

Abstract
Background. The licensing of herpes zoster vaccine has demonstrated that therapeutic vaccination can help control chronic viral infection. Unfortunately, human trials of immunodeficiency virus (HIV) vaccine have shown only marginal efficacy.
Methods. In this double-blind study, 17 HIV-infected individuals with viral loads of <50 copies/mL and CD4+ T-cell counts of >350 cells/µL were randomly assigned to the vaccine or placebo arm. Vaccine recipients received 3 intramuscular injections of HIV DNA (4 mg) coding for clade B Gag, Pol, and Nef and clade A, B, and C Env, followed by a replication-deficient adenovirus type 5 boost (1010 particle units) encoding all DNA vaccine antigens except Nef. Humoral, total T-cell, and CD8+ cytotoxic T-lymphocyte (CTL) responses were studied before and after vaccination. Single-copy viral loads and frequencies of latently infected CD4+ T cells were determined.
Results. Vaccination was safe and well tolerated. Significantly stronger HIV-specific T-cell responses against Gag, Pol, and Env, with increased polyfunctionality and a broadened epitope-specific CTL repertoire, were observed after vaccination. No changes in single-copy viral load or the frequency of latent infection were observed.
Conclusions. Vaccination of individuals with existing HIV-specific immunity improved the magnitude, breadth, and polyfunctionality of HIV-specific memory T-cell responses but did not impact markers of viral control.
Clinical Trials Registration. NCT00270465
Keywords: HIV, vaccination, therapy, cytotoxic T lymphocytes, humoral immunity, viral latency
The herpes zoster vaccine has shown that boosting cellular immunity by therapeutic vaccination can help control a chronic viral infection [1]. CD8+ cytotoxic T-lymphocytes (CTLs) play an important role in the control of human immunodeficiency virus (HIV) disease progression [2–5] and appear to be responsible for long-term viral suppression in some individuals [4, 6, 7]. Therefore, vaccination to boost existing CTLs and/or induce new CTL responses in individuals receiving effective antiretroviral therapy is a potentially useful adjunctive treatment [8–11]. Unfortunately, no human therapeutic vaccination trial for HIV has shown convincing evidence for vaccine-induced suppression of viral replication [12–19]. Early studies suffered from the use of weak immunogens; more-recent studies have been conducted with more-potent immunogens, but characterization of vaccine-induced CTL responses has been limited [12]. As a result, little is known about the effect of vaccination on preexisting immunity or the induction of new T-cell clonotypes directed against new epitopes. A better understanding of the effects of vaccination with a potent immunogen on existing HIV-specific immunity would facilitate the design of future therapeutic trials.
We have previously shown that a DNA prime, recombinant adenovirus type 5 (rAd5) boost vaccination induces strong CTL responses in healthy HIV-uninfected volunteers [20–22]. CTL responses induced by this vaccine are detectable in most vaccinees, broadly directed, and polyfunctional, a characteristic that is associated with long-term viral control in individuals with chronic HIV infection [23]. This vaccine is currently being tested in a phase IIb preventive trial in the United States (clinical trials registration NCT00865566). Here, we report the results of a phase I, double-blind, placebo-controlled trial in which the vaccine was given to 12 individuals who were HIV infected and virologically suppressed to a VL <50 copies/ml by antiretroviral therapy before, during and after vaccination. We performed an extensive characterization of the magnitude, phenotype, function, epitope specificity, and clonotypic structure of the HIV-specific CTL response before and after vaccination.
METHODS
Study Participants
The Vaccine Research Center 101 study (VRC 101; NIH 06-I-0056; clinical trials registration NCT00270465) was a double-blind, placebo-controlled vaccine trial approved by the National Institute of Allergy and Infectious Diseases (NIAID) Institutional Review Board and reviewed by the Intramural NIAID Data and Safety Monitoring Board in accordance with Title 45, Part 46, of the US Code of Federal Regulations. The Division of Acquired Immunodeficiency Syndrome of the NIAID sponsored the Investigational New Drug application. The VRC conducted the study.
All subjects signed informed consents. Highly active antiretroviral therapy (HAART)–treated HIV-infected individuals who were 18–50 years old, had CD4+ T-cell counts of >350 cells/μL, and had undetectable HIV loads (<50 copies/mL) were randomly assigned in a 2:1 ratio to receive vaccine or placebo. HIV infection was confirmed using an HIV-1/HIV-2 enzyme-linked immunosorbent assay (ELISA). Individuals with nadir CD4+ T-cell counts of <100 cells/μL or chronic active hepatitis were excluded, as were pregnant women and individuals taking systemic immunosuppressive drugs. No samples were available from subjects prior to the initiation of therapy.
Randomization and Masking
Study participants were randomly assigned to the vaccine or placebo arm on the basis of the protocol design (Figures 1A and 1B) by means of a computer-generated block-randomization procedure. The randomization code was generated and maintained by the study statistician and the pharmacists.
Figure 1.

Vaccination regimen for Vaccine Research Center 101 study (VRC 101). Seventeen individuals were enrolled in VRC 101. Twelve individuals received 3 DNA prime vaccinations, with the first vaccination administered at enrollment and the second and third DNA priming vaccinations administered 1 and 2 months, respectively, after enrollment. A replication-deficient adenovirus type 5 (Ad5) boost vaccination was given at month 6. Five individuals received placebo injections. Blood draws used for immunologic assessment are shown by x's. Individuals who agreed to apheresis underwent this procedure in the month preceding vaccination and 1 month after recombinant Ad5 (rAd5) boosting. Primary immunologic end points were the differences between human immunodeficiency virus (HIV)–specific interferon γ responses at enrollment; at week 10, 2 weeks after the last DNA priming vaccination; and at week 28, 4 weeks after rAd5 boosting. Abbreviation: Vax, vaccination.
Vaccine and Vaccination Schedule
Vaccine recipients received 4-mg VRC-HIVDNA016-00-VP priming vaccinations on day 0, week 4, and week 8, followed by 1010 particle units of VRC-HIVADV014-00-VP as a boost vaccination at month 6. Both vaccines have been described previously [24, 25]. All vaccinations were given intramuscularly in a 1-mL volume. The Biojector 2000 injection system was used to administer VRC-HIVDNA016-00-VP; a needle and syringe were used to administer VRC-HIVADV014-00-VP. Phosphate-buffered saline (PBS) was used as placebo for VRC-HIVDNA016-00-VP vaccinations. Final formulation buffer was used as placebo for VRC-HIVADV014-00-VP. Study supplies were manufactured under current Good Manufacturing Practices conditions. Safety evaluations included physical examination and monitoring of laboratory parameters. Local (pain, swelling, or redness) and systemic (fever, malaise, myalgia, headache, chills, or nausea) reactogenicity symptoms were recorded on 5-day diary cards following each vaccination.
Peptides
All peptides used had a purity of >70%. Peptides were pooled according to antigen (EnvA, EnvB, EnvC, Gag, Pol, and Nef) or in a separate matrix format for epitope mapping.
Measurement of Antibody Responses and Epitope Mapping
Standardized research ELISAs were performed to delineate antibody responses to viral antigens encoded within the vaccine [25]. Ninety percent Ad5 neutralization titers were measured as described previously [26]. Raw peptide microarray data were processed and analyzed as described elsewhere and used to determine changes in epitope-specific antibody responses [27].
ELISpot Assays
Longitudinal frequencies of T-cell responses to vaccine antigens were determined using a validated ELISpot assay [28]. ELISpot assays used to identify HIV-derived epitopes were performed as described previously [29]. All assays used cryopreserved cells.
Antibodies
Anti-CD4 Cy55PE, anti-CD8 Qdot 705, anti-CD14 Pacific Blue, and anti-CD19 Pacific Blue were from Invitrogen. Anti-CD27 Cy5PE and anti-CD45RO TRPE were from Beckman Coulter. Anti-CD107a Alexa680 and anti-HLA DR Alexa680 were conjugated in our laboratory (available at: http://drmr.com/abcon/index.html). Other antibodies were purchased from BD Biosciences.
CD8+ T-Cell Epitope Mapping
To identify HIV-specific CD8+ T-cell epitopes using minimal sample, a 5-dimensional matrix was constructed containing 869 different peptides in 75 different peptide pools. Each pool contained 60 different 15mers [30]. Each peptide was represented 5 times in these pools. Pre- and postvaccination interferon γ (IFN-γ) ELISpot matrices were run in parallel. Candidate epitopes were identified by matrix response pattern. For each candidate epitope, a second-round pre- and postvaccination determination was performed to determine the actual peptide epitope. Intracellular cytokine staining assays identified CD4+ and CD8+ T-cell epitopes.
Flow Cytometry
Cryopreserved peripheral blood mononuclear cells (PBMCs) were thawed and rested in R10 for 2 hours prior to use. For peptide epitope identification, PBMCs were stimulated overnight in a 96-well V-bottomed plate. Permeabilized cells were stained with anti-CD3, anti-CD4, anti-CD8, anti–IFN-γ, and anti–tumor necrosis factor α [31]. The limit of detection was 0.02%.
CD8+ T cell functional and activation profiles were characterized as described previously [23, 32]. Data were collected on a LSR II flow cytometer (BD Immunocytometry Systems). Analysis was performed using FlowJo software (Tree Star, Inc).
Tetrameric Antigen Complexes
Recombinant peptide-major histocompatibility complex class I (pMHCI) tetramers were produced as described previously [33].
Tetramer Staining and Cell Sorting
Frozen PBMCs were thawed, rested, and stained with LIVE/DEAD Violet Viability/Vitality Dye (Invitrogen). After washing with PBS, pretitered amounts of tetramer were added and incubated at 37°C for 10 minutes. Cells were then washed and stained with anti–CD3 H7APC, anti–CD4 Cy5.5PE, and anti–CD8 PE for 10 minutes at room temperature. Viable CD3+CD4−CD8+tetramer+ cells were sorted directly into RNAlater (Ambion), using a modified FACS Aria flow cytometer (BD Immunocytometry Systems), and stored at −80°C.
Clonotypic Analysis
HIV antigen–specific CD8+ T-cell repertoires were characterized using an unbiased template-switch anchored reverse transcription polymerase chain reaction to amplify TRB gene products as described previously with minor modifications, including the addition of a barcode to the 5′ RACE primer [34]. Sequences were analyzed using Sequencher 4.9 and BioEdit 7.0.5.3. Nonproductive rearrangements and sequences determined to be products of resampled complementary DNA template were disregarded.
Determination of Viral Loads and CD4+ T-Cell Counts
CD4+ T-cell counts and clinical HIV loads were determined by a CLIA-certified laboratory. Ultrasensitive single-copy sensitive viral loads were measured as described previously [35].
Determination of Frequency of Latently Infected CD4+ T Cells
CD3+CD4+ T cells with an HLA DR− phenotype were purified from frozen PBMCs by flow cytometry, diluted 5-fold, and cultured as described previously [36]. Frequencies of infected cells were determined in infectious units per million (IUPM) on the basis of maximum likelihood methods [37]. Pre- and postvaccination purified CD4+ T cells from the same individual were cultured simultaneously on the same pool of PBMC-irradiated feeders to minimize experimental variation.
Statistics
Safety and tolerability of the vaccine regimen were the primary end points of this study. Secondary objectives were comparisons of the HIV-specific T-cell frequency, breadth, phenotype, and function before vaccination and 4 weeks after Ad5 boosting. All values are reported as medians, with ranges in parentheses. Statistical comparisons were performed using Prism statistical programs (GraphPad software). Pre- and postvaccination comparisons were performed using the Wilcoxon signed rank test. All tests were 2 tailed. Half-maximal functional sensitivities were determined by fitting the data to a nonlinear sigmoidal model, using Prism.
RESULTS
Subjects
Seventeen HIV-positive subjects were enrolled into VRC 101. All were white males infected in the United States and were assumed to have HIV clade B infection. Twelve volunteers were randomly assigned to the DNA prime, rAd5 boost arm; 5 volunteers were randomly assigned to the placebo arm (Figure 1B). CD4+ T-cell counts and Ad5 neutralization titers were similar in both arms (Table 1). The median age was greater in the placebo recipient group than in the vaccine recipient group (49 years [range, 45–50 years] vs 44 years [range, 26–50 years]; P < .05). The median time since diagnosis was not significantly different between placebo recipients and vaccine recipients (7 years [range, 1–17 years] and 4.5 years [range, 1–16 years], respectively). The median duration of treatment was not significantly different between placebo recipients and vaccine recipients (7 years [range, 1–16 years] and 4.5 years [range, 1–11 years], respectively). All volunteers were HLA typed; no imbalance in class I HLA types was apparent.
Table 1.
Demographic and Clinical Characteristics of Study Participants
| Characteristic | Vaccine Recipients (n = 12) | Placebo Recipients (n = 5) | All Subjects (n = 17) |
|---|---|---|---|
| Sex | |||
| Male | 12 | 5 | 17 |
| Female | 0 | 0 | 0 |
| Age, y, median | 39.5 (26–50) | 49 (45–50)a | 44 (26–50) |
| Race | |||
| White | 12 | 5 | 17 |
| Others combined | 0 | 0 | 0 |
| CD4+ T-cell count at entry, cells/μL | 660.5 (469–1674) | 769 (554–1182) | 677 (469–1674) |
| HIV load at entry, copies/mL | <50 | <50 | <50 |
| Ad5 titer at entryb | 243.5 (<12 to >8748) | 234 (<12 to >8748) | 234 (<12 to >8748) |
| Time since HIV diagnosis, y | 4.5 (1–16) | 7 (1–17) | 5 (1–17) |
| Duration of ART, y | 4.5 (1–11) | 7 (1–16) | 5 (1–16) |
| Nadir CD4+ T-cell count, cells/μL | 350 (147–800) | 399 (280–800) | 350 (147–800) |
Data are no. of participants or median value (range).
Abbreviations: Ad5, adenovirus type 5; HIV, human immunodeficiency virus;
a P < .05.
b Data are reciprocal 90% neutralization titer.
Vaccine Safety
Vaccination was well tolerated. For DNA vaccinations and placebo injections, local reactogenic events (pain/tenderness, swelling, or redness) were mild at most. Among the 12 vaccine recipients, 5 reported mild and 4 reported moderate systemic reactogenicity at least once during the 5 days following DNA vaccination; moderate symptoms included malaise and myalgia (Supplementary Table 1). One report of severe myalgia and malaise was caused by a work-related fracture. Among the 5 placebo recipients, 1 reported mild systemic symptoms at least once. After rAd5 boost injections, local reactogenic events in both vaccine and placebo recipients were mild at most. No serious vaccine-related adverse effects were reported during this trial. One subject each from the vaccine and placebo groups was withdrawn from the vaccination schedule because of adverse events (urticaria and ventricular bigeminy, respectively) assessed as unlikely to be related to study injection.
Vaccine Boosted T-Cell Responses
Vaccination resulted in a significantly stronger HIV-specific T-cell responses. Compared with the response frequency before vaccination, Gag- (P < .005), Pol- (P < .05) clade A (P < .005), clade B (P < .005), and clade C (P < .05) Env-specific ELISpot responses were all significantly increased 1 month after boosting (Figure 2A). The frequency of Nef ELISpot responses, which were not encoded in the rAd5 vaccine used in this trial, was not significantly increased; furthermore, there was no change in the frequency of ELISpot responses observed in sham-vaccinated individuals. A significant increase in the frequency of HIV-specific ELISpot responses was not seen 3 or 6 months after rAd5 boosting. There was no apparent effect of Ad5 seropositivity or nadir CD4+ T-cell count on the boosting of HIV-specific responses.
Figure 2.

Vaccination increased the frequency of HIV-specific CD8 T cells. A, Frequency of interferon γ (IFN-γ)–producing cells per million peripheral blood mononuclear cells (PBMCs) after overnight incubation with vaccine-matched 15mer peptides overlapped by 11 residues, corresponding to the human immunodeficiency virus (HIV) clade B Gag, clade B Pol, clade B Nef, and clade A, B, and C Env gene products. Box plots represent the second and third quartiles; horizontal bars indicating median values. Whiskers indicate the 90% range for the data. Dots represent individual data outside the 10%–90% range. Blue bars show data from ELISpot analyses done using PBMCs prepared from blood drawn on day 1 before vaccination; red bars represent data from PBMCs prepared from blood draws 1 month after recombinant adenovirus type 5 (rAd5) vaccination. Significant differences in IFN-γ ELISpot frequencies before and 1 month after vaccination are indicated by horizontal lines. In vaccinees, incubation of PBMCs with clade B Gag, clade B Pol, and clade A, B, and C Env 15mers resulted in significantly greater response frequencies after vaccination. No significant difference was observed in the Nef responses after vaccination No significant difference responses to any peptide pools was observed in the placebo group pre- and post-vaccination. B, Frequency of IFN-γ–producing ELISpots per million PBMCs after overnight incubation with individual 15mers either before or after vaccination. In vaccinees, the frequency of postvaccination IFN-γ responses was significantly higher after vaccination (P < .001). No significant difference was observed in incubations containing PBMCs from placebos. C, Frequency of IFN-γ–producing CD8+ T cells measured by intracellular cytokine staining in response to 6-hour incubation with specific 15mers identified as inducing IFN-γ production either before or after vaccination. Responses are shown for Gag, Pol, Nef, and Env 15mers. No Env epitopes were identified in the placebo group. Postvaccination IFN-γ production was observed significantly more frequently in vaccinees after vaccination than before vaccination (P < .05). When grouped by HIV gene product, only Gag-specific responses were significantly increased by vaccination (P < .05). No significant difference was observed in the placebo group. Abbreviation: Vax, vaccination.
Epitope Mapping
To further define the effects of vaccination, epitope mapping using vaccine-matched overlapping 15mers was performed for 8 vaccine recipients and 3 placebo recipients who volunteered for apheresis the month preceding the first DNA vaccine or placebo-injection and one month after the rAd5 boost vaccination or placebo-injection. Epitopes were first identified using ELISpot analysis, and epitope-induced IFN-γ production by CD8+ T cells was confirmed by intracellular cytokine staining. This effort identified 48 vaccine-matched epitopes in the 8 vaccinees. By ELISpot analysis, the frequencies of postvaccination responses to these epitopes were significantly higher than those before vaccination (P < .001). In the 3 placebo recipients, 11 peptide epitopes were identified. No significant differences were found between the frequencies of pre- and postvaccination responses to these epitopes (Figure 2B). Epitope-specific CD8+ T cells were also more frequent after vaccination than they were before vaccination, when intracellular cytokine staining analysis was used (P < .05); no difference was observed in placebo recipients. When the response measured by intracellular cytokine staining was separated into Gag-, Pol-, Env-, and Nef-specific gene products (Figure 2C), only Gag peptide-specific responses were significantly increased (P < .05).
Function and Maturation of CD8+ T-Cell Responses
Optimized 8–10mer epitopes were determined on the basis of the vaccine recipient's class I HLA type, published epitopes, or class I binding motifs and the subject's response to candidate peptides. Vaccine-specific responses were characterized using 6 optimized epitopes (Supplementary Table 2). The median increase in CD8+ T-cell responses to these epitopes after vaccination was 2.01-fold (range, 1.51–7.24-fold). Five functions (surface mobilization of CD107a and intracellular production of IFN-γ, tumor necrosis factor α, interleukin 2, and macrophage inflammatory protein 1β) were quantified for these epitope-specific responses; the median number of simultaneous functions increased from 1.65 (range, 1.17–2.17) before vaccination to 2.19 (range, 1.5–2.95) after vaccination (P < .05; Figure 3A). The maturational phenotype of these epitope-specific CD8+ T cells was also determined (Supplementary Figure 1A). In general, epitope-specific responses were either slightly more mature (based on loss of CD27 expression) or unchanged after vaccination, compared with before vaccination (Figure 3B). The exception was the CD8+ T-cell response to the B51-restricted epitope YI9: expression of CD27 on antigen-specific CD8+ T cells increased from 20% before vaccination to 66.5% after vaccination. This response also increased from barely detectable expression before vaccination to expression on 0.63% of CD8+ T cells after vaccination.
Figure 3.

Functional, maturation, and activation profile of pre- and postvaccination CD8+ T-cell responses for different peptide epitopes. A, Pre- and postvaccination functional profiles in response to 6-hour incubation with 2 μg/mL optimized peptide epitope, showing the frequencies of memory CD8+ T cells displaying the depicted combinations of surface mobilization of CD107a (7) and intracellular production of interferon γ (IFN-γ; g), interleukin 2 (2), macrophage inflammatory protein 1β (M), and tumor necrosis factor α (T). Individual data points are represented by a blue dot, for prevaccination values, and a red dot, for postvaccination values. Six different peptide epitopes were tested using peripheral blood mononuclear cells (PBMCs) prepared from 4 different vaccinees (Supplementary Table 3). Bars represent the range of the second and third quartiles, with horizontal lines representing the median values for each of the 31 functional subgroups. The median prevaccination response frequency among memory CD8+ T cells was 0.09% (range, 0.04%–0.61%). The median postvaccination response frequency among memory CD8+ T cells was 0.37% (range, 0.1%–0.92%). A median 2-fold increase in response (range, 1.51–7.24-fold) was observed after vaccination. Median polyfunctionality was significantly increased, from 1.65 (range, 1.17–2.7) to 2.19 (range, 1.5–2.95), as determined by a Wilcoxon signed ranks test (P < .05). B, Maturational profile of IFN-γ–producing memory CD8+ T cells for both pre- and postvaccination responses, as determined by surface expression of CD27 and CD57. The red circles represent the maturation profile for the response to the B51-optimized epitope YI9. C, Activation profile of IFN-γ–producing memory CD8+ T cells for both pre- and postvaccination responses, based on surface expression of CCR5, HLA DR, and CD38 and intracellular expression of Ki67. Red circles represent the activation profile for the response to the B51-optimized epitope YI9.
These peptide epitopes were also used to measure the activation state of vaccine-specific CD8+ T cells both before and after vaccination (Supplementary Figure 1B). All but 1 response remained unchanged or shifted to a slightly more activated state. Once again, the epitope with the largest increase in response, YI9, showed a pattern that differed from the patterns of the other 5 epitopes. Surface staining for CD38 on the YI9-specific CD8+ T cells increased from 0% before vaccination to 84% after vaccination. Ki67 staining also increased from 0% to 59%, suggesting continued vaccine-induced stimulation of these CD8+ T cells, 1 month after vaccination (Figure 3C).
T-Cell Receptor Use
T-cell receptor use was determined in 5 separate epitope-specific CD8+ T-cell responses, all of which were increased by vaccination and for which class I tetramers were available (Supplementary Table 2). Of note, these epitopes were well conserved across clades, except for YI9, which was only represented in clade A. The median prevaccination frequency of the 5 tetramer-positive CD8+ T cells studied in this this group was 0.17% (range, 0.013%–0.33%). After vaccination, the median frequency of tetramer-positive CD8+ T cells was 0.48% (range, 0.26%–0.98%). The median increase in the frequency of tetramer-positive CD8+ T cells was 2.2-fold (range, 1.4–78-fold). In general, the T-cell receptors used by these epitope-specific T cells tended to remain stable, with little change in clonal hierarchy despite the overall increase in response frequency. The one exception was the YI9-specific CD8+ T-cell population, which exhibited a complete change in T-cell receptor clonotype use after vaccination (Figure 4). The epitope that these T cells responded to was an A clade epitope, YPPIQGVI. The YI9 epitope–specific CTLs also showed a change to a less mature phenotype and higher frequency of CD38 and Ki67 expression after vaccination. To test whether the A clade YI9–specific clonotypes cross-reacted with the analogous B clade 9mer, YAPPISGQI, the frequency of IFN-γ–producing cells, as determined by intracellular cytokine assay, was determined after stimulation with each peptides at different peptide concentrations. The half-maximal functional sensitivity was 13.8 nM for the vaccine matched A clade YI9 peptide and 882 nM for the homologous B clade 9mer (Figure 5). There was no response to the B clade 9mer prior to vaccination.
Figure 4.

Clonotypic analysis of antigen-specific CD8+ T cells before and after vaccination show evidence of vaccine induced T-cell clonotypes. Pre- and postvaccination histographs showing tetramer-positive CD8+ T-cell populations for 3 different optimized epitopes and graphs showing the frequency of specific T-cell receptor clonotypes, as defined by CDR3β sequence, for each tetramer-defined population. Prevaccination clonotype frequencies are shown by blue bars; postvaccination clonotype frequencies are shown by red bars. A, Tetramer-specific T-cell receptor frequency for the optimized B57 epitope KF11. Seventy-seven clones were sequenced before vaccination, and 82 clones were sequenced after vaccination. B, Tetramer-specific T-cell receptor frequency for the positive responses to the optimized B08 epitope YL8. Eighty-six clones were sequenced before vaccination, and 85 clones were sequenced after vaccination. C, Tetramer-specific T-cell receptor frequencies for the optimized B51 epitope YI9. Seventy-three clones were sequenced before vaccination, and 92 clones were sequenced after vaccination.
Figure 5.
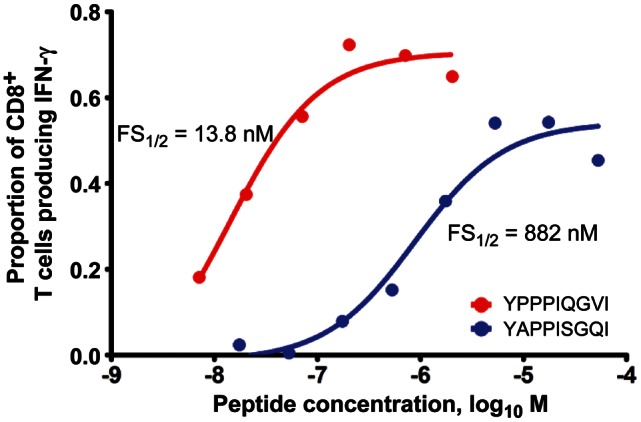
Plot showing the functional sensitivity (FS) of a vaccine-induced response to the A clade epitope YI9 and the homologous B clade epitope YAPPISGQI. Sigmoidal fit of the frequency of interferon γ–producing cells in response to stimulation with different concentrations of peptide. Half-maximal concentrations for each 9mer peptide are shown on the graph.
The Effect of Vaccination on Viral Load, CD4+ T-Cell Count, Frequency of Latently Infected CD4+ T-Cells, and Antibody Response
Vaccination has been reported to activate CD4+ T cells and transiently increase viral load [38, 39]. We found no effect of vaccination on CD4+ T-cell counts (Figure 6A) or HIV load, measured by either standard clinical methods (Figure 6B) or an ultrasensitive single copy assay (Figure 6C). Frequency of latently infected resting CD4+ T cells was also unaffected by vaccination (Figure 6D). No significant change in pooled clade A, B, or C envelope antibody titers was observed between the serum samples obtained before and 1 month after vaccination. Vaccination did appear to cause a statistically nonsignificant increase in binding to peptides in the V3 loop. On the basis of the magnitude of response, it appears that the response was more specific to subtypes A, B, and CRF02 (data not shown).
Figure 6.

Vaccination did not affect human immunodeficiency virus (HIV) load or CD4+ T-cell count. A, CD4+ T-cell counts during the Vaccine Research Center 101 study. Weeks after enrollment are indicated on the x-axis. Time of vaccination is indicated by text. Median CD4+ T-cell count is indicated for placebo recipients (blue circles) and vaccinees (red squares). Bars indicate the limit of data for the second and third quartile. B, Viral loads are shown for vaccine recipients (red dots) and placebo recipients (blue dots). The open bars shown for one of the vaccine recipients indicate an interruption in the trial, caused by the release of the results of the STEP trial. C, Single-copy HIV RNA levels for placebo recipients (blue dots) and vaccine recipients (red dots) on the day of enrollment prior to the first DNA priming vaccination, immediately prior to recombinant adenovirus type 5 (rAd5) boosting, 1 month after Ad5 boosting, and 3 months after Ad5 boosting. Horizontal bars indicate median values for vaccine recipients (red) and placebo recipients (blue). Values less than the lower limit of detection are shown below the black dashed line. D, Log plots of frequency of latently infected resting CD4+ T cells for both vaccine and placebo recipients, both before vaccination and 1 month after vaccination. Determinations in which the frequency of latently infected cells were less than the lower limit of detection are shown as open circles denoting the limit of detection. Abbreviation: Vax, vaccination.
DISCUSSION
In this study, vaccination of HIV-infected individuals receiving effective antiretroviral therapy with a DNA prime, rAd5 boost vaccine regimen was safe and well tolerated and effectively expanded HIV-specific T-cell responses. Although the majority of HIV-specific CD8+ T cells induced by the vaccination represented expansions of preexisting responses whose maturational phenotype and clonotypic hierarchy did not change, we demonstrated increased polyfunctionality among the boosted populations and the induction of a new CD8+ T-cell response to an epitope that was not otherwise targeted during infection. We did not detect any change in either the median single-copy assay–determined viral load or viral latency after vaccination.
The rationale for vaccinating HIV-infected individuals receiving HAART is to improve the quality and/or magnitude of HIV-specific immune responses, thereby leading to better immune control. Numerous HIV-induced immune defects are at least partially reversed by HAART. Total and antigen-specific CD4+ T-cell numbers increase, allowing cessation of secondary prophylaxis for cytomegalovirus infection, Pneumocystis jirovecii pneumonia, and toxoplasmosis [40]. The frequency and surface density of exhaustion markers such as PD-1 and CD160 are also reduced by treatment, a change associated with improved functional and proliferative capacity of total and antigen-specific CD8+ T cells [41–44]. In addition, chronic immune activation decreases with treatment. The immune system is therefore more likely to respond to vaccination in the presence of HAART. However, merely boosting the frequency of HIV-specific CTLs is not sufficient to improve viral control [45]. It is reasonable to speculate that therapeutic vaccination will need to change the functional capacity or broaden epitope recognition within the CTL compartment if it is to improve virologic control. The induction of a CTL response directed against a B clade epitope by an A clade Env sequence shown here is encouraging, particularly considering that previous B clade Env epitopes have not been identified at this site [46]. Induction of new, relevant HIV-specific epitopes using slightly mismatched antigens may offer a means of broadening the immune response in chronically infected individuals; in this case, however, a single newly induced response with a relatively low avidity is likely to have a minimal impact on viral control.
Instituting treatment interruption to judge the efficacy of a therapeutic intervention is still both possible and important, but it has been made more difficult primarily because of the increased emphasis on the benefit of early therapy initiation and the importance of continuous therapy. These considerations have made it difficult to recruit volunteers who are willing to interrupt treatment during a study. Nonetheless, this study has shown that it is possible to increase the magnitude and quality of the HIV-specific CTL response by therapeutic vaccination, albeit in the absence of evidence of efficacy. We examined 2 possible treatment-interruption surrogates previously used to assess treatment efficacy: ultrasensitive single-copy viral load [47] and quantitation of latent viral load [48]. Although limited by small sample size and by undetectable plasma virus and latently infected CD4+ T cells, neither method showed evidence of vaccine efficacy. In the absence of treatment-interruption studies, these techniques most likely give some indication of vaccine efficacy. However, only limited data are available comparing these methods to the effect of vaccination on control of viral replication in chronic infection. Accordingly, these methods should not be accepted as a replacement for treatment interruption in the assessment of vaccine efficacy.
Therapeutic vaccination of individuals treated with HAART during acute rather than chronic HIV infection may result in improved virologic control. In these individuals, vaccination would be conducted in the context of a larger CD4+ T-cell repertoire and less exhausted CD4+ and CD8+ T-cell compartments. Escape mutations are also less likely in early infection. During HAART, vaccination could boost nascent responses and potentially induce new HIV-specific CD8+ T-cell clonotypes, resulting in improved immunologic control. Furthermore, induction of HIV-specific CD8+ T-cell responses with a vaccine that produces a low level of antigen may induce higher-avidity T-cell clonotypes than those induced during high-level viremia [49]. Whether the observed vaccine-elicited changes in the immune response are sufficient to alter the subsequent course of disease in individuals with chronic HIV infection could not be tested with treatment interruption in this trial. Although sample size hampers interpretation of these data, the absence of any significant change in either single-copy viral load or the frequency of latent infection may suggest that a more potent immunogen is needed or that therapeutic vaccination alone is not sufficient to improve viral control in chronically infected individuals. Nonetheless, the changes in CTL function reported here have been associated previously with improved virologic control in chronic infection. It is therefore possible that therapeutic vaccination in combination with other treatment modalities may constitute a viable treatment strategy in chronically infected individuals. Adjunctive options include treatment with histone deacetylase inhibitors or other agents to stimulate HIV out of latency, thereby rendering these cells more susceptible to killing by CD8+ T cells, and the use of PD-1–blocking antibodies to further revitalize exhausted CTLs [50]. Such combined modality treatment interventions in conjunction with therapeutic vaccination may lead to enhanced immune control and a decrease in the latent viral reservoir.
VRC 101 STUDY TEAM
The VRC 101 Team consists of LaSonji Holman, Cynthia Hendel, Sarah Plummer, Laura Novik, Brenda Larkin, Steve Rucker, Pamela Costner, Trishna Goswami, Sarah Read, Susan Leitman, Hope Decederfelt, Judith Starling, Tiffany Alley, Richard Jones, Diane Johnson, Sara Jones, Sandra Sitar, LaChonne Stanford, Rhonda Washington-Lewis, Ericka Thompson, Kathy Rhone, and Phyllis Zaia.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the study volunteers, for their participation in this study; Woody Dubois, PhD, and Michelle Conan-Cibotti, PhD, for their work in managing the IND; and Phil Gomez, Rebecca Sheets, and Judy Stein, for their work in developing and producing the vaccine.
J. C., R. K., and B. G. were responsible for the conduct of this study. J. C. was responsible for the accumulation and analysis of data and held final responsibility for submission of the manuscript for publication.
Financial support. This work was supported by the Bench-to-Bedside Program, National Institutes of Health (NIH), and by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases (NIAID), NIH. Latent HIV reservoir studies were funded by the NIAID, NIH (R01 AI062446-01A2 to D. P.). Tetramer studies were funded by the UK Medical Research Council (MRC; G0501963 to D. A. P.); D. A. P. is a MRC senior clinical fellow. Single-copy viral load measurements were supported by the Intramural Research Program of the HIV Drug Resistance Program, National Cancer Institute, NIH (to F. M. M.). Antibody specificity studies were supported by the Bill and Melinda Gates Foundation (OPP1032317 to R. G.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis. 2010;51:197–213. doi: 10.1086/653605. [DOI] [PubMed] [Google Scholar]
- 2.Goulder PJ, Phillips RE, Colbert RA, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–7. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 3.Koup RA, Safrit JT, Cao Y, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. Journal of Virology. 1994;68:4650–5. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Migueles SA, Sabbaghian MS, Shupert WL, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A. 2000;97:2709–14. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salazar-Gonzalez JF, Salazar MG, Keele BF, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med. 2009;206:1273–89. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Autran B, Descours B, Avettand-Fenoel V, Rouzioux C. Elite controllers as a model of functional cure. Curr Opin HIV AIDS. 2011;6:181–7. doi: 10.1097/COH.0b013e328345a328. [DOI] [PubMed] [Google Scholar]
- 7.Pereyra F, Jia X, McLaren PJ, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–7. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadogan M, Dalgleish AG. HIV immunopathogenesis and strategies for intervention. Lancet Infect Dis. 2008;8:675–84. doi: 10.1016/S1473-3099(08)70205-6. [DOI] [PubMed] [Google Scholar]
- 9.Trono D, Van Lint C, Rouzioux C, Verdin E, Barre-Sinoussi F, Chun TW, Chomont N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science. 2010;329:174–80. doi: 10.1126/science.1191047. [DOI] [PubMed] [Google Scholar]
- 10.McMichael AJ. HIV vaccines. Annu Rev Immunol. 2006;24:227–55. doi: 10.1146/annurev.immunol.24.021605.090605. [DOI] [PubMed] [Google Scholar]
- 11.Autran B, Carcelain G, Combadiere B, Debre P. Therapeutic vaccines for chronic infections. Science. 2004;305:205–8. doi: 10.1126/science.1100600. [DOI] [PubMed] [Google Scholar]
- 12.Schooley RT, Spritzler J, Wang H, et al. AIDS clinical trials group 5197: a placebo-controlled trial of immunization of HIV-1-infected persons with a replication-deficient adenovirus type 5 vaccine expressing the HIV-1 core protein. J Infect Dis. 2010;202:705–16. doi: 10.1086/655468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markowitz M, Jin X, Hurley A, et al. Discontinuation of antiretroviral therapy commenced early during the course of human immunodeficiency virus type 1 infection, with or without adjunctive vaccination. J Infect Dis. 2002;186:634–43. doi: 10.1086/342559. [DOI] [PubMed] [Google Scholar]
- 14.Dorrell L, Yang H, Ondondo B, et al. Expansion and diversification of virus-specific T cells following immunization of human immunodeficiency virus type 1 (HIV-1)-infected individuals with a recombinant modified vaccinia virus Ankara/HIV-1 Gag vaccine. J Virol. 2006;80:4705–16. doi: 10.1128/JVI.80.10.4705-4716.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinloch-de Loes S, Hoen B, Smith DE, et al. Impact of therapeutic immunization on HIV-1 viremia after discontinuation of antiretroviral therapy initiated during acute infection. J Infect Dis. 2005;192:607–17. doi: 10.1086/432002. [DOI] [PubMed] [Google Scholar]
- 16.Yang H, Guimaraes-Walker A, Hibbs S, et al. Interleukin-10 responses to therapeutic vaccination during highly active antiretroviral therapy and after analytical therapy interruption. AIDS. 2009;23:2226–30. doi: 10.1097/QAD.0b013e328331a424. [DOI] [PubMed] [Google Scholar]
- 17.Goujard C, Marcellin F, Hendel-Chavez H, et al. Interruption of antiretroviral therapy initiated during primary HIV-1 infection: impact of a therapeutic vaccination strategy combined with interleukin (IL)-2 compared with IL-2 alone in the ANRS 095 Randomized Study. AIDS Res Hum Retroviruses. 2007;23:1105–13. doi: 10.1089/aid.2007.0047. [DOI] [PubMed] [Google Scholar]
- 18.Kilby JM, Bucy RP, Mildvan D, et al. A randomized, partially blinded phase 2 trial of antiretroviral therapy, HIV-specific immunizations, and interleukin-2 cycles to promote efficient control of viral replication (ACTG A5024) J Infect Dis. 2006;194:1672–6. doi: 10.1086/509508. [DOI] [PubMed] [Google Scholar]
- 19.Tubiana R, Carcelain G, Vray M, et al. Therapeutic immunization with a human immunodeficiency virus (HIV) type 1-recombinant canarypox vaccine in chronically HIV-infected patients: The Vacciter Study (ANRS 094) Vaccine. 2005;23:4292–301. doi: 10.1016/j.vaccine.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Churchyard GJ, Morgan C, Adams E, et al. A phase IIA randomized clinical trial of a multiclade HIV-1 DNA prime followed by a multiclade rAd5 HIV-1 vaccine boost in healthy adults (HVTN 204) PLoS One. 2011;6:e21225. doi: 10.1371/journal.pone.0021225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kibuuka H, Kimutai R, Maboko L, et al. A phase 1/2 study of a multiclade HIV-1 DNA plasmid prime and recombinant adenovirus serotype 5 boost vaccine in HIV-Uninfected East Africans (RV 172) J Infect Dis. 2010;201:600–7. doi: 10.1086/650299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koup RA, Roederer M, Lamoreaux L, et al. Priming immunization with DNA augments immunogenicity of recombinant adenoviral vectors for both HIV-1 specific antibody and T-cell responses. PLoS One. 2010;5:e9015. doi: 10.1371/journal.pone.0009015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Betts MR, Nason MC, West SM, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–9. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catanzaro AT, Koup RA, Roederer M, et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J Infect Dis. 2006;194:1638–49. doi: 10.1086/509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catanzaro AT, Roederer M, Koup RA, et al. Phase I clinical evaluation of a six-plasmid multiclade HIV-1 DNA candidate vaccine. Vaccine. 2007;25:4085–92. doi: 10.1016/j.vaccine.2007.02.050. [DOI] [PubMed] [Google Scholar]
- 26.Sprangers MC, Lakhai W, Koudstaal W, et al. Quantifying adenovirus-neutralizing antibodies by luciferase transgene detection: addressing preexisting immunity to vaccine and gene therapy vectors. J Clin Microbiol. 2003;41:5046–52. doi: 10.1128/JCM.41.11.5046-5052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haynes BF, Gilbert PB, McElrath MJ, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366:1275–86. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham BS, Koup RA, Roederer M, et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 DNA candidate vaccine. J Infect Dis. 2006;194:1650–60. doi: 10.1086/509259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geldmacher C, Metzler IS, Tovanabutra S, et al. Minor viral and host genetic polymorphisms can dramatically impact the biologic outcome of an epitope-specific CD8 T-cell response. Blood. 2009;114:1553–62. doi: 10.1182/blood-2009-02-206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roederer M, Koup RA. Optimized determination of T cell epitope responses. J Immunol Methods. 2003;274:221–8. doi: 10.1016/s0022-1759(02)00423-4. [DOI] [PubMed] [Google Scholar]
- 31.Lamoreaux L, Roederer M, Koup R. Intracellular cytokine optimization and standard operating procedure. Nat Protoc. 2006;1:1507–16. doi: 10.1038/nprot.2006.268. [DOI] [PubMed] [Google Scholar]
- 32.Maenetje P, Riou C, Casazza JP, et al. A steady state of CD4+ T cell memory maturation and activation is established during primary subtype C HIV-1 infection. J Immunol. 2010;184:4926–35. doi: 10.4049/jimmunol.0903771. [DOI] [PubMed] [Google Scholar]
- 33.Price DA, Brenchley JM, Ruff LE, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med. 2005;202:1349–61. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quigley MF, Almeia JR, Price DA, Douek DC. Unbiased molecular analysis of T cell receptor expression using template-switch anchored RT-PCR. Curr Protoc Immunol. 2011 doi: 10.1002/0471142735.im1033s94. 94:10.33.1–10.33.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmer S, Wiegand AP, Maldarelli F, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41:4531–6. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siliciano JD, Siliciano RF. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol Biol. 2005;304:3–15. doi: 10.1385/1-59259-907-9:003. [DOI] [PubMed] [Google Scholar]
- 37.Taswell C. Limiting dilution assays for the determination of immunocompetent cell frequencies. III. Validity tests for the single-hit Poisson model. J Immunol Methods. 1984;72:29–40. doi: 10.1016/0022-1759(84)90430-7. [DOI] [PubMed] [Google Scholar]
- 38.Gunthard HF, Wong JK, Spina CA, et al. Effect of influenza vaccination on viral replication and immune response in persons infected with human immunodeficiency virus receiving potent antiretroviral therapy. J Infect Dis. 2000;181:522–31. doi: 10.1086/315260. [DOI] [PubMed] [Google Scholar]
- 39.Ho DD. HIV-1 viraemia and influenza. Lancet. 1992;339:1549. doi: 10.1016/0140-6736(92)91321-x. [DOI] [PubMed] [Google Scholar]
- 40.Benson CA, Kaplan JE, Masur H, Pau A, Holmes KK. Treating opportunistic infections among HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association/Infectious Diseases Society of America. MMWR Recomm Rep. 2004;53(RR-15):1–112. [PubMed] [Google Scholar]
- 41.Day CL, Kauffman DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 42.Petrovas C, Casazza JP, Brenchley JM, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–92. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto T, Price DA, Casazza JP, et al. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood. 2011;117:4805–15. doi: 10.1182/blood-2010-11-317297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oxenius A, Price DA, Gunthard HF, et al. Stimulation of HIV-specific cellular immunity by structured treatment interruption fails to enhance viral control in chronic HIV infection. Proc Natl Acad Sci U S A. 2002;99:13747–52. doi: 10.1073/pnas.202372199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yusim K, Korber BT, Brander C, et al. HIV molecular immunology. Los Alamos, NM: Los Alamos National Laboratory, Theoretical Biology and Biophysics; 2009. [Google Scholar]
- 47.Maldarelli F. Targeting viral reservoirs: ability of antiretroviral therapy to stop viral replication. Curr Opin HIV AIDS. 2011;6:49–56. doi: 10.1097/COH.0b013e32834134ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen L, Siliciano RF. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol. 2008;122:22–8. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davenport MP, Riberio RM, Zhang L, Wilson DP, Perelson AS. Understanding the mechanisms and limitations of immune control of HIV. Immunol Rev. 2007;216:164–75. doi: 10.1111/j.1600-065X.2006.00485.x. [DOI] [PubMed] [Google Scholar]
- 50.Deeks SG, Autran B, Berkhout B, et al. Towards an HIV cure: a global scientific strategy. Nat Rev Immunol. 2012;12:607–14. doi: 10.1038/nri3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.