

Abstract
Natural products of the caged Garcinia xanthones (CGX) family are characterized by a unique chemical structure, potent bioactivities and promising pharmacological profiles. We have developed a Claisen/Diels-Alder reaction cascade that, in combination with a Pd(0)-catalyzed reverse prenylation, provide rapid and efficient access to the CGX pharmacophore, represented by the structure of cluvenone. To further explore this pharmacophore, we have synthesized various A-ring oxygenated analogues of cluvenone and have evaluated their bioactivities in terms of growth inhibition, mitochondrial fragmentation, induction of mitochondrial-dependent cell death and Hsp90 client inhibition. We found that installation of an oxygen functionality at various positions of the A-ring influences significantly both the site-selectivity of the Claisen/Diels-Alder reaction and the bioactivity of these compounds, due to remote electronic effects.
Introduction
Tropical plants of the genus Garcinia have led to the identification of an intriguing family of xanthone-derived natural products that have interesting bioactivities and a documented value in traditional Eastern medicine.i Collectively referred to as caged Garcinia xanthones (CGXs),ii these compounds are structurally defined by an unusual architecture in which a 4-oxatricyclo[4.3.1.03,7] dec-8-en-2-one scaffold has been built onto the C-ring of a xanthone backbone. This motif is further decorated via substitutions at the A-ring and peripheral oxidations to produce a variety of related subfamilies. Gambogic acid (1, GA),iii the archetype of this family, has been studied as a potent antitumor and anti-inflammatory agent and has entered phase I clinical trials in China.iv Various biological studies have suggested that 1 can induce cell apoptosis by activating the mitochondrial pathway via mechanisms that are partly dependent on the Bcl-2 family of proteins.v Although the primary direct molecular target of 1 is still under investigation, recent studies have suggested that it can bind with low micromolar kd to the heat shock protein Hsp90,vi thereby reducing the expression levels of Hsp90-dependent clients that are involved in cell growth, apoptosis, angiogenesis and metastasis.vii
Intrigued by the unusual chemical motif and therapeutic promise of CGXs, we sought to design a general synthetic strategy that would allow us to further explore and optimize its biological properties.viii,ix These studies led to the identification of cluvenone (2, CLV), a structurally simplified CGX that retains the biological activities of 1.x Along these lines, we have shown that CLV parallels the activity of GA in inducing cell stress and apoptosis in various cancer lines at low micromolar concentration. Fluorescent labeling studies have indicated that both 1 and 2 localize in mitochondria and induce significant changes in the morphology and structure of this organelle, thereby leading to cell apoptosis.xi It has been proposed that the C9-C10 enone functionality of these compounds contributes to their bioactivity, presumably by acting as a conjugate electrophile.9a,b To further explore and optimize the CGX pharmacophore, we synthesized A-ring oxygenated xanthones 3 and 4 and compared their activities to those of 1, 2 and 5.10 Here we show that that installation of an oxygen group at C6 or C18 positions of the CGX motif (gambogic acid numbering) affects significantly both the synthesis and the bioactivity of these compounds.
Results and Discussion
Synthesis of A-ring hydroxylated caged Garcinia xanthones
The synthesis of caged Garcinia xanthones containing a protected phenolic group at the C6 and C18 positions of the A ring is highlighted in Schemes 1 and 2. Acyl chloride 7a, prepared via oxalyl chloride/DMF-mediated chlorination of commercially available 2,6-dimethoxybenzoic acid (6a), was subjected to an AlCl3-catalyzed Friedel-Crafts acylation with pyrogallol trimethyl ether (8) (Scheme 1). The resulting benzophenone intermediate was treated with NaOH to produce xanthone 9a in 90% combined yield. Demethylation of 9a with 48% HBr in AcOH gave rise to hydroxylated xanthone 10a (95% yield). Protection of the C-ring catechol of 9a as the corresponding diphenylketal, followed by alkylation of the C6 phenol with MOMCl/NaH and deprotection of the ketal functionality yielded xanthone 11a (3 steps, 56% overall yield).9d Pd(0)-catalyzed reverse prenylation of 11a with carbonate 12 produced diallyl ether 13a in 63% yield.10 In a similar manner, 13b was produced from 2,4-dimethoxybenzoic acid (6b) in 7 steps and 17% overall yield.
Scheme 1.

Reagents and conditions: (a) 5.0 equiv. oxalyl chloride, DMF (cat.), DCM, 16 h, 25 °C; (b) 3.0 equiv. AlCl3, Et2O, 18 h, 25 °C then 9.0 equiv NaOH, MeOH:H2O (1.5:1), 48 h, 110 °C, 90% for 9a, 58% for 9b; (c) 48% HBr: HOAc (1:2), 12 h, 120 °C, 95% for 10a, 81% for 10b; (d) 1.6 equiv. Ph2CCl2, Ph2O, 4 h, 175 °C; (e) 2.5 equiv. MOMCl, 2.5 equiv NaH, acetone, 12 h, 25 °C; (f) H2, 10% Pd/C, THF:MeOH (3:1), 18 h, 25 °C, 56% for 11a; 43% for 11b (over 3 steps); (g) 10 equiv. 12, 10 mol% Pd(PPh3)4, THF, 2 h, 5 °C, 63% for 13a, 84% for 13b.
Scheme 2.

Reagents and conditions: (a) 1.0 equiv. ZnCl2 (0.5 M in THF), THF, 2-24 h, 40 °C, 93% for 14, 0% for 18, 75% for 4 and 75% for 21; (b) DMF, 120 °C, 2 h, 100%.
Conversion of 13a and 13b to the corresponding caged structures was achieved via the tandem Claisen/Diels-Alder reaction cascade (Scheme 2).xii To this end, heating of 13a (DMF, 120 °C, 2 h) produced two isomeric caged xanthones: the “regular” caged structure 15 (80% yield) that is formed upon initial Claisen migration of the C12 allyl ether at the C13 position, and the “neo” caged structure 16 (20% yield) that is formed upon initial Claisen migration of the C13 allyl ether at the C12 position (gambogic acid numbering). On the other hand, ZnCl2-induced cleavage of the MOM group (40 °C, 2 h) of 13a formed 14 (93% yield) that, upon heating, underwent the Claisen/Diels-Alder reaction to form regular caged xanthone 3 (92% yield) together with the neo caged compound 17 (8% yield). In a similar manner, heating of 13b (DMF, 2 h, 120 °C) produced the regular caged structure 19 (80% yield) together with the neo structure 20 (20% yield). To our surprise, removal of the MOM group from 13b under ZnCl2 conditions proved to be problematic and led to partial or full loss of the dimethylallyl ethers. Thus, this deprotection was accomplished after conversion of 13b to the corresponding caged compounds 19 and 20. To this end, treatment of 19 and 20 with ZnCl2 (40 °C, 24 h) afforded caged xanthones 4 and 21 respectively in ca 75% yield.xiii
The products distribution of the Claisen/Diels-Alder reaction deserves some comments. We have previously postulated that the site selectivity of this reaction can be rationalized by considering the electronic deficiency of the B-ring carbonyl group, that, being para to the C12 allyl ether, facilitates its rupture during the inaugural Claisen rearrangement.13 In turn, Claisen migration of this group at the C13 position leads to a site selective Diels-Alder reaction ultimately forming the regular caged motif as the major product. In contrast, the competing Claisen migration of the C13 dimethylallyl ether at the C12 position is not electronically favored. Thus, the corresponding neo caged motif is formed only as the minor product.xiv,xv The results of our study with compounds 13a, 13b and 14 further support this proposal. Specifically, the Claisen/Diels-Alder reaction of 14 produces the regular and the neo caged compounds 3 and 17 in a 92:8 ratio. This enhanced site selectivity can be explained by considering that the C6 hydroxyl group increases the electron deficiency of the B-ring carbonyl group via hydrogen bonding and thus facilitates the rupture of the C12 allyl ether, leading predominantly to the regular caged motif of 3. In contrast, the presence of a MOM ether at C6 or C18 (i.e. 13a or 13b) donates electron density at the B-ring carbonyl group thus attenuating the site selectivity of the Claisen/Diels-Alder reaction (regular/neo motifs: 80/20).
Cell proliferation studies of A-ring hydroxylated caged Garcinia xanthones
The ability of the synthesized CGXs to inhibit cancer cell growth was evaluated in the T-cell acute lymphoblastic leukemia cell line (CEM). Based on our previous studies, this cell line was found to be one of the most sensitive.8 The inhibitory concentrations at 50% inhibition (IC50) of the active CGXs ranged from 0.12-1.0 μM. Compound 510 was found to be relatively inactive with an IC50 greater than 5 μM (Table 1 and Figure 2).
Table 1.
Inhibition of cell proliferation by CGXs in CEM cells
| Compound | IC50 (μM) |
|---|---|
| 1 | 0.12 |
| 2 | 0.18 |
| 3 | 0.3 |
| 4 | 1.0 |
| 5 | 5.1 |
| 15 | 0.5 |
| 19 | 0.5 |
Figure 2.

Inhibition of 3H-thymidine incorporation by selected CGXs in CEM cells. Cells were treated with increasing concentrations of various CGXs for 48 h and pulsed with 3H-thymidine prior to harvesting.
It is interesting to relate the activities of these CGXs to their structure, in particular as they relate to the reactivity of the C9-C10 enone as a conjugate electrophile (Table 1 and Figure 2). Comparison of 3 with 15 suggests that the presence of a free phenolic group at the C6 position of the A ring (i.e. compound 3) increases the potency of this compound, while its protection as a MOM ether (i.e. compound 15) decreases its potency. This modulation of potency parallels the effect of the C6 oxygen on the activation of the C9-C10 enone. Specifically, in compound 3, the C6 free phenolic functionality can create a hydrogen bond with the adjacent carbonyl group, thereby enhancing the electrophilicity of the C10 center. In contrast, the MOM ether functionality at C6 of 15 enriches the electron density of the B-ring carbonyl group (vinylogous carbonyl system), thereby reducing the electrophilicity of the C10 center. In a similar manner, compound 19 (containing a MOM ether at C18) is twice as potent as 4 (that contains a free phenolic group at C18). This is consistent with the prediction that a free phenolic group para to the B ring carbonyl would act as a strong electron donor, thereby reducing the electrophilicity of the C10 center. Along these lines, compound 5 (containing a methyl group at C10) is about twenty times less potent than 2 in which the C10 center is not substituted and thus more accessible for a conjugate addition reaction with a bionucleophile. Overall, these comparative studies support the notion that increasing the electrophilicity of the C10 enone enhances the potency of the CGX motif.
Effect of GA and CLV on mitochondria
We have previously reported that CLV (2) and GA (1) localize in mitochondria, and induce mitochondrial fragmentation leading to activation of caspase-3 (C3) ultimately resulting in cell apoptosis.11 Despite having similar phenotypes, the cellular effects of 1 and 2 have not been compared. To this end, we began by analyzing the timing of induction of the apoptotic pathway, by western blot (WB) analysis of active caspase-9, a known initiator caspase,xvi and caspase-3, the known downstream executioner of apoptosis.xvii Caspase-9 (C9) was examined due to its well established connection to the mitochondrial apoptotic pathway and its subsequent activation of C3. HeLa cells were treated with 2 μM GA (Figure 3a) or CLV (Figure 3b) from 0 min (control cells, incubated with equal volume of DMSO) to 180 min. Both compounds induced similar activation of C9 and C3 after about 120 min of incubation. In fact, C9 appears to be activated right before C3 since it is visible after about 90 min of incubation.
Figure 3.

Comparison between GA and CLV with respect to mitochondrial fragmentation and induction of apoptosis. Cells treated with GA (a) or CLV (b) for the indicated amounts of time (from 0 min to 180 min) were processed for WB analysis to show activation of the mitochondria-dependent apoptosis pathway, represented by caspase-9 (C9) and by the downstream executioner caspase-3 (C3). Cells treated with GA for 0 min (c), 30 min (d) and 60 min (e) or with CLV for 0 min (f), 30 min (g) and 60 min (h) were analyzed by IF to reveal the status of the mitochondria. Mitochondria are in red, nuclei are in blue. Tubulin (bottom line) was used as loading control.
We then evaluated the status of the mitochondrial network by immunofluorescence (IF). As shown in Figure 3 (g-h), both GA and CLV induced a similar degree of mitochondrial disruption. Both compounds were found to induce a first degree of fragmentation after 30 min incubation, at which time the mitochondrial network appeared fragmented in larger discrete elements (Figure 3d for GA and 3g for CLV). At longer incubation times (1h treatment for both compounds) the mitochondria appeared collapsed in the perinuclear area and the mitochondrial network was no longer visible (Fig. 3e and 3h).
Comparison of the IF results with the WB analysis supports the notion that mitochondrial fragmentation (visible after 30 min incubation) appears before the activation of the initiatory C9 (visible after 90 min incubation). This observation confirms that both GA and CLV induce cell death via the intrinsic pathway, in which mitochondrial damage leads initially to C9 activation that subsequently activates C3. These results also suggest that GA and CLV act with a similar timing to activate the mitochondrial-dependent (intrinsic) apoptotic pathway.
Effect of A-ring hydroxylated caged Garcinia xanthones on apoptosis and mitochondria
We then evaluated the ability of compounds 3, 4 and 5 to induce mitochondrial fragmentation and cell death in HeLa cells. Cells incubated for 60 to 180 min with CLV and derivatives 3, 4 and 5 were analyzed to assess induction of apoptosis by WB analysis of active caspase-3 (C3). As shown in Figure 4, 2 and 3 were able to induce apoptosis after 90 min incubation (compare lanes 3 and 7). On the other hand, compound 4, which carried the phenolic group at C18 was less effective, inducing C3 activation only after 120 min treatment (lane 12). Moreover, compound 5 proved to be inactive at the concentrations tested (2 μM). Even at much higher concentrations (up to 80 μM), 5 proved to be the least effective of the compounds, requiring 180 min to induce comparable levels of apoptosis. The results parallel the potency profile of these compounds and provide additional arguments in favor of the role of A ring hydroxylation in modulating the reactivity of the C9-C10 enone as an electrophile.
Figure 4.
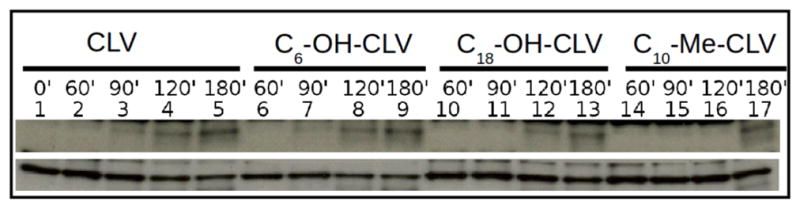
Effect of selected CGXs on apoptosis. Cells were treated for 60 min, 90 min, 120 min, 180 min with 2 μM CLV (lanes 2–5), 2 μM 3 (lanes 6–9), 2 μM 4 (lanes 10–13) and 80 μM 5 (lanes 14–17). Control cells treated with equal volume of DMSO are in lane 1. The cells were then collected and analyzed by WB to measure induction of apoptosis by caspase-3 (C3) activation. Both CLV and 3 show active caspase-3 starting at 90 min (lanes 3 and 7 respectively); 4 requires 120 min incubation to achieve comparable levels of activation; 5 induced caspase-3 activation only after 180 min incubation at 80 μM (lane 17). Tubulin (bottom line) was used as loading control.
We also tested compounds 3, 4 and 5 for their ability to induce mitochondrial fragmentation. Control untreated cells are shown in Figure 5a. Compound 3 induced mitochondrial disruption after 30 min of incubation (Figure 5b), while extensive levels of fragmentation were reached after 60 minutes of incubation (Figure 5c). The action of compound 3 was therefore similar to that of CLV at identical time points (compare with Figures 3g and 3h respectively). To achieve similar levels of fragmentation, compound 4 had to be incubated for 60 min (Figure 5e) and 90 min (Figure 5f). Moreover, compound 5 showed the lowest levels of fragmentation, being able to induce partial mitochondrial disruption only after 120 min incubation at 80 μM concentration (Figure 5i). Overall, the findings suggest that the extent of mitochondrial fragmentation parallels the activation of the C9-C10 enone as an electrophile.
Figure 5.

Effect of 3, 4 and 5 on mitochondria. Cells treated with compounds 3, 4 and 5 were imaged to visualize mitochondrial structure by IF. DMSO-treated control cells are in (a). Top panel: treatment with 3 for 30 min (b) and 60 min (c). Middle panel: treatment with 4 for 30 min (d), 60 min (e) and 90 min (f). Bottom panel: treatment with 5 for 60 min (g), 90 min (h), 120 min (i). Mitochondria are shown in red; nuclei are in blue. Intact mitochondria are shown in a, d, g and h. Initial level of fragmentation is observed in b, e, and i. More extensive fragmentation is observed in c and f.
The caged Garcinia xanthones induce irreversible mitochondrial fragmentation
Inspired by the above findings we sought to evaluate if the effect of CGXs on mitochondria can be reversed upon removal of these compounds from the incubation media. We begun by treating HeLa cells with 2μM of GA or CLV for 0 min, 15 min and 30 min. The cells were then allowed to recover for 90 min in fresh medium and subsequently were fixed and processed for IF. As shown in Figure 6, 30 min incubation with GA (Figure 6c) and CLV (Figure 6f) were necessary and sufficient to induce a permanent mitochondrial fragmentation, while shorter time point (15 min) proved to be not effective and mitochondrial integrity was recovered (Figure 6b for GA and Figure 6e for CLV).
Figure 6.

Irreversibility of mitochondrial fragmentation by GA and CLV (a-f) Cells were assessed by IF for their ability to regain mitochondrial integrity after 90 min of recovery in fresh medium following treatment with GA for 0 min (a), 15 min (b) and 30 min (c) or CLV for 0 min (d), 15 min (e) and 30 min (f). Recovery after 30 min of treatment for both GA (c) and CLV (f) could not restore mitochondrial integrity. Mitochondria are shown in red; nuclei are in blue. (g-h) Western Blot analysis was used to measure the degree of irreversibility by caspase-3 (C3) activation. As shown in (g) (GA) and (h) (CLV), while incubation times shorter than 90 min did not induce C3 activation (lanes 1-4 and lanes 10–12), after recovery in the absence of the drugs, C3 was shown to be active once the compounds had been present on the cells for more than 30 min (lanes 7–9 and 15–17). Tubulin (bottom line) was used as loading control.
To assess whether the phenotype of the irreversible mitochondrial fragmentation corresponded to an irreversible effect on apoptosis we incubated cells with GA and CLV for 0 min, 30 min, 45 min, 60 min and 90 min, and then allowed them to recover for 90 min in fresh medium. The cells were then analyzed by western blot in order to visualize caspase-3 activation. As shown in Figure 6 (g, h), no signs of apoptosis (measured by caspase-3 activation) were visible prior to 90 min incubation with either GA (Figure 6g, lanes 1–5) or CLV (Figure 4h, lanes 10–13). However, when cells were treated for 45 min and then allowed to recover for 90 min (Figure 6g lane 7 and Figure 6h lane 15), caspase-3 was active, indicating that, during 45 min of incubation, both GA and CLV were inducing irreversible morphological changes to mitochondrial structure leading to the irreversible activation of the apoptotic pathway.
We then tested the irreversibility of mitochondrial fragmentation upon exposure to compounds 3, 4 and 5 (Figure 7). The cells were treated as in Figure 6; after the incubation with CGXs for different amounts of time, the cells were subjected to 90 min recovery in fresh medium and were then processed for IF and WB analysis to detect mitochondria fragmentation and caspase-3 activation.
Figure 7.
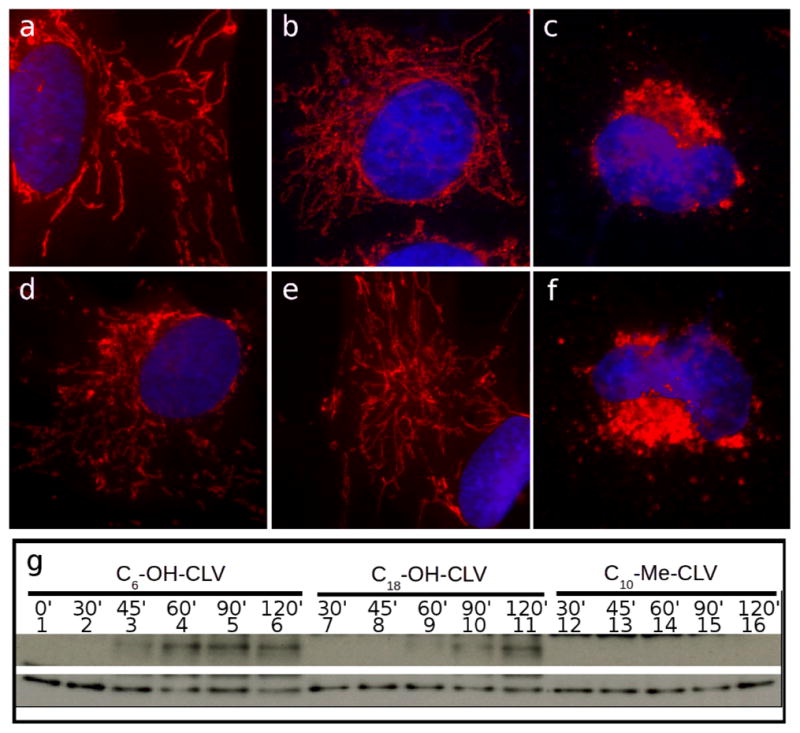
Irreversibility of mitochondrial fragmentation by 3, 4 and 5. Cells were incubated with CGXs for the specified amounts of time, then subjected to 90 min recovery in fresh medium and processed for IF. Mitochondria are in red, nuclei are in blue. Top panel: Treatment with 3 prior to recovery for 0 min (a), 15 min (b) and 30 min (c). Bottom panel: Treatment with 4 prior to recovery for 15 min (d), 30 min (e) and 45 min (f). Sign of irreversibility (indicated by the failure to recover intact mitochondrial network) are shown in (c) and (f). WB analysis indicating caspase-3 activation is shown in (g). Compound 3 needs a minimum of 45 min to induce activation of caspase-3 (lane 3). Compound 4 needs at least 60 min (lane 9), while 5 proved to be reversible up to 120 min incubation (data not shown). Tubulin (bottom line) was used as loading control.
All the compounds were incubated at 2 μM (with the exception of 5 that was used at 80 μM) for times ranging from 0 min to the minimun time necessary to induce caspase-3 activation, at 15 min intervals. This time varies from each compound, being equal to 90 min for 3, 120 min for 4 and 180 min for 5. After washing of the compounds, the cells were allowed to recover for 90 min and processed for IF. Figure 7 shows the results for compounds 3 and 4. Compound 5 proved to have a reversible effect at the times and concentrations tested (data not shown). Compound 3 behaved similarly to CLV, since 30 min incubation were sufficient to induce an irreversible mitochondrial fragmentation (Figure 7c). On the other hand, compound 4 proved to be less effective, requiring 45 min (Figure 7f) to induce the same level of mitochondrial disruption. We then performed WB analysis to confirm that the irreversible mitochondria fragmentation corresponded to the activation of caspase-3. As shown in Figure 7g, compound 3 induced caspase activation after 45 min of incubation, reproducing the results obtained for both CLV and GA. Analogue 4 was once again less effective, inducing caspase-3 activation following 45 min of treatment. Compound 5 was ineffective for up to 120 min of incubation.
Effect of A-ring hydroxylated caged Garcinia xanthones on modulation of Hsp90
Hsp90 is a nodal protein that plays critical role in protein folding and is essential to cell proliferation.7 In cancer cells this protein is overexpressed and also exists in a high-affinity conformation that facilitates malignant progression.xviii, As such, it represents an important target for the development of anticancer agents.xix Treatment of different cancer cell lines with known Hsp90 inhibitors is known to induce degradation of kinases, transcription factors and other protein clients of Hsp90.xx Encouraged by recent studies suggesting that Hsp90 is a major target of GA,6 we evaluated the effect of CLV and its A-ring hydroxylated derivatives 3 and 4 on this protein in SKBR3 cells, a breast cancer cell line. The cells were treated with different concentrations of GA, CLV, 3 and 4 or just the solvent (DMSO) for 24 hours and analyzed for several different client kinases, as well as Hsp70 and Hsp90. As seen on Figure 8, the active form of the survival kinase Akt, pAkt (anti-phospho Ser473) disappears faster than the total Akt (tAkt). The kinase Raf-1 that functions in the MAPK/ERK signaling pathway controlling cell growth degrades in a concentration dependent manner for all compounds tested. Also, Her2 kinase, a well established therapeutic target for breast cancer, proves to be extremely sensitive to treatment with GA, CLV and 3 (starts degrading at the lowest dose tested) and less sensitive to compound 4 (degradation happens only at the higher concentration used). Additionally, the mass induction of Hsp70 and the less prominent induction of Hsp90, are indicators of Hsp90 inhibition, upon which the heat shock factor (HSF1) becomes activated and leads to transcriptional induction of the heat shock genes (e.g. Hsp70). Comparisons of the amounts of client degradation and induction of Hsp70 using the different compounds, suggests that both CLV and 3 behave similarly and they resemble the client degradation rates and Hsp70 induction observed with GA.
Figure 8. Gambogic acid (GA), CLV, 3 and 4 induce degradation of Hsp90 client proteins.

SKBR3 cells were treated with different concentrations of the drugs or DMSO (control) for 24 hours. Cell lysates were fractionated by SDS-PAGE and analyzed for pAkt (S473), tAkt (total Akt), Raf-1, Her2 (indicated by an arrowhead; the asterisk depicts a non specific band), Hsp70 and Hsp90. GAPDH was used as a loading control.
In order to test the SKBR3 cells for induction of apoptosis, after treating the cells with the same concentrations of GA, CLV, 3 and 4 that lead to degradation of Hsp90 clients, we analysed the cell lysates for cleavage of PARP, a DNA polymerase involved in DNA repair and cell death (Figure 9). Again, 24 hours post treatment, even at the low concentration of 0.5 μM both GA and CLV induce cleavage of PARP (~MW 89-lower band) which clearly indicates that the cells are already undergoing apoptosis. Comparison between CLV and 3, shows that both compounds are able to induce apoptosis at similar rates (compare CLV and 3 at 2 μM). As expected, compound 4 did not induce significant apoptosis at the concentrations used.
Figure 9. GA, CLV, 3 and 4 induce apoptosis in a concentration dependent manner in SKBR3 cells.
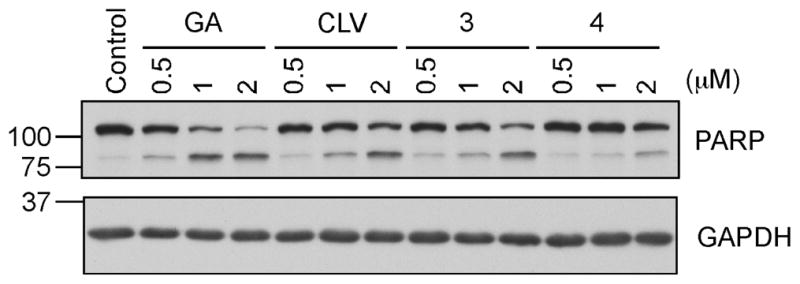
SKBR3 cells, after treatment with the indicated concentrations of the compounds or DMSO (control) for 24 hours, were lysed and analyzed for cleavage of PARP by SDS-PAGE. GAPDH was used as a control for equal loading.
Conclusions
Inspired by the biological potential of cluvenone (CLV) and related caged Garcinia xanthones (CGXs), we synthesized selected A-ring hydroxylated analogues and evaluated their effect on cell growth, mitochondrial fragmentation, mitochondriotoxicity and Hsp90 client protein degradation. We found that both the C6 and C18 hydroxylated cluvenones inhibit growth of CEM cells at low micromolar concentrations and induce cell death via the mitochondrial pathway. In addition, gambogic acid, CLV and the hydroxylated cluvenones induce Hsp 90-dependent protein client degradation at low micromolar concentrations. Interestingly, the site of A ring hydroxylation significantly influences the site selectivity of the Claisen/Diels-Alder cascade. For instance, introduction of a MOM ether group at C6 or C18 (e.g. compound 4) increases via resonance the electron density of the B ring carbonyl group leading to formation of both regular and neo caged motifs (4:1 ratio in favor of the regular caged structure). In contrast, the presence of a free phenolic group at C6 (i.e. compound 3) decreases the electron density of the B ring carbonyl, thus favoring formation of the regular motif (produced in 11.5:1 ratio over the neo caged motif). Importantly, the site of A ring hydroxylation also affects the bioactivity of the CGX motif. Specifically, 3 is approximately 3 times more active than 4 in inhibiting cell growth of CEM cells. A similar trend is observed in the mitochondrial fragmentation, the caspase activation and the Hsp90 client inhibition assays. We postulate that this trend stems from tuning the reactivity of the C9-C10 enone as a conjugate electrophile. In support of this postulate, methylation at C10 (i.e. compound 5) significantly limits the reactivity of this enone thereby decreasing the overall bioactivity of the CGX motif. Overall, our studies indicate that the A-ring of the caged Garcinia xanthones is amenable to functionalization in a manner that modulates not only the site selectivity of the Claisen/Diels-Alder reaction cascade but also their biological potency.
Supplementary Material
Figure 1.

Structures of selected caged Garcinia xanthones (CGXs).
Acknowledgments
Financial support from the National Institutes of Health (CA 133002) is gratefully acknowledged. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968. We also thank Dr. Anthony Mrse (UCSD NMR Facility) and Dr. Yongxuan Su (UCSD MS Facility).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedues for all reactions and biological assays and copies of 1H and 13C NMR spectra for compounds 3, 4 and 9-21.
Notes and references
- i.For selected general references on this topic see: Ollis WD, Redman BT, Sutherland IO, Jewers K. J Chem Soc Chem Commun. 1969:879–880.Kumar P, Baslas RK. Herba Hungarica. 1980;19:81–91.
- ii.For selected recent reviews on caged Garcinia xanthones see: Chantarasriwong O, Batova A, Chavasiri W, Theodorakis EA. Chem Eur J. 2010;16:9944–9962. doi: 10.1002/chem.201000741.Pouli N, Marakos P. Anti-Cancer Agents Med Chem. 2009;9:77–98. doi: 10.2174/187152009787047699.Han Q-B, Xu HX. Curr Med Chem. 2009;16:3775–3796. doi: 10.2174/092986709789104993.
- iii.(a) Yates P, Karmarkar SS, Rosenthal D, Stout GH, Stout VF. Tetrahedron Lett. 1963;4:1623–1629. [Google Scholar]; (b) Anthony A, Desiraju GR. Supramol Chem. 2001;13:11–23. [Google Scholar]; (c) Weakley TJR, Cai SX, Zhang HZ, Keana JFW. J Chem Crystallogr. 2001;31:501–505. [Google Scholar]; (d) Ren Y, Yuan C, Chai H, Ding Y, Li XC, Ferreira D, Kinghorn AD. J Nat Prod. 2011;74:460–463. doi: 10.1021/np100422z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iv.(a) Guo Q, Qi Q, You Q, Gu H, Zhao L, Wu Z. Basic & Clin Pharmacol & Toxicol. 2001;31:178–184. doi: 10.1111/j.1742-7843.2006.pto_485.x. [DOI] [PubMed] [Google Scholar]; (b) Wu X, Cao S, Goh S, Hsu A, Tan BKH. Planta Med. 2002;68:198–203. doi: 10.1055/s-2002-23142. [DOI] [PubMed] [Google Scholar]; (c) Guo Q, Qi Q, Gu H, Wu Z. Basic & Clin Pharmacol & Toxicol. 2006;99:178–184. doi: 10.1111/j.1742-7843.2006.pto_485.x. [DOI] [PubMed] [Google Scholar]; (d) Yang Y, Yang L, You Q-D, Nie F-F, Gu H-Y, Zhao L, Wang X-T, Guo Q-L. Cancer Lett. 2007;256:259–266. doi: 10.1016/j.canlet.2007.06.014. [DOI] [PubMed] [Google Scholar]; (e) Zhou ZT, Wang JW. Chin J New Drugs. 2007;16:79–82. [Google Scholar]; (f) Qi Q, You Q, Gu H, Zhao L, Liu W, Lu N, Guo Q. J Ethnopharmacol. 2008;117:433–438. doi: 10.1016/j.jep.2008.02.027. [DOI] [PubMed] [Google Scholar]; (g) Gu H, You Q, Liu W, Yang Y, Zhao L, Qi Q, Zhao J, Wang J, Lu N, Ling H, Guo Q, Wang X. Intern Immunopharmacol. 2008;8:1493–1502. doi: 10.1016/j.intimp.2008.05.013. [DOI] [PubMed] [Google Scholar]; (h) Qiang L, Yang Y, You QD, Ma YJ, Yang L, Nie FF, Gu HY, Zhao L, Lu N, Qi Q, Liu W, Wang XT, Guo QL. Biochem Pharmacol. 2008;75:1083–1092. doi: 10.1016/j.bcp.2007.10.033. [DOI] [PubMed] [Google Scholar]; (i) Zhao L, Zhen C, Wu Z, Hu R, Zhou C, Guo Q. Drug & Chem Toxicol. 2010;33:88–96. doi: 10.3109/01480540903173534. [DOI] [PubMed] [Google Scholar]
- v.(a) Liu W, Guo QL, You QD, Zhao L, Gu HY, Yuan ST. World J Gastroenterol. 2005;11:3655–3659. doi: 10.3748/wjg.v11.i24.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhai D, Jin C, Shiau C-W, Kitada S, Satterthwait AC, Reed JC. Mol Cancer Ther. 2008;7:1639–1646. doi: 10.1158/1535-7163.MCT-07-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xu X, Liu Y, Wang L, He J, Zhang H, Chen X, Li Y, Yang J, Tao J. Int J Dermatol. 2009;48:186–192. doi: 10.1111/j.1365-4632.2009.03946.x. [DOI] [PubMed] [Google Scholar]
- vi.(a) Zhang L, Yi Y, Chen J, Sun Y, Guo Q, Zheng Z, Song S. Biochem & Biophys Res Commun. 2010;403:282–287. doi: 10.1016/j.bbrc.2010.11.018. [DOI] [PubMed] [Google Scholar]; (b) Davenport J, Manjarrez JR, Peterson L, Krumm B, Blagg BS, Matts RL. J Nat Prod. 2011;74:1085–1092. doi: 10.1021/np200029q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.For selected reviews on this topic see: Sreedhar AS, Csermely P. Pharmacol & Therapeut. 2004;101:227–257. doi: 10.1016/j.pharmthera.2003.11.004.Taipale M, Jarosz DF, Lindquist S. Nature Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918.Hartl FU, Bracher A, Hayer–Hartl M. Nature. 2011;475:324–332. doi: 10.1038/nature10317.McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036.Da Silva VCH, Ramos CHI. J Proteomics. 2012;75:2790–2802. doi: 10.1016/j.jprot.2011.12.028.Whitesell L, Lindquist SL. Nature Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716.Pearl LH, Prodromou C. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738.
- viii.Batova A, Lam T, Wascholowski V, Yu AL, Giannis A, Theodorakis EA. Org Biomol Chem. 2007;5:494–500. doi: 10.1039/b612903j. [DOI] [PubMed] [Google Scholar]
- ix.(a) Zhang HZ, Kasibhatla S, Wang Y, Herich J, Guastella J, Tseng B, Drewe J, Cai SX. Bioorg Med Chem. 2004;12:309–317. doi: 10.1016/j.bmc.2003.11.013. [DOI] [PubMed] [Google Scholar]; (b) Kuemmerle J, Jiang S, Tseng B, Kasibhatla S, Drewe J, Cai SX. Bioorg Med Chem. 2008;16:4233–4241. doi: 10.1016/j.bmc.2008.02.084. [DOI] [PubMed] [Google Scholar]; (c) Li NG, You QD, Huang XF, Wang JX, Guo QL, Chen XG, Li Y, Li HY. Chin Chem Lett. 2007;18:659–662. [Google Scholar]; (d) Wang J, Zhao L, Hu Y, Guo Q, Zhang L, Wang X, Li N, You Q. Eur J Med Chem. 2009;44:2611–2620. doi: 10.1016/j.ejmech.2008.09.034. [DOI] [PubMed] [Google Scholar]
- x.Chantarasriwong O, Cho WC, Batova A, Chavasiri W, Moore C, Rheingold AL, Theodorakis EA. Org Biomol Chem. 2009;7:4886–4894. doi: 10.1039/b913496d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xi.(a) Guizzunti G, Batova A, Chantarasriwong O, Dakanali M, Theodorakis EA. ChemBioChem. 2012;13:1191–1198. doi: 10.1002/cbic.201200065. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guizzunti G, Theodorakis EA, Yu AL, Zurzolo C, Batova A. Invest New Drugs. 2012;30:1841–1848. doi: 10.1007/s10637-011-9745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xii.Tisdale EJ, Slobodov I, Theodorakis EA. Proc Natl Acad Sci USA. 2004;101:12030–12035. doi: 10.1073/pnas.0401932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiii.(a) Tisdale EJ, Chowdhury C, Vong BG, Li H, Theodorakis EA. Org Lett. 2002;4:909–912. doi: 10.1021/ol017278w. [DOI] [PubMed] [Google Scholar]; (b) Tisdale EJ, Li H, Vong BG, Kim SH, Theodorakis EA. Org Lett. 2003;5:1491–1494. doi: 10.1021/ol034276y. [DOI] [PubMed] [Google Scholar]; (c) Tisdale EJ, Vong BG, Li H, Kim SH, Chowdhury C, Theodorakis EA. Tetrahedron. 2003;59:6873–6887. [Google Scholar]; (d) Tisdale EJ, Slobodov I, Theodorakis EA. Org Biomol Chem. 2003;1:4418–4422. doi: 10.1039/b311833a. [DOI] [PubMed] [Google Scholar]
- xiv.An alternative explanation for the observed site selectivity could be that the Claisen rearrangement is reversible and that the rate of the Diels-Alder reaction controls the product selection. For computational studies in support of this proposal see: Hayden AE, Xu H, Nicolaou KC, Houk KN. Org Lett. 2006;8:2989–2992. doi: 10.1021/ol060917o.
- xv.For related synthetic studies see: Nicolaou KC, Li J. Angew Chem Int Ed. 2001;40:4264–4268. doi: 10.1002/1521-3773(20011119)40:22<4264::AID-ANIE4264>3.0.CO;2-1.Nicolaou KC, Sasmal PK, Xu H, Namoto K, Ritzen A. Angew Chem Int Ed. 2003;42:4225–4229. doi: 10.1002/anie.200351805.Nicolaou KC, Sasmal PK, Xu H. J Am Chem Soc. 2004;126:5493–5501. doi: 10.1021/ja040037+.Nicolaou KC, Xu H, Wartmann M. Angew Chem Int Ed. 2005;44:756–761. doi: 10.1002/anie.200462211.
- xvi.For a recent review on this topic see: Wuerstle ML, Laussmann MA, Rehm M. Exper Cell Res. 2012;318:1213–1220. doi: 10.1016/j.yexcr.2012.02.013.
- xvii.For selected reviews on this topic see: Porter AG, Jaenicke RU. Cell Death and Different. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476.Crawford ED, Wells JA. Annu Rev Biochem. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639.
- xviii.Kamal A, Thao L, Sensitaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- xix.(a) Jhaveri K, Taldone T, Modi S, Chiosis G. Biochim Biophys Acta. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Solit DB, Chiosis G. Drug Discov Today. 2008;13:38–43. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]; (c) Travers J, Sharp S, Workman P. Drug Discov Today. 2012;17:242–252. doi: 10.1016/j.drudis.2011.12.021. [DOI] [PubMed] [Google Scholar]
- xx.(a) Ame JC, Spenlehauer C, de Murcia G. BioEssays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]; (b) Schiewer MJ, Goodwin JF, han S, Brenner JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P, Chinnaiyan AM, McCue P, Gomella LG, Raj GV, Dicker AP, Brody JR, Pascal JM, Centenera MM, Butler LM, Tilley WD, Feng FY, Knudsen KE. Cancer Discov. 2012;2:1134–1149. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.