

Abstract
Although multiple cellular mechanisms have been proposed to explain endocrine resistance in breast cancer, the genomics events promoting the dysregulation of gene expression pattern are not clearly understood. Because chromatin plays a dynamic role in the estrogen receptor α (ERα) transcriptional program, we herein review signaling pathways implicated in endocrine resistance and try to merge them with recent epigenetic studies.
Keywords: breast cancer, chromatin, endocrine resistance, epigenetic, genomic, transcription factor
Breast Cancer And Development Of Anti-Estrogen Resistance
Estrogens, in particular 17β-estradiol (E2), regulate many essential physiological processes but also play an important role in the development and progression of hormone-dependent breast cancer. The functions of these hormones are mainly mediated by the nuclear receptors ERα and ERβ, which act as ligand-dependent transcription factors.1,2 ERβ function in breast cancer is not clearly understood but seems to be associated with good prognosis because of its antiproliferative properties. In contrast, the role of ERα in promoting proliferation of breast cancer cells is well characterized and will be the main focus of this review.
Approximately 70% of breast cancers are classified as ERα-positive breast cancers. ERα expression is a crucial determinant for the patients to be able to benefit from anti-estrogen (AE) therapies. Tamoxifen (TAM) and fulvestrant/ICI182780 (ICI) are the mainstay AE therapies for ER-positive breast cancers and have different mechanisms of action. Tamoxifen competes with E2 binding on the ER receptor and induces conformational changes leading to inactivation and stabilization of the receptor. ICI is indicated for late stage or metastasized breast cancer during or after anti-estrogen initial treatment or in relapse. ICI inhibits ERα DNA binding, inducing its degradation in a proteasomal dependent pathway.1,3
Despite the fact that these therapies allow a significant decrease of breast cancer mortality, a large number of patients fail to respond to initial therapy (de novo resistance) or develop resistance after prolonged treatment (acquired resistance) that limit the usefulness of theses drugs.4-6 AE-resistant breast cancers present an E2-independent proliferation characterized notably by a dysregulation of the cell cycle and apoptotic gene expression program. Several molecular mechanisms have been proposed to explain this phenomenon, especially in the more documented case of resistance to tamoxifen that we discuss below.
Molecular Mechanisms And Signaling Pathways Associated With AE-Resistance
Different molecular mechanisms have been proposed to elucidate resistance to endocrine therapy. Polymorphism of CYP2D6, a member of the cytochrome P450 family which converts tamoxifen in its active metabolites, results in alleles with different levels of activity of this enzyme and consequently a poor tamoxifen-response.7 Because ERα is a predictive marker for endocrine responses, loss of its expression could partly explain AE-response failure. Mutation of ERα and expression of a truncated variant named ERα36 have also been reported.8,9
ERα activity is not only regulated by its ligand but also by posttranslational modifications such as phosphorylation. ERα phosphorylation is known to be associated with resistance to tamoxifen.10,11 For example, phosphorylation of serine residues S118, S167 and S305 influence interaction with other proteins such as transcriptional coregulators and reduce sensitivity to tamoxifen in vitro. Only S305 phosphorylation seems to have clinical relevance12,13 and is partly responsible for AE-resistance. This modification leads to an altered orientation between ERα and the coactivator SRC-1, which renders the transcription complex active in the presence of tamoxifen.14 Likewise, overexpression and/or posttranslational modifications can occur on coregulators that modulate interactions with nuclear receptors and thereby their activity. For example, phosphorylation of the methyltransferase CARM1 allows it to bind ERα directly and mediate activation of ERα in a ligand-independent manner.15 The constitutive interaction of theses proteins observed in tamoxifen resistance context preserves ERα transcriptional activity even in the presence of TAM.
These posttranslational modifications ensue from activation of different signaling pathways and the corresponding increased activity of signal transducing kinases. Overexpression of HER2 (ERBB2), a member of the epidermal growth factor receptor family, is one of the best described mechanisms of AE-resistance.4,5,16 Near to 30% of breast cancers show HER2 alterations which are associated with poor prognosis. Furthermore, EGFR and IGFR are also known to be involved in AE-resistance and all the pathways, such as PI3K/AKT and MAPK/ERK, acting downstream from theses membrane receptors are responsible for the phosphorylation of ERα and its coregulators. PI3K/AKT signaling pathway is involved in proliferation, survival, growth and motility and also appears to have an important role in the development of resistance in antitumor therapies.17 Indeed, it has been shown that AKT overexpression in breast cancer cells confers estrogen independency and TAM and ICI resistance. Similarly, cells resistant to TAM present an increased expression of AKT.
Another pathway recently associated with AE-resistance involves the G-protein coupled receptor GPR30, a membrane-bound estrogen receptor that mediates rapid nongenomic effects of estrogen.18,19 Interestingly, TAM and ICI were found to be able to bind GPR30, resulting in cell proliferation through EGFR-MAPK or cAMP signaling pathways, mimicking, rather than antagonizing, estrogen actions. These results suggest an important role for GPR30 receptor signaling in the development of AE resistance. All these signaling pathways described here are integrated at the nuclear level to regulate specific gene expression programs.
Transcriptional Mechanisms In AE-Resistance
In breast cancer cells, ERα not only controls gene expression indirectly via nongenomic activation of various signaling cascades as discussed above, but mostly acts directly through genomic regulation of gene transcription. E2-induced ERα gene regulation involves chromatin binding through estrogen-responsive elements (ERE) or by a tethering mechanism involving other transcription factors such as AP-1, Sp1 or NFκB and the binding of coactivator proteins allowing the recruitment of the transcriptional machinery. Increased AP-1 and NFκB transcriptional activity is known to be associated with endocrine resistance. Alterations in coregulatory proteins such as overexpression or posttranslational modifications have also been associated with aberrant transcriptional programs and endocrine resistance. As previously mentioned, CARM1 phosphorylation by cAMP/PKA pathway potentiates ligand-independent activation of ERα. MED1, a subunit of mediator coactivator complex acting as a link between transcription factors and RNA polymerase II, was shown to be upregulated and phosphorylated by persistent ERK pathway activation during acquired TAM-resistance development.20
Overexpression of MED1 is associated with aggressiveness and decrease of recurrence free survival in breast cancer patients. Depletion of MED1 expression in a TAM-resistant cell model restored TAM sensitivity, allowing the inhibition of proliferation and a decrease of ERα target genes such as EBAG9 and TFF1. Overexpression and increased phosphorylation of AIB1 (amplified in breast cancer 1; also called NCOA3 or SRC3) have been observed in breast cancer and leads to constitutive ERα-mediated transcription.21 This coactivator competes with the paired box 2 gene product (PAX2), a mediator of ERα-dependent repression of HER2 by tamoxifen. Consequently, loss of PAX2 allows HER2 transcription and activation of this pathway, resulting in alteration of the tamoxifen response and is associated with a poor outcome.22 Overall, mechanisms of AE-resistance often involve modifications of transcription factors and coregulators expression and activity, resulting in altered gene expression. AE-resistance can also stem from upstream signaling events, where increased expression and/or activation of transduction signal pathways driving cell cycle and cell survival remodels transcriptional programs.
Aberrant expression of ERα target genes is often associated with AE-resistance. E2 act on cell proliferation via genomic and nongenomic pathways by increasing expression of cell cycle regulators and in particular cyclin D1, an important protein involved in G1/S phase transition. Anti-estrogens TAM or ICI reduce cell proliferation by inducing G1 phase-specific growth arrest. In AE-resistant breast cancers, expression and function of cyclin D1 and also cyclin E, c-Myc or cyclin dependant kinase (CDK) inhibitors p21waf1 and p27kip1 are frequently altered.4,6,23 CCND1 is overexpressed in 50% of breast cancers and overexpression of CCND1 in cellular models confers resistance to tamoxifen. This resistance is due to different mechanisms such as constitutive action of ERα, modification of coregulators activity, increased HER2 signaling and transcription factors activity such as SP1/AP1. AE can either downregulate cell cycle activators and/or upregulate inhibitors such as p21waf1 or p27kip1. Modulation of expression of these targets prevents the growth inhibitory effects of AE. For example, the commonly aberrant c-Myc expression observed in breast cancers can contribute to AE resistance by decreasing p21waf1 expression and HER2 overexpression reduces p27kip1 level. Furthermore posttranslational modification of p21waf1 and p27kip1 via AKT-dependent phosphorylation leads to cytoplasmic relocation and loss of their inhibitor role on cyclin/CDK complexes. AE treatments can also induce cellular stress response and apoptosis in breast cancer. At the transcriptional level, poor response to tamoxifen is associated with increase of survival gene expression such as the antiapoptotic protein Bcl2 and decrease of death factors such as the proapoptotic molecules BAK, BIK or caspases. This involves a direct action of ERα on transcription and/or PI3K/AKT, HER2 and NFκB signaling pathways. In AE-resistant cell lines, Bcl2, which is overexpressed, presents a constitutive activation and thus alters apoptotic pathway.
Recent advances in RNA research have allowed the identification of miRNAs, a new class of small noncoding regulatory RNAs. MiRNAs are involved in the regulation of gene expression at the posttranscriptional level, silencing target genes after binding to the 3′ untranslated region (UTR) of mRNA. In breast cancers, miRNAs expression is often dysregulated and an aberrant miRNAs expression signature is associated with endocrine resistance.24,25 Several miRNAs are known to be involved in resistance and target genes implicated in cell cycle/apoptosis or in signaling pathways. For example miR-221, miR-222 and miR-206 are known to target ERα and let-7 targets the truncated variant ERα36.26,27 Genes involved in cell cycle (CCND1, p27kip1) and apoptosis (BCL2) seem to be direct targets of miR-15a/16 and miR-221/222, whose expression is often altered in breast cancers overexpressing HER2.28,29 PTEN, a known tumor suppressor often found mutated in a large number of cancers, is a target of miR-301 and miR-101 and decreased PTEN expression allows AKT pathway activation.30,31 Increased expression of miR-26a and miR-181a inhibits progesterone receptor PgR expression, a key actor in estrogen signaling.32 Because miRNAs have the potential to deliver clear clinical benefits as biomarkers and therapeutics in the treatment of breast cancer, a better characterization of their role in the pathogenesis of breast cancer is highly warranted.
Epigenetic Modifications Associated With AE-Resistance
Recent technological advances in genomic studies have enabled a rapid increase in our understanding of transcriptional mechanisms. Chromatin structure, which control DNA accessibility, generally has a repressive effect on the recruitment of the transcriptional machinery, including transcription factors such as ERα. The chromatin structure can be rendered more permissive to transcription by the 3 following mechanisms: ATP-dependent chromatin remodeling, modification of nucleosome composition by replacement of canonical histones with histone variants and modification of epigenetic marks. For example, the histone variant H2A.Z is clearly implicated in the regulation of ERα-dependent genes and in the stabilization of nucleosomes at the promoter, an essential step in the recruitment of the transcriptional machinery.33 Furthermore, H2A.Z overexpression increases growth in a cancer cell model and is associated with poor prognosis for breast tumors, suggesting that it could be an important factor in the establishment of endocrine resistance.34,35 The alteration of epigenetic marks is another mechanism influencing transcription by posttranslational modifications of histones. Recent studies have shown that histone methylation and acetylation are involved in carcinogenesis and could be biomarkers of recurrent breast cancers. It has recently shown that methylation of histone H3 lysine 27 (H3K27) imposes ligand-dependent regulation of the ERα-dependent apoptotic response via Bcl-2 in breast cancer.36 In hormone responsive breast cancer cells, the histone demethylase JMJD3 and ERα act together to remove the repressive H3K27me3 mark from enhancer region, allowing gene activation and antiapoptotic effect of Bcl-2. In AE-resistant cells, this pathway is altered and Bcl-2 is expressed constitutively due to a lack of H3K27 methylation caused by inactivation of the methyltransferase EZH2.
An emerging class of transcription factors, defined as pioneer factors, dictates global chromatin structure and directs recruitment of transcription factors to establish lineage-specific transcriptional programs.37,38 The forkhead protein FOXA1 has been increasingly described as one such pioneer factor and is an important driver of ERα binding to the chromatin. FOXA1 binding is favored by the presence of nucleosome-harboring H3K4me1/me2, an active epigenetic mark. Expression of FOXA1 predicts a more favorable outcome in breast cancer patients. In TAM-resistant breast cancer cells, ER-binding events acquired during resistance correlate with a gain in FOXA1 binding. Silencing of FOXA1 in these cells decreases proliferation and ERα binding demonstrating that FOXA1 is required for tamoxifen action in both AE-sensitive and AE-resistant contexts.39 Similar results have been obtained by the same team in primary breast cancers from patients with different clinical outcome. Differential ERα- binding program observed in tumors was due to the redistribution of FOXA1 binding events correlating with the outcome of the patients.40 PBX1, another pioneer factor, has been recently described as a driver of ERα-mediated transcriptional response in breast cancer cells capable of guiding ERα recruitment to a specific subset of sites. PBX1 transcriptional program is associated with poor outcome in breast cancer patients and its expression is associated with a high risk of metastasis.41 Although a link with AE-resistance has not been directly established, PBX1 could have an important role in the development of drug resistance in breast cancer. Taken together, these data suggest that PBX1 and FOXA1 could be used as prognostic biomarkers and therapeutic targets.
DNA methylation at the promoter regions is a known important feature in cancer development. Hypermethylation of CpG islands in tumor suppressor gene promoters often leads to inactivation of transcription by altering chromatin conformation and local histone marks. Hypomethylation of oncogene promoters confers resistance to endocrine therapies. Indeed, comparison of gene expression and DNA methylation profiles in estrogen-responsive breast cancer cells and AE-resistant derivatives have shown significant changes in downstream ERα target gene networks contributing to the acquisition of resistance.42 For example, the PTEN promoter is methylated by increased expression of the DNA methyltransferase DNMT1 in TAM- resistant breast cancer cells. This results in the downregulation of PTEN expression leading to increased AKT phosphorylation.43 DNA methyltransferase 3B (DNMT3B) is recruited by the transcription factor Twist to the ERα promoter in TAM-resistant cells, inducing ERα promoter hypermethylation and reducing ERα expression.44 This example illustrates one of the mechanisms causing loss of ERα activity often observed in breast cancers which could contribute to the development of hormone resistance.
Integration Of Signaling Pathways To Chromatin Events In AE-Resistance
ERα can be activated by signaling pathways involving growth factors, such as EGF, in the absence of estrogen in breast cancer cells. Lupien et al. have shown that EGF treatment induces a transcriptional program distinct from estrogen treatment.45 This different ERα cistrome specifically regulates genes found overexpressed in HER2-positive human breast cancers, explaining endocrine therapy resistance observed in ERα-positive breast cancers that overexpress HER2. This suggests that for optimal treatment it is important to target both pathways, i.e. ERα signaling and HER2 signaling, to have a stronger growth inhibition response. Moreover, overexpression of the methyltransferase EZH2 is a marker of advanced and metastatic stages in prostate and breast cancers. Elevated AKT levels are associated with low levels of H3K27me3. This effect is due to direct interaction of EZH2 with AKT. Phosphorylation of EZH2 by AKT inhibits its interaction with histone H3 and decreases H3K27me3 expression causing target gene reactivation. As described above, we have previously shown that in AE- resistant cancer cells, Bcl-2 expression is altered due to the demethylation of H3K27me3, caused by inactivation of the methyltransferase EZH2.36 This result presents the first evidence of a connection between signaling pathways and genomic events in an endocrine resistance context.
Conclusion
Although the implication of multiple signaling pathways in the acquisition of AE-resistance have been well described, the crosstalk driving downstream events at the transcriptional and chromatin level remain poorly understood. In this review, we depict the complexity of the mechanisms involved in endocrine resistance (Fig. 1), bringing to light recent results obtained from epigenetic and genomic studies. The use and development of genome-wide approaches will extend our understanding on how perturbation of transcriptional mechanisms and epigenetic modifications may influence the response of breast cancer toward AE therapies.
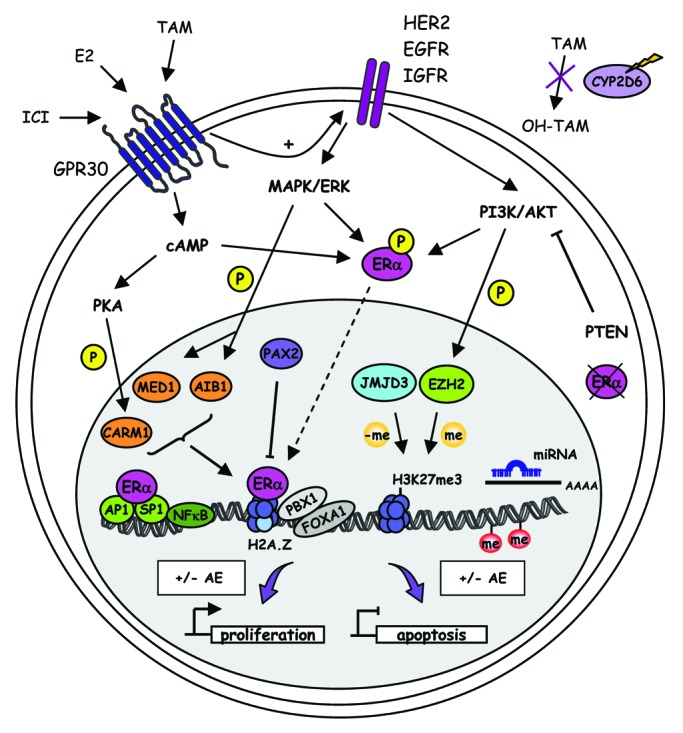
Figure 1. Molecular mechanisms in endocrine resistance. De novo resistance in breast cancer is characterized by loss of ERα expression and CYP2D6 deficiency that reduces tamoxifen to be metabolized into the active metabolite 4-hydroxytamoxifen (OH-TAM). Acquired resistance to AE involves alteration in signal transduction mediated by increased expression and/or activity of receptor tyrosine kinase family members such as HER2, EGFR and IGFR or G coupled-protein receptor GPR30. These events result in aberrant activation of downstream signaling pathways including cAMP/PKA, MAPK/ERK and PI3K/AKT. These activated kinases drive the phosphorylation of ERα and some of its coactivators, such as AIB1, MED1 or CARM1, which are responsible for constitutive ERα-mediated transcription and inhibition of AE effects on proliferation and apoptosis gene targets. Dysregulation of miRNA transcription is also involved in AE resistance. At the genomic level, substitution of canonical histones with the histone variant H2A.Z, or modification of DNA and histone methylation levels, exert an important influence on gene transcription. Pioneer factors FOXA1 and PBX1 drive ERα recruitment to specific genomic sites. In AE-resistance, the binding profile of these factors is often altered, resulting in aberrant ERα cistrome and gene expression program.
Acknowledgments
We thank Elaine Beaulieu and Alain Lavigueur for critical reading of the manuscript. This work was supported with funds from the Canadian Institute of Health Research (CIHR). N.G. holds a Chercheur boursier (junior 1) award from the FRSQ. S.B. was the recipient of a postdoctoral fellowship from Réseau Québécois en Reproduction. We apologize to those whose papers are not cited here due to space limitations.
Glossary
Abbreviations:
- AE
anti-estrogen
- ER
estrogen receptor
- E2
17β-estradiol
- TAM
tamoxifen
- ICI
ICI182780/fulvestrant
Footnotes
Previously published online: www.landesbioscience.com/journals/transcription/article/20496
References
- 1.Badve S, Nakshatri H. Oestrogen-receptor-positive breast cancer: towards bridging histopathological and molecular classifications. J Clin Pathol. 2009;62:6–12. doi: 10.1136/jcp.2008.059899. [DOI] [PubMed] [Google Scholar]
- 2.Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597–608. doi: 10.1038/nrc3093. [DOI] [PubMed] [Google Scholar]
- 3.Normanno N, Di Maio M, De Maio E, De Luca A, de Matteis A, Giordano A, et al. NCI-Naple Breast Cancer Group Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer. 2005;12:721–47. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 4.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–43. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 5.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH. Pathways to tamoxifen resistance. Cancer Lett. 2007;256:1–24. doi: 10.1016/j.canlet.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh MS, Francis PA, Michael M. Tamoxifen, cytochrome P450 genes and breast cancer clinical outcomes. Breast. 2011;20:111–8. doi: 10.1016/j.breast.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Barone I, Brusco L, Fuqua SA. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin Cancer Res. 2010;16:2702–8. doi: 10.1158/1078-0432.CCR-09-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao J, Jiang X, Wang Y, Chen B. Advances in the understanding of the structure and function of ER-α36,a novel variant of human estrogen receptor-alpha. J Steroid Biochem Mol Biol. 2011;127:231–7. doi: 10.1016/j.jsbmb.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 10.de Leeuw R, Neefjes J, Michalides R. A role for estrogen receptor phosphorylation in the resistance to tamoxifen. Int J Breast Cancer. 2011;2011:232435. doi: 10.4061/2011/232435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev. 2011;32:597–622. doi: 10.1210/er.2010-0016. [DOI] [PubMed] [Google Scholar]
- 12.Holm C, Kok M, Michalides R, Fles R, Koornstra RH, Wesseling J, et al. Phosphorylation of the oestrogen receptor alpha at serine 305 and prediction of tamoxifen resistance in breast cancer. J Pathol. 2009;217:372–9. doi: 10.1002/path.2455. [DOI] [PubMed] [Google Scholar]
- 13.Kok M, Zwart W, Holm C, Fles R, Hauptmann M, Van’t Veer LJ, et al. PKA-induced phosphorylation of ERα at serine 305 and high PAK1 levels is associated with sensitivity to tamoxifen in ER-positive breast cancer. Breast Cancer Res Treat. 2011;125:1–12. doi: 10.1007/s10549-010-0798-y. [DOI] [PubMed] [Google Scholar]
- 14.Zwart W, Griekspoor A, Berno V, Lakeman K, Jalink K, Mancini M, et al. PKA-induced resistance to tamoxifen is associated with an altered orientation of ERalpha towards co-activator SRC-1. EMBO J. 2007;26:3534–44. doi: 10.1038/sj.emboj.7601791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carascossa S, Dudek P, Cenni B, Briand PA, Picard D. CARM1 mediates the ligand-independent and tamoxifen-resistant activation of the estrogen receptor alpha by cAMP. Genes Dev. 2010;24:708–19. doi: 10.1101/gad.568410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eccles SA. The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int J Dev Biol. 2011;55:685–96. doi: 10.1387/ijdb.113396se. [DOI] [PubMed] [Google Scholar]
- 17.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29:4452–61. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat. 2011;128:457–66. doi: 10.1007/s10549-011-1584-1. [DOI] [PubMed] [Google Scholar]
- 19.Ignatov A, Ignatov T, Roessner A, Costa SD, Kalinski T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat. 2010;123:87–96. doi: 10.1007/s10549-009-0624-6. [DOI] [PubMed] [Google Scholar]
- 20.Nagalingam A, Tighiouart M, Ryden L, Joseph L, Landberg G, Saxena NK, et al. Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis. 2012;33:918–30. doi: 10.1093/carcin/bgs105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao W, Zhang Q, Kang X, Jin S, Lou C. AIB1 is required for the acquisition of epithelial growth factor receptor-mediated tamoxifen resistance in breast cancer cells. Biochem Biophys Res Commun. 2009;380:699–704. doi: 10.1016/j.bbrc.2009.01.155. [DOI] [PubMed] [Google Scholar]
- 22.Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, et al. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663–6. doi: 10.1038/nature07483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caldon CE, Sutherland RL, Musgrove E. Cell cycle proteins in epithelial cell differentiation: implications for breast cancer. Cell Cycle. 2010;9:1918–28. doi: 10.4161/cc.9.10.11474. [DOI] [PubMed] [Google Scholar]
- 24.Andorfer CA, Necela BM, Thompson EA, Perez EA. MicroRNA signatures: clinical biomarkers for the diagnosis and treatment of breast cancer. Trends Mol Med. 2011;17:313–9. doi: 10.1016/j.molmed.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Fu SW, Chen L, Man YG. miRNA Biomarkers in Breast Cancer Detection and Management. J Cancer. 2011;2:116–22. doi: 10.7150/jca.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2011;30:1082–97. doi: 10.1038/onc.2010.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y, Deng C, Lu W, Xiao J, Ma D, Guo M, et al. let-7 MicroRNAs Induce Tamoxifen Sensitivity by Downregulation of Estrogen Receptor α Signaling in Breast Cancer. Mol Med. 2011;17:1233–41. doi: 10.2119/molmed.2010.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cittelly DM, Das PM, Salvo VA, Fonseca JP, Burow ME, Jones FE. Oncogenic HER2Delta16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis. 2010;31:2049–57. doi: 10.1093/carcin/bgq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. 2008;283:29897–903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sachdeva M, Wu H, Ru P, Hwang L, Trieu V, Mo YY. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene. 2011;30:822–31. doi: 10.1038/onc.2010.463. [DOI] [PubMed] [Google Scholar]
- 31.Shi W, Gerster K, Alajez NM, Tsang J, Waldron L, Pintilie M, et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res. 2011;71:2926–37. doi: 10.1158/0008-5472.CAN-10-3369. [DOI] [PubMed] [Google Scholar]
- 32.Maillot G, Lacroix-Triki M, Pierredon S, Gratadou L, Schmidt S, Bénès V, et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009;69:8332–40. doi: 10.1158/0008-5472.CAN-09-2206. [DOI] [PubMed] [Google Scholar]
- 33.Gévry N, Hardy S, Jacques PE, Laflamme L, Svotelis A, Robert F, et al. Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev. 2009;23:1522–33. doi: 10.1101/gad.1787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Svotelis A, Gévry N, Gaudreau L. Regulation of gene expression and cellular proliferation by histone H2A.Z. Biochem Cell Biol. 2009;87:179–88. doi: 10.1139/O08-138. [DOI] [PubMed] [Google Scholar]
- 35.Svotelis A, Gévry N, Grondin G, Gaudreau L. H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle. 2010;9:364–70. doi: 10.4161/cc.9.2.10465. [DOI] [PubMed] [Google Scholar]
- 36.Svotelis A, Bianco S, Madore J, Huppé G, Nordell-Markovits A, Mes-Masson AM, et al. H3K27 demethylation by JMJD3 at a poised enhancer of anti-apoptotic gene BCL2 determines ERα ligand dependency. EMBO J. 2011;30:3947–61. doi: 10.1038/emboj.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magnani L, Eeckhoute J, Lupien M. Pioneer factors: directing transcriptional regulators within the chromatin environment. Trends Genet. 2011;27:465–74. doi: 10.1016/j.tig.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–41. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magnani L, Ballantyne EB, Zhang X, Lupien M. PBX1 genomic pioneer function drives ERα signaling underlying progression in breast cancer. PLoS Genet. 2011;7:e1002368. doi: 10.1371/journal.pgen.1002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006;66:11954–66. doi: 10.1158/0008-5472.CAN-06-1666. [DOI] [PubMed] [Google Scholar]
- 43.Phuong NT, Kim SK, Lim SC, Kim HS, Kim TH, Lee KY, et al. Role of PTEN promoter methylation in tamoxifen-resistant breast cancer cells. Breast Cancer Res Treat. 2011;130:73–83. doi: 10.1007/s10549-010-1304-2. [DOI] [PubMed] [Google Scholar]
- 44.Vesuna F, Lisok A, Kimble B, Domek J, Kato Y, van der Groep P, et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-α. Oncogene. 2011 doi: 10.1038/onc.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lupien M, Meyer CA, Bailey ST, Eeckhoute J, Cook J, Westerling T, et al. Growth factor stimulation induces a distinct ER(alpha) cistrome underlying breast cancer endocrine resistance. Genes Dev. 2010;24:2219–27. doi: 10.1101/gad.1944810. [DOI] [PMC free article] [PubMed] [Google Scholar]