.1745-7262.2008.00386.x. [DOI] [PubMed] [Google Scholar]
- 148.Ribarski I, Lehavi O, Yogev L, et al. USP26 gene variations in fertile and infertile men. Hum Reprod. 2009;24:477–484. doi: 10.1093/humrep/den374. [DOI] [PubMed] [Google Scholar]
- 149.Miyagawa Y, Nishimura H, Tsujimura A, et al. Single-nucleotide polymorphisms and mutation analyses of the TNP1 and TNP2 genes of fertile and infertile human male populations. J Androl. 2005;26:779–786. doi: 10.2164/jandrol.05069. [DOI] [PubMed] [Google Scholar]
- 150.Miyamato T, Sato H, Yogev L, et al. Is a genetic defect in Fkbp6 a common cause of azoospermia in humans? Cell Mol Biol Lett. 2006;11:557–569. doi: 10.2478/s11658-006-0043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Thangaraj K, Joshi MB, Reddy AG, et al. Sperm mitochondrial mutations as a cause of low sperm motility. J Androl. 2003;24:388–392. doi: 10.1002/j.1939-4640.2003.tb02687.x. [DOI] [PubMed] [Google Scholar]
- 152.Dieterich K, Soto Rifo R, Faure AK, et al. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat Genet. 2007;39:661–665. doi: 10.1038/ng2027. [DOI] [PubMed] [Google Scholar]
- 153.Tang CJ, Lin CY, Tang TK. Dynamic localization and functional implications of Aurora-C kinase during male mouse meiosis. Dev Biol. 2006;290:398–410. doi: 10.1016/j.ydbio.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 154.Dam AH, Koscinski I, Kremer JA, et al. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet. 2007;81:813–820. doi: 10.1086/521314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lu L, Lin M, Xu M, et al. Gene functional research using polyethylenimine-mediated in vivo gene transfection into mouse spermatogenic cells. Asian J Androl. 2006;8:53–59. doi: 10.1111/j.1745-7262.2006.00089.x. [DOI] [PubMed] [Google Scholar]
- 156.Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Phys Rev. 2007;87:1–28. doi: 10.1152/physrev.00009.2006. [DOI] [PubMed] [Google Scholar]
- 157.Jobling MA, Tyler-Smith C. The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet. 2003;4:598–612. doi: 10.1038/nrg1124. [DOI] [PubMed] [Google Scholar]
- 158.Hopps CV, Mielnik A, Goldstein M, et al. Detection of sperm in men with Y chromosome microdeletions of the AZFa, AZFb and AZFc regions. Hum Reprod. 2003;18:1660–1665. doi: 10.1093/humrep/deg348. [DOI] [PubMed] [Google Scholar]