

Abstract
Target site choice is a complex and poorly understood aspect of DNA transposition despite its importance in rational transposon-mediated gene delivery. Thoughmost transposons choose target sites essentially randomly or with some slight sequence or structural preferences, insertion sequence IS608 from Helicobacter pylori, which transposes using single-stranded DNA, always inserts just 3′ of a TTAC tetranucleotide. Our results from studies on the IS608 transposition mechanism demonstrated that the transposase recognizes its target site by co-opting an internal segment of transposon DNA and utilizes it for specific recognition of the target sites through base-pairing. This suggested a way to redirect IS608 transposition to novel target sites. As we demonstrate here, we can now direct insertions in a predictable way into a variety of different chosen target sequences, both in vitro and in vivo.
INTRODUCTION
Target site selection by transposable elements is generally poorly understood (Craig, 1997) yet a crucial aspect of transposition given its importance to the ultimate fate of the mobile element, which survives only if it does not integrate into a deleterious genomic location. It is also key in efforts to exploit transposition systems to deliver genes at precise places in a genome. Most transposons appear to insert randomly into DNA. Others insert preferentially into sites governed by aspects of DNA structure such as bent DNA (Hallet et al., 1991; Pribil and Haniford, 2003), single-nucleotide mismatches (Yanagihara and Mizuuchi, 2002), nucleosomes (Pryciak and Varmus, 1992), or specific chromatin-associated proteins such as Sir4 (Zhu et al., 2003). Yet others insert specifically into predetermined sites; the bacterial Tn7 transposon, in its TnsD-dependent pathway in Escherchia coli, inserts between the phoS and glmS genes, a site conserved in many bacteria (Parks and Peters, 2009). Others insert into very short sequences, such as targeting by Tc1/mariner family elements of the dinucleotide TA (Plasterk et al., 1999) or IS903, which uses a 21 base pair palindromic sequence (Hu et al., 2001).
We have investigated the insertion specificity of the Helicobacter pylori insertion sequence, IS608, and have shown that it is determined by an unusual mechanism whereby the target site is not recognized directly by the transposase but through base-pairing interactions with nucleotides within the transposon DNA (Barabas et al., 2008).
IS608 is active in E. coli (Kersulyte et al., 2002) and carries a transposase gene, tnpA, and another gene, tnpB, of unknown function dispensable for transposition (Figure 1A). It is fundamentally different from classical ISs because it transposes using single-stranded (ss) DNA (Guynet et al., 2008). This essential dissimilarity is also reflected in its structure and organization; its transposase, TnpA, resembles rolling circle replication initiators and conjugative plasmid relaxases with an HUH (histidine-hydrophobic-histidine) motif and a catalytic tyrosine rather than the classic DDE transposases. Cleavage of the transposon ends and the target site creates covalent 5′ phosphotyrosine enzyme-substrate intermediates (Ton-Hoang et al., 2005). IS608 has no terminal inverted repeats; rather, both the left end (LE) and right end (RE) include imperfect subterminal palindromes (IPL and IPR) some distance from the cleavage sites (CL and CR) (Figure 1B). A circular ssDNA transposon intermediate with abutted RE and LE (the transposon or RE-LE junction) is generated by precise excision of the “top” strand (Figure 1C) and inserts specifically into a single-strand target 3′ to a TTAC tetranucleotide (CT) (Figures 1D and 1E; Kersulyte et al., 2002; Guynet et al., 2008) also required for subsequent transposition (Ton-Hoang et al., 2005). Excision is accompanied by reclosure of the DNA originally flanking the excised strand, generating a donor joint and preserving the target TTAC without DNA gain or loss (Ton-Hoang et al., 2005). The entire transposition cycle has been reconstituted in vitro with purified TnpA (Guynet et al., 2008).
Figure 1. Organization of IS608 and Overall Transposition Pathway.
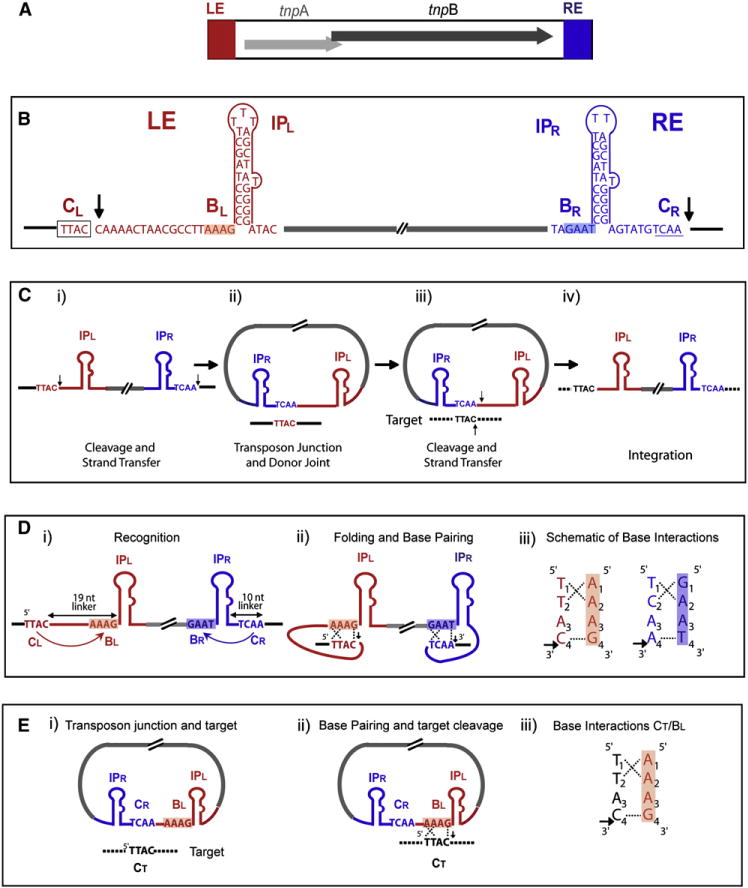
(A) Organization. tnpA and tnpB open reading frames (light and dark arrows, respectively). Left end (LE) and right end (RE) (red and blue boxes, respectively).
(B) Sequence of LE and RE. Sequence and secondary structures, IPL and IPR, at the LE and RE IS608 are shown. Left and right tetranucleotide cleavage sites are boxed in black (CL) and underlined in blue (CR). CR forms part of IS608, whereas CL does not. BL and BR are shown on a red and blue background, respectively. Position of cleavage and of formation of the 5′ phosphotyrosine TnpA-DNA intermediate (vertical arrows).
(C) Transposition pathway. (i) Schematized IS608 with IPL and IPR, left (TTAC; CL) and right (TCAA; CR) cleavage sites. (ii) Formation of a single-strand transposon circle intermediate with abutted left and right ends. The transposon junction (TCAA) and donor joint (TTAC) are shown. (iii) Pairing with the target (TTAC) and cleavage (vertical arrows). (iv) Inserted transposon with new left and right flanks (dotted black lines).
(D) Cleavage site recognition showing asymmetry of the IS608 ends. (i) The distance between the IP foot and the cleavage position, the linker, is 19 nt at LE and 10 nt at RE. Transposon DNA between LE and RE is indicated in gray. (ii) Tetranucleotide cleavage sites (CL, red and CR, blue) are recognized by the respective tetranucleotide boxes (BL and BR) 5′ to the hairpin foot by folding and base-pairing (dotted lines). (iii) Enlargement of base interactions observed in the crystal structures (Barabas et al., 2008). Horizontal arrow indicates position of cleavage.
(E) Transposition pathway tointegration. (i) The ssDNA transposon circle including the RE-LE transposon junction and the target DNA. (ii) Recognition of the target tetranucleotide by BL. (iii) Engagement with target DNA carrying the TTAC tetranucleotide CT (dotted line) together with transposon junction cleavage and target cleavage (vertical black arrows). Arrangement of base interactions in the crystal structures. Horizontal arrow indicates position of cleavage.
TnpA is dimeric, with two sites on one face of the dimer to which IPL and IPR bind and two active sites on the opposite face composed of amino acids provided by each monomer. These shared active sites are formed by juxtaposition of an α helix (αD) carrying the catalytic tyrosine (Y127) supplied by one monomer and the two histidines of the HUH motif (that coordinate the catalytically essential divalent metal ion) supplied by the other.
An important question is how TnpA recognizes its cleavage sites. At LE, the cleavage site is 19 nucleotides 5′ to the foot of IPL preceded by a flanking tetranucleotide sequence TTAC (CL), whereas, at RE, the cleavage site, TCAA (CR), is only 10 nucleotides 3′ to the foot of IPR (Figures 1B and 1D). Crystal structures revealed that three of the four CL nucleotides interact with a short sequence, BL, just 5′ of the foot of IPL, and three of the four CR nucleotides interact with BR, a short sequence just 5′ of the foot of IPR (Figure 1C). Cleavages create 5′ phosphotyrosine bonds between TnpA and substrates at both ends: with the 5′ end of LE but with the 5′ end of the RE DNA flank (Figure 1B).
A key implication is that changes in BL should resultinchanges in interaction with CL. Because CL is identical to CT (as it was the target of the preceding insertion) (Figures 1C and 1D), target choice should therefore also involve base-pairing between CT and BL (Barabas et al., 2008). The RE-LE junction of the ssDNA transposon circle does not include CL. Therefore, BL is available to interact with CT (Figure 1E). Changes in BL should, therefore, lead to predictable changes in IS608 target specificity.
We show here that, by taking advantage of the unique opportunity provided by this ssDNA transposition system, it is possible to redirect its integration specificity in a predictable way. We have analyzed the effect of 11 different BL sequences carrying single-or multiple-nucleotide changes. At least nine resulted in a change in the target sequence CT, both in vivo and in vitro. Importantly, these changes could be predicted based on the wild-type (WT) CT/BL pairing pattern observed in the crystal structures.
RESULTS
Role of LE and RE boxes BL and BR in Cleavage, Strand Transfer, and Target Choice
Cleavage and Strand Transfer In Vitro with Isolated RE and LE Derivatives
Structural studies (Barabas et al., 2008) suggested that excision, requiring the cleavage at each transposon end, is facilitated by base-pairing between the cleavage sites CL and CR and three of four nucleotides in BL and BR (Figure 1E). To determine the effect of these interactions on IS608 excision, we examined the consequence of exchanging BL and CL for BR and CR on cleavage and strand transfer of LE in vitro (Figure 2A). Note that the convention used here is that, unless otherwise indicated, the C and B tetranucleotides associated with LE and RE are wild-type. Only changes are shown. LE with a correct spacer but carrying both CR and BR (LECR-BR) (Figure 2A, lane 4) underwent cleavage (Figure 2A, lane 5), generating a donor joint with WT RE (Figure 2A, lane 6) at similar levels to WT LE (Figure 2A, lanes 1–3). Similarly, RE carrying CL and BL with the correct spacer (RECL-BL) (Figure 2A, lane 10) underwent correct cleavage (Figure 2A, lane 11) and junction formation with WT LE (Figure 2A, lane 12) similar to WT RE (Figure 2A, lanes 7–9). Moreover, LECR-BR underwent strand transfer with RECL-BL (Figure 2A, lane 13). Thus, LE and RE cleavage specificity can be exchanged, and the derivatives can successfully undergo strand exchange to form a donor joint and a transposon junction.
Figure 2. Redirecting RE-LE Junction Integration: Exchanging BL, BR, CL, and CR.
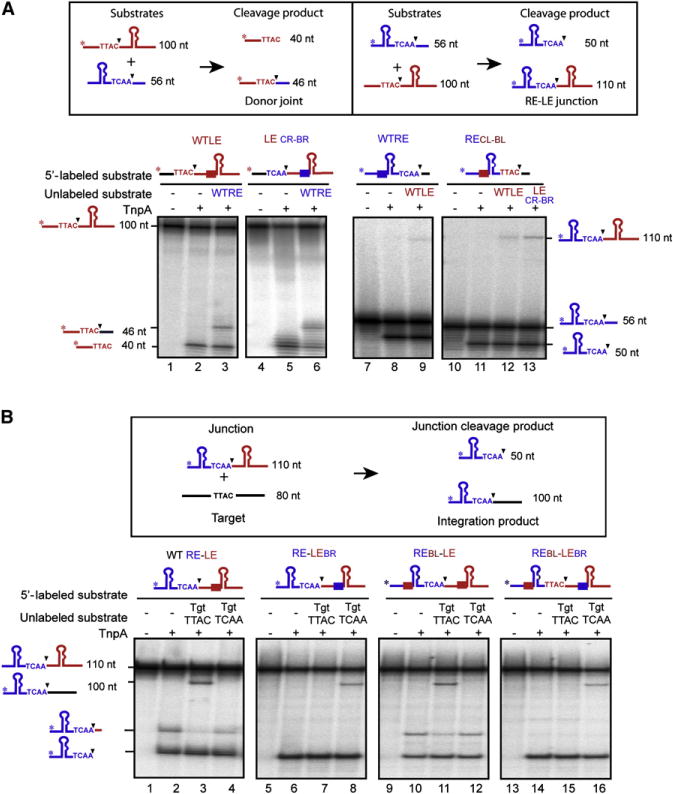
(A) Cleavage and strand transfer of individual exchanged ends. BR and BL are shown as blue and red boxes, respectively. Labeled WT LE, LECR-BR, WT RE, and RECL-BL (lanes 1, 4, 7, and 10) incubated with TnpA alone (lanes 2, 5, 8, and 11) or with unlabeled WT RE (lanes 3 and 6) reveal formation of donor joint or WT LE (lanes 9 and 12) or, with unlabeled LECR/BR (lane 13), reveal the formation of RE-LE junction.
(B) Junction cleavage and strand transfer. Labeled junctions (lanes 1, 5, 9, and 13) incubated with TnpA alone (lanes 2, 6, 10, and 14) or with unlabeled TTAC or TCAA target oligonucleotides (lanes 3 and 4, 7 and 8, 11 and 12, and 15 and 16).
However, exchanging BL or BR without the corresponding changes in CL or CR had a number of diverse effects on cleavage and strand transfer, including a large reduction in cleavage activity compared to that of WT LE or RE, with a tendency to use additional cleavage sites (data not shown). This was presumably because the base-pairing needed for recognition was perturbed.
Cleavage and Integration of RE-LE Junction: Choice of Target Sequence
Because target choice also involves base-pairing between BL in the RE-LE junction and CT (Figures 1D and 1E), changing BL in the junction sequence should result in a predictable change in insertion specificity.
The results obtained with a series of hybrid RE-LE junctions are shown in Figure 2B. The WT junction (Figure 2B, lane 1) underwent robust cleavage, generating a 50 nt RE (Figure 2B, lane 2) as well as cleavage at a secondary site (see Guynet et al., 2008). In the presence of the WT target TTAC, the expected RE target recombination product was generated (Figure 2B, lane 3), and cleavage at the secondary site was suppressed, whereas there was no reaction with a TCAA target (representing CR) (Figure 2B, lane 4). Exchanging BL for BR (RE-LEBR) (Table S1 available online and Figure 2B, lane 6) had little effect on the major cleavage product, although it eliminated the secondary cleavage activity. However, RE-LEBR no longer recombined with the TTAC target (Figure 2B, lane 7) but had acquired the ability to recombine with a TCAA target (Figure 2B, lane 8). Thus, the sequence at BL clearly determines the specificity of insertion of the junction intermediate. Although cleavage was reduced, REBL-LE, in which BR had been substituted by BL, generated a similar pattern to that observed with the WT RE-LE, with a product of the expected size (Figure 2B, lanes 9–12). However, the observation that REBL-LE is cleaved at all suggests that additional factors may be involved in determining cleavage. Finally, RECL-BL-LEBR, in which BR and BL and the TCAA junction cleavage site had been exchanged (Figure 2B, lane 13), underwent robust cleavage (Figure 2B, lane 14) and failed to undergo recombination with the normal TTAC target (Figure 2B, lane 15) but generated recombination products with TCAA (Figure 2B, lane 16). These results demonstrate that the junction BL sequence determines the target.
Integration Activity In Vivo of IS608 with Modified LE
To establish whether the change in insertion specificity seen in vitro is also observed in vivo, we performed standard mating out assays (Galas and Chandler, 1982) using the conjugative F-derived plasmid pOX38Km as a target and an artificial IS608 derivative composed of a CmR gene flanked by a WT RE and various LEderivatives with modified CL and BL (see Experimental Procedures). The results are presented in Figure 3 (lines 1–4). The two hybrid LE derivatives (LECR-BL and LECL-BR) (Figure 3, lines 2 and 3) exhibited 103-fold lower transfer frequencies than the WT (Figure 3, line 1). Moreover, the sequence of individual exconjugant clones for each derivative donor transposon did not identify a transposon-target junction but revealed the transposon donor plasmid (with its resident IS608) itself in the exconjugants. Such low-frequency transfer of “nonmobilizable” plasmids by the conjugative F plasmid has been observed previously (O’Connor and Malamy, 1984). Aberrant cleavages as observed in vitro (data not shown) would presumably be abortive in vivo and would not give rise to productive transposition.
Figure 3. Redirecting Insertions.

Columns 1: line number as in text; 2: name of LE derivative as used in text and in Table S1 (note that a WT RE was used as the second IS end in all cases); 3: schematic of LE showing CL and BL tetranucleotides and LE secondary structure, IPL; 4: interactions between individual CL and BL as suggested by Barabas et al. (2008); 5: predicted target site based on the interactions shown in column 4; 6 and 7: observed target site and number of examples identified; 8: transfer frequency of the IS608-associated CmR marker.
*Experiments in which candidate exconjugants were prescreened for loss of the donor plasmid marker ApR. Mating results are the mean of at least four independent experiments.
On the other hand, although the transposition frequency of the LECR-BR derivative was only 10-fold higher than the hybrid LE derivatives (Figure 3, line 4), 7 of the 12 exconjugant samples carried an IS copy inserted into the predicted TCAA target sequence. The other five examples revealed transfer of the donor plasmid.
Thus, initial junction cleavage is independent of whether the cleavage site is CL (TTAC) or CR (TCAA), but the target chosen for integration (a strand transfer reaction) depends critically on the sequence present in the RE-LE junction. When this is the wild-type sequence, integration occurs into the wild-type target site (TTAC). However, when replaced with BR (normally present in RE), integration occurs into TCAA and not the TTAC target.
Changing Insertion Specificity: Effect ofPoint Mutations in CL and BL
To examine target sequence choice in more detail, we determined the insertion specificity of a series of IS608 derivatives in which individual bases had been changed within CL and BL, rather than the entire tetranucleotide. These complementary changes were based on predictions from interactions between CL and BL in the cocrystal structures with the WT sequences and the transposase (Barabas et al., 2008). This is schematized in Figures 1D and 1E.
Transposition Activity In Vivo of IS608 with Modified LE
The results of mating out assays with the IS608 derivatives carrying various mutations in LE with compensatory mutations to retain base-paring potential are shown in Figure 3.
Transfer frequencies of the IS608 CmR marker (Figure 3) ranged over three orders of magnitude between different mutants. Therefore, the changes introduced have widely different effects on transposition efficiency. Exconjugants of those with diminished frequencies were generally enriched for donor plasmid DNA, again reflectingalow level of activity of F-mediated donor plasmid transfer and low transposition activity. We initially tried to identify insertion sites in all exconjugants. However, because these were rare in mutants with diminished transfer frequencies, we subsequently first screened these mutants for exconjugants that did not exhibit the ApR phenotype (i.e., ampicillin-sensitive exconjugants) associated with the transposon donor plasmid and that had, therefore, not acquired the donor plasmid by F-mediated donor plasmid transfer. Derivatives isolated in this way are marked with an asterisk in Figure 3.
In almost every case (Figure 3, lines 5–14), derivatives of IS608 with changes in the sequence of BL (and accompanying changes in CL) could be directed to a corresponding predicted tetranucleotide target sequence. In three cases (lines 7, 9, and 13), a secondary target site varying by a single nucleotide was also revealed as a minor product. In the two cases in which insertions into the pOX38Km target were not observed (Figure 3, lines 12 and 14), the IS608 mutant derivatives exhibited very low transposition activities approaching background F-mediated donor plasmid transfer. More extensive screening might, therefore, have identified IS608 insertions with the predicted insertion specificity.
Thus, the insertion specificity of IS608 can be varied in a predictable way by introducing concomitant changes in CL and BL. These changes resulted in large changes intransposition activities that are, at present, unpredictable.
Effects of Point Mutations in CL and BL on Cleavage, Strand Transfer, and Junction Integration Activity In Vitro
The effects of base changes in CL and BL could occur at several steps: strand cleavage and transfer leading to excision, donor joint and transposon circle formation, or cleavage and strand transfer associated with insertion and formation of LE and RE transposon target junctions. To examine strand cleavage and donor joint formation in vitro, we used 5′ end-labeled LE derivatives in standard in vitro reactions (Guynet et al., 2008). We also examined cleavage of the equivalent synthetic RE-LE junctions and integration into the predicted target sequences (see Figure 3).
Representative examples are shown in Figure 4. Results of cleavage and strand transfer of LE derivatives (Figure 4A) and of cleavage and recombination of the corresponding RE-LE junctions with a corresponding target sequence (Figure 4B) can be broadly divided into two classes. Cleavage and strand transfer activities of mutants with near WT transposition frequencies in vivo (Figure 3, lines 9 and 11) also appeared similar to WT in vitro (Figure 4A, compare lanes 1–3 with lanes 4–6 and 7–9, respectively). Integration of the corresponding RE-LE junctions into the predicted target sequences (TGAC and CTAC) was also relatively efficient (Figure 4B, lanes 4–6 and 7–9). However, those derivatives with lower transposition activity (Figure 3, lines 7, 13, 12, and 14) tended to generate additional secondary cleavage products (Figure 4A, lanes 10–12, 13–15, and 19–21) or failed to undergo strand transfer (Figure 4A, lanes 16–18). Among these, only the least active in transposition, when in the form of an RE-LE junction (Figure 4B), did not generate additional bands in the presence of target DNA (Figure 4B, lanes 16–18 and 19–21), in agreement with the in vivo results (Figure 3, lines 14 and 12, respectively).
Figure 4. In Vitro Activity of Point-Mutated BLand CL.

(A) Donor joint formation. Labeled oligonucleotides representing LE derivatives (lanes 1, 4, 7, 10, 13, 16, and 19) incubated with TnpA alone (lanes 2, 5, 8, 11, 14, 17, and 20) or with unlabeled WT RE (lanes 3, 6, 9, 12, 15, 18, and 21). (Asterisks) Strand transfer products.
(B) RE-LE junction integration. Labeled oligonucleotides representing RE-LE junction derivatives (lanes 1, 4, 7, 10, 13, 16, and 19) incubated with TnpA alone (lanes 2, 5, 8, 11, 14, 17, and 20) or with unlabeled respective target (lanes 3, 6, 9, 12, 15, 18, and 21). (Asterisks) Strand transfer products.
LE BLA3 Does Not Affect Target Choice but Activates TnpA Catalysis
Structural studies demonstrated that three of the BL nucleotides interact with three of the four CL nucleotides (Figure 1D). The penultimate A, BLA3, does not appear to be involved, as it occupies a pocket that sequesters the tyrosine nucleophile when TnpA is in an inactive conformation (Barabas et al., 2008). BRA3 plays an identical role. To test whether BLA3 is involved in target choice, we analyzed the effect of various base changes at this position on transposition activity in vivo.
Transposition assays (Figure 5) showed that the various mutants exhibited 10- to 102-fold reduced activity compared to the WT sequence. A BLA3T mutation (Figure 5, line 2) combined with a WT RE reduced transposition 10-fold compared to WT (Figure 5, line 1) but retained the WT sequence specificity (TTAC). A derivative carrying both the LE mutation and an equivalent mutation in RE (BRA3T) showed 10-fold lower activity than did WT (Figure 5, line 3) but also retained WT insertion specificity (TTAC). Mutations were also introduced into CL and BL to generate CLA3G/BLA3G, CLA3C/BLA3C, and CLA3T/BLA3T (Figure 5, lines 4, 5, and 6). These also had a 102-fold reduced activity but retained TTAC insertion specificity. Thus, the nature of the base at the penultimate position affects transposition efficiency, but not specificity.
Figure 5. Effect of BLA3.
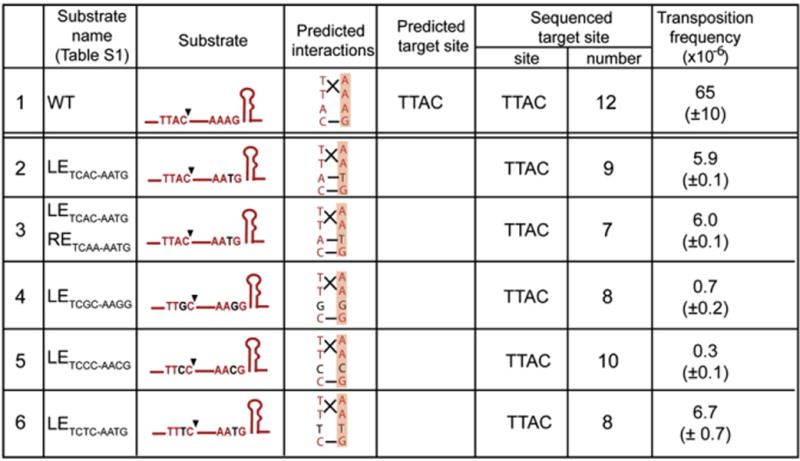
Columns 1: line number as in text; 2: name of LE derivative as used in text and in Table S1 (note that a WT RE was used as the second IS end in all cases); 3: schematic of LE showing CL and BL tetranucleotides and LE secondary structure, IPL; 4: interactions between individual CL and BL as suggested by Barabas et al. (2008); 5: predicted target site based on the interactions shown in column 4; 6 and 7: observed target site and number of examples identified; 8: transfer frequency of the IS608-associated CmR marker.
These in vivo results imply that noncanonical CL sequences carrying mutations in both BL and CL (e.g., lines 4–6) are recognized and cleaved in the excision reaction in the context of LE, and the resulting transposon junctions retain their ability to integrate into a TTAC target. Experiments in vitro also confirmed that these three LE substrates are cleaved but do not participate in strand transfer with a target molecule carrying the mutated CL (data not shown). Thus, BLA3 does not participate in target choice.
DISCUSSION
The ability to direct amobile elementto a precise, predetermined location in a genome would have important implications across many fields of biology. We have demonstrated here that we can redirect the integration of one particular insertion sequence, IS608, away from its native target site (TTAC) and into novel DNA sites. This is possible only because of two highly unusual features of IS608 transposition. The first is that it occurs exclusively using ssDNA substrates (Guynet et al., 2008) in strict contrast to the double-strand transposition pathways of other known DNA transposons (Craig, 1997). The second crucial feature is that its target site, CT, is not recognized directly by the transposase but by base pairs with corresponding BL tetranucleotides within the insertion sequence itself. This form of recognition is permitted only because of the ssDNA nature of the transposition pathway. Recognition mediated by base-pairing is also the basis for the target specificity of mobile group II introns, or targetrons. In such systems, nucleic acid complementarity guides the integration of an excised intron RNA into a DNA target; targetrons can be redirected to novel sites that are generally computationally selected to optimize the constraints imposed by the intron-encoded protein (Perutka et al., 2004).
Previous attempts to change or induce specificity to mobile elements have generally been limited by the need to balance changes within the protein component of the element with corresponding changes in the desired target site. For example, this standard logic led to the attempt to render HIV integration site specific by creating a fusion between the viral integrase and the lambda repressor to target lambda operators (Bushman, 1994). Alternatively, elaborate protein evolution strategies have been applied to redirect target specificity, as in the case of another recombinase, Cre, in which active mutated forms have been evolved that target a site within the HIV LTR rather than the native loxP site (Sarkar et al., 2007).
Our previous studies suggested that straightforwardly changing the IS608 transposon BL tetranucleotide sequence should lead to a change in target specificity. Indeed, of 11 tetranucleotide sequences tested, 9 resulted in a predictable target sequence change (Figure 3). The targets chosen were based on the major interactions observed between BL and CL (or CT) in a structure of TnpA with bound LE (Barabas et al., 2008; Figures 1D and 1E). Transposition frequency varied considerably between the different sequences. At low frequencies, it was confounded by the level of transfer of the transposon donor plasmid itself, presumably by F plasmid-mediated transfer of the “nonmobilizable” transposon donor plasmid, as originally observed for pBR322 (O’Connor and Malamy, 1984). We have not yet determined the reasons for these large variations but assume that they are related to the ability to form correct interactions with the required stability between CL and BL at the excision step or between CT and BL at the integration step. However, because base-pairing is not colinear (Figures 1D and 1E) and other as yet unknown interactions may occur, these effects are, at present, difficult to predict. In addition, spontaneous excision of IS608 is observed in overnight cultures (Ton-Hoang et al., 2005), and the proportion of such plasmids within the donor plasmid population might also affect observed transposition frequencies.
Members of the IS200/IS605 family are widely distributed among bacteria and archaea. They have been identified in more than 100 species, and each IS exhibits its own distinct target sequence variant and potential IPL and IPR secondary structures (ISfinder: http://www-is.biotoul.fr). Of particular interest are those ISs that have longer target sequences, such as ISDra2, which recognizes a pentanucleotide target sequence (Islam et al., 2003). These studies may provide a way to target specific insertions. However, in order for such targeting to be useful, it will, of course, be necessary to extend the length of the recognized sequence to increase specificity. Further understanding of the detailed mechanism of transposition of these elements will be necessary to design experiments to investigate possible ways of accomplishing this. The in vitro and in vivo studies presented here demonstrate the value of a parallel approach to investigating retargeting, as in general, the results obtained from in vitro analysis both of cleavage and strand transfer reactions with independent LE and RE derivatives and with various mutant combinations incorporated into RE-LE junctions correlate with the results observed in vivo. Low transposition activity in vivo is associated with aberrant cleavage and low strand transfer activity, and target site changes observed in vivo can be reproduced in vitro. This correlation provides us a pathway to rapidly investigate the fundamental properties of modified IS200 transposons.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Media
JS219 (MC1061, recA1, and lacIq) (Cam et al., 1988) was used as a donor in mating out experiments and in all other transposition-related assays. The recipient for mating out experiments was MC240 (XA103, F−, ara, del(lac pro), gyrA (nalR), metB, argEam, rpoB, thi, supF). LB grown cultures were supplemented, where necessary, with ampicillin (Ap, 100 μg/ml), kanamycin (Km, 25 μg/ml), streptomycin (Sm, 20 μg/ml), spectinomycin (Sp, 30 μg/ml), rifampicin (50 μg/ml), nalidixic acid (Nal, 20 μg/ml), or chloramphenicol (Cm, 30 μg/ml). Selection was on L plates supplemented with appropriate antibiotics.
Mating Out Assays
The frequency of IS608 transposition was determined by a standard mating out assay (Galas and Chandler, 1982) using the conjugal plasmid pOX38Km as a target replicon (Chandler and Galas, 1983). JS219/pOX38Km, containing a p15A derivative (pBS121) as a source of transposase under control of plac, was transformed with the different transposon donor plasmids. The standard IS donor plasmid was pBS102 with a pBR322-based replicon carrying an ApR gene and a synthetic transposon composed of 143 bp of the left IS608 end and 79 bp of the right flanking a CmR cassette (Ton-Hoang et al., 2005). Relevant mutations were introduced into LE or RE by inverse PCR using the primers shown in Table S1. Overnight cultures grown from a single colony in L broth supplemented with Sp, Sm, Ap, Cm, and Km at 37°C were diluted into L broth with 0.1 mM IPTG (to induce transposase expression) but without antibiotics, and growth was continued to an OD600 of ~0.6, followed by 1 hr incubation without agitation. The donor cells were mixed at an ~1:1 ratio with the recipient MC240 (NalR, RifR) and incubated for 1 hr at 37°C. Suitable dilutions were plated on L agar plates supplemented with Nal, Rif, and Km (for scoring transfer) and Nal, Rif, and Cm (for scoring integration).
DNA Procedures
Standard techniques were used for DNA manipulation and cloning. Restriction and DNA-modifying enzymes were purchased from New England Biolabs. Plasmid DNA was extracted using miniprep or maxiprep kits (QIAGEN). Mutagenesis was performed on pBS102 (Ton-Hoang et al., 2005) by inverse PCR (Weinerand Costa, 1994). PCR reactions were carried out with Phusion polymerase (Promega, 20 cycles: 1 min 94°C, 1 min 55°C, and 5 min 72°C). The final PCR product was digested with DpnI (New England Biolabs) and self-ligated.
TnpA Purification
TnpA was purified, and the in vitro cleavage and strand reactions were as described (Ton-Hoang et al., 2005; Guynet et al., 2008).
Oligonucleotide Cleavage and Strand Transfer Reactions In Vitro
Oligonucleotides were purchased as purified products from Sigma or Eurogentech. Their purity was monitored prior to use, and if necessary, they were further gel purified. Oligonucleotides were 5′ labeled with [γ-32P] ATP (5000 Ci/mmol, Amersham Inc.) using T4 kinase (NEB Inc.). Cleavage and strand transfer reactions were performed as previously described (Guynet et al., 2008). In brief, 28 nM of a 5′ labeled oligonucleotide was incubated with 10 μM TnpA-His6 (60 min, 37°C, in 20 mM HEPES [pH 7.5], 160 mM NaCl, 5 mM MgCl2, 10 mM DTT, 20 μg/ml BSA, 0.5 μg of poly-dIdC, and 20% glycerol). For strand transfer reactions, samples contained an excess of one or two additional unlabeled oligonucleotide(s) at a final concentration of 1 μM. Products were separated on a 10% denaturing gel and analyzed by phosphorimaging. Sequences of oligonucleotides used in the in vitro study are presented in Table S1.
Insertion Site Sequencing
Insertion sites within pOX38Km were sequenced by arbitrary PCR. PCR reactions were carried out on single colonies with Taq polymerase (Biolabs). Each colony was suspended in 100 μl H2O. The first PCR cycles were performed with B181 (B181: 5′TTGTGCATGA TATTGACTCT, which is complementary to coordinates 101 and 121 at the left end) and ARB1 (5′GGCCACGCGTCGACTAGTAC(N)10GACTG) in a final volume of 50 μl (38 μl H2O, 5 μl 10 × Taq buffer, 1 μl dNTP 10 μM, 2 μl B181 20 μM, 2 μl ARB1 20 μM, 2 μl bacterial suspension, and 0.2 μl Taq) with the following conditions: 5 min 95°C, 6 × (30 s 95°C, 30 s 30°C, 1 min 30 s 72°C), 30 × (30 s 95°C, 30 s 45°C, 2 min 72°C). The second round of PCR used B182 (5′TTCGCCTTGTATCCCCATAG, complementary to coordinates 60 to 40) and ARB2 (5′GGCCACGCGTCGACTAGTAC) in a final volume of 50 μl (38 ml H2O, 5 μl 10 × Taq buffer, 1 μl dNTP 10mM, 2 μl B182 20 μM, 2 μl ARB2 20 μM, 2 μl of first PCR, and 0.2 μl Taq) with the following conditions: 5 min 95°C, 30 × (30 s 95°c, 30 s 52°C, 2 min 72°C). The PCR products were purified (QIAGEN) and sequenced using B181.
SUPPLEMENTAL DATA
Supplemental Data include one table and can be found with this article online at http://www.cell.com/molecular-cell/supplemental/S1097-2765(09)00350-5.
Supplementary Material
Acknowledgments
We would like to thank members of the Chandler lab (G. Duval-Valentin, P. Kichenaradja, B. Marty, P. Rousseau, P. Siguier, O. Walisko, and E. Simmonot). The Chandler lab was supported by continuous intramural funding from the CNRS (France), by European contract LSHM-CT-2005-019023, and partially by ANR grant MOBIGEN. C.G. received a training grant from the French MENRT, and A.A. was supported by European contract LSHM-CT-2005-019023. The Dyda lab was supported by the NIH NIDDK intramural program.
References
- Barabas O, Ronning DR, Guynet C, Hickman AB, Ton-Hoang B, Chandler M, Dyda F. Mechanism of IS200/IS605 Family DNATransposases: Activation and Transposon-Directed Target Site Selection. Cell. 2008;132:208–220. doi: 10.1016/j.cell.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD. Tethering human immunodeficiency virus 1 integrase to a DNA site directs integration to nearby sequences. Proc Natl Acad Sci USA. 1994;91:9233–9237. doi: 10.1073/pnas.91.20.9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam K, Bejar S, Gil D, Bouche JP. Identification and sequence of gene dicB: translation of the division inhibitor from an in-phase internal start. Nucleic Acids Res. 1988;16:6327–6338. doi: 10.1093/nar/16.14.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler M, Galas DJ. Cointegrate formation mediated by Tn9. II. Activity of IS1 is modulated by external DNA sequences. J Mol Biol. 1983;170:61–91. doi: 10.1016/s0022-2836(83)80227-7. [DOI] [PubMed] [Google Scholar]
- Craig NL. Target site selection in transposition. Annu Rev Biochem. 1997;66:437–474. doi: 10.1146/annurev.biochem.66.1.437. [DOI] [PubMed] [Google Scholar]
- Galas DJ, Chandler M. Structure and stability of Tn9-mediated cointegrates. Evidence for two pathways of transposition. J Mol Biol. 1982;154:245–272. doi: 10.1016/0022-2836(82)90063-8. [DOI] [PubMed] [Google Scholar]
- Guynet C, Hickman AB, Barabas O, Dyda F, Chandler M, Ton-Hoang B. In Vitro Reconstitution of a Single-Stranded Transposition Mechanism of IS608. Mol Cell. 2008;29:302–312. doi: 10.1016/j.molcel.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Hallet B, Rezsohazy R, Delcour J. IS231A from Bacillus thuringiensis is functional in Escherichia coli: transposition and insertion specificity. J Bacteriol. 1991;173:4526–4529. doi: 10.1128/jb.173.14.4526-4529.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WY, Thompson W, Lawrence CE, Derbyshire KM. Anatomy of a preferred target site for the bacterial insertion sequence IS903. J Mol Biol. 2001;306:403–416. doi: 10.1006/jmbi.2000.4421. [DOI] [PubMed] [Google Scholar]
- Islam SM, Hua Y, Ohba H, Satoh K, Kikuchi M, Yanagisawa T, Narumi I. Characterization and distribution of IS8301 in the radioresistant bacterium Deinococcus radiodurans. Genes Genet Syst. 2003;78:319–327. doi: 10.1266/ggs.78.319. [DOI] [PubMed] [Google Scholar]
- Kersulyte D, Velapatino B, Dailide G, Mukhopadhyay AK, Ito Y, Cahuayme L, Parkinson AJ, Gilman RH, Berg DE. Transposable element ISHp608 of Helicobacter pylori: nonrandom geographic distribution, functional organization, and insertion specificity. J Bacteriol. 2002;184:992–1002. doi: 10.1128/jb.184.4.992-1002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor MB, Malamy MH. Role of the F factor oriV1 region in recA-independent illegitimate recombination. Stable replicon fusions of the F derivative pOX38 and pBR322-related plasmids. J Mol Biol. 1984;175:263–284. doi: 10.1016/0022-2836(84)90348-6. [DOI] [PubMed] [Google Scholar]
- Parks AR, Peters JE. Tn7 elements: Engendering diversity from chromosomes to episomes. Plasmid. 2009;61:1–14. doi: 10.1016/j.plasmid.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutka J, Wang W, Goerlitz D, Lambowitz AM. Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J Mol Biol. 2004;336:421–439. doi: 10.1016/j.jmb.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Plasterk RH, Izsvak Z, Ivics Z. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999;15:326–332. doi: 10.1016/s0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- Pribil PA, Haniford DB. Target DNA bending is an important specificity determinant in target site selection in Tn10 transposition. J Mol Biol. 2003;330:247–259. doi: 10.1016/s0022-2836(03)00588-6. [DOI] [PubMed] [Google Scholar]
- Pryciak PM, Varmus HE. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell. 1992;69:769–780. doi: 10.1016/0092-8674(92)90289-o. [DOI] [PubMed] [Google Scholar]
- Sarkar I, Hauber I, Hauber J, Buchholz F. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007;316:1912–1915. doi: 10.1126/science.1141453. [DOI] [PubMed] [Google Scholar]
- Ton-Hoang B, Guynet C, Ronning DR, Cointin-Marty B, Dyda F, Chandler M. Transposition of ISHp608, member of an unusual family of bacterial insertion sequences. EMBO J. 2005;24:3325–3338. doi: 10.1038/sj.emboj.7600787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner MP, Costa GL. Rapid PCR site-directed mutagenesis. PCR Methods Appl. 1994;4:S131–S136. doi: 10.1101/gr.4.3.s131. [DOI] [PubMed] [Google Scholar]
- Yanagihara K, Mizuuchi K. Mismatch-targeted transposition of Mu: a new strategy to map genetic polymorphism. Proc Natl Acad Sci USA. 2002;99:11317–11321. doi: 10.1073/pnas.132403399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Dai J, Fuerst PG, Voytas DF. Controlling integration specificity of a yeast retrotransposon. Proc Natl Acad Sci USA. 2003;100:5891–5895. doi: 10.1073/pnas.1036705100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.