

Abstract
Design of appropriate inhaled carriers with adequate aerodynamic properties, drug release, biodegradation and evasion of macrophage uptake is a major challenge for controlled release pulmonary drug delivery. In this study, PEG graft copolymerized onto N-phthaloyl chitosan (NPHCs) was synthesized then characterized using FTIR, EA, DSC and 2D-XRD. The resulting PEG-g-NPHCs copolymers were self-assembled into drug loaded nanoparticles and encapsulated in respirable/swellable sodium alginate semi-IPN hydrogel microspheres as novel biodegradable carriers for controlled release pulmonary drug delivery. The developed nano-/microspheres carrier systems were formed via spray drying followed by ionotropic crosslinking in mild aqueous medium. The size of the developed self-assembled nanoparticles and the microspheres was measured using dynamic light scattering and laser diffraction, respectively. Morphology, moisture content, in-vitro biodegradation and dynamic swelling studies were also investigated for the developed carriers. A model protein was entrapped and the in-vitro release profiles were determined in PBS, pH 7.4 at 37°C. A dry powder aerosolization study was conducted using a Next Generation Impactor (NGI). The developed microspheres had suitable aerodynamic diameters (1.02–2.63 μm) and an excellent fine particle fraction, FPF of 31.52%. The microspheres showed also a very fast initial swelling within the first 2 min and started to enzymatically degrade within the first two hours. Moreover, the microspheres entrapped up 90% of the model drug and showed promising in-vitro sustained release profiles as compared to the control formulation.
Keywords: Nanoparticles, microspheres, chitosan, PEG, pulmonary, sustained, lung, drug delivery
1. Introduction
One of the major shortcomings in pulmonary drug delivery is the present inability to control the pharmacokinetics of inhaled therapeutics beyond a few hours. To date, there are no effective sustained release pulmonary drug delivery systems available due to the efficient clearance of therapeutics from the deep lung either through phagocytosis in the alveolar region or via the rapid absorption of delivered therapeutics (Smyth and Hickey, 2005; Grenha, et al., 2007). Therefore, development of an appropriate drug-loaded vehicle system with adequate aerodynamic characteristics that can confer sustained release of drug once deposited in the lung is considered one of the major challenges in inhalation therapy (Ahsan, et al., 2002). In particular, the drug-loaded carrier particles, targeted to the deep lung, should be aerodynamically small enough to pass through the mouth, throat and conducting airways and reach the deep lung, but not so small that they fail to deposit and are exhaled again (Grenha, et al., 2007). The most appropriate range of aerodynamic diameters of these particles to be respirable is around 0.5–5 μm. However, microparticles of this aerodynamic respirable size range are also of a size that is cleared most rapidly from the lung by alveolar macrophages. Ahsan et al. and others (Ahsan, et al., 2002; Makino, et al., 2003; Martonen, et al., 2005) have reported that increasing the size of microparticles tends to reduce their clearance by macrophages. However, increasing microparticle size is generally impractical for pulmonary drug delivery as this would minimize the respirable fraction that can be delivered. Larger low density particles can be delivered in this way but controlling drug release from these particles may be problematic. Therefore, in our earlier studies (El-Sherbiny, et al., 2010a; El-Sherbiny and Smyth, 2010), a promising new strategy was suggested. This new strategy involves the developing of new swellable microparticles that have respirable aerodynamic sizes when dry but large geometric sizes when swollen after deposition in the moist lung. This enables the delivery systems to evade macrophage uptake and at same time confer sustained drug release through a controlled polymeric architecture.
Ultra small particles with geometric diameters less than a couple hundred nanometers, (nanoparticles, NPs) have showed an obstinate residency in the lungs (Oberdorster, 2001). Once deposited, these NPs may stay in the lung lining fluid until their dissolution, evading phagocytosis and mucociliary clearance (Krenis and Strauss, 1961; Kawaguchi, et al., 1986; Rudt and Muller, 1992). In addition, drug loaded NPs embedded within hydrogels can allow for additional control of drug release (Cascone, et al., 2002; Kim and Martin, 2006). Therefore, using NPs embedded in swellable and respirable microparticles may represent a promising approach for controlling drug action and release, not only in alveolar region where macrophage clearance occurs but also throughout the lumen of the lungs. NPs must be encapsulated or formulated into microparticles for pulmonary drug delivery due to their small aerodynamic size which, as discussed above, leads to lower deposition in the airways and their extensive exhalation from the lungs following inspiration (Heyder and Rudolf, 1984). In addition, NPs-based carrier systems may aggregate in both dry powder and liquid forms causing them to form microparticles suitable for rapid phagocytosis and clearance (Kabbaj and Phillips, 2001; Tsapis, et al., 2002).
In this contribution, a new carrier system was developed for controlling pulmonary drug delivery and combines the benefits of both NPs and the respirable/swellable hydrogel microparticles suggested in our previous studies (El-Sherbiny, et al., 2010a; El-Sherbiny and Smyth, 2010) while avoiding their drawbacks. The developed carrier system comprised of semi-interpenetrating polymeric network (semi-IPN) microspheres. This semi-IPN is based on spray dried ionotropically-crosslinked alginate microspheres containing homogeneously entrapped self-assembled NPs. The NPs were prepared from the N-phthaloyl chitosan (NPHCs) graft copolymerized with poly(ethylene glycol) (PEG).
Sodium alginate is a natural non-toxic biodegradable polyanionic copolymer. It consists of 1,4-linked β-D-mannuronic acid (M) and α-L-guluronic acid (G) residues arranged either as consecutive blocks or in a random distribution. Alginate has a unique ability to form hydrogels via ionotropic crosslinking in presence of divalent cations such as calcium and barium ions (El-Sherbiny, 2010) and the hydrogels based on calcium-crosslinked alginate have been widely investigated for drug delivery purposes (Zahoor, et al., 2005; Murata, et al., 2009; El-Sherbiny, 2010; El-Sherbiny, et al., 2010b).
Chitosan (Cs) is a cationic biopolymer obtained through alkaline N-deacetylation of chitin. Cs has numerous desirable characteristics including biodegradability, non-toxicity and biocompatibility (Majeti and Kumar, 2000). Also, Cs showed a good ability to improve drug absorption via protection of the drug against enzymatic degradation (Parka, et al., 2006). Moreover, Cs was found to have a significant role in enhancing the drug absorption in lung tissues which was attributed to its effect in opening the intercellular tight junction of the lung epithelium (Parka, et al., 2006). Graft copolymerization of various synthetic polymers onto Cs and its derivatives can further enhance their characteristics and consequently expands their potential applications. One of these synthetic polymers that commonly grafted onto Cs is poly(ethylene glycol) (PEG). PEG is a water soluble biocompatible and non toxic polymer (Ohya, et al., 2000). Owing to the characteristics of PEG, development of polymeric vehicles in form of micro-and nano-hydrogel particles based on PEG graft copolymerized onto either Cs or the N-phthaloyl derivative of Cs (NPHCs) have received recently a growing interest as novel carriers for drugs (Maoa, et al., 2005; Opanasopit, et al., 2006; Prego, et al., 2006; Opanasopit, et al., 2007; Yao, et al., 2007; Zhang, et al., 2008).
In this study, PEG-g-NPHCs was synthesized and characterized then self-assembled into NPs. The suspensions of these NPs in aqueous sodium alginate solutions were spray dried to produce microspheres followed by ionotropic crosslinking in mild aqueous medium to form semi-IPN hydrogel microspheres. The resulting respirable/swellable microspheres were then evaluated as novel biodegradable carriers that can confer sustained release of drugs once deposited in the lung.
2. Materials and methods
2.1. Materials
Chitosan (medium MW, %N-deacetylation; about 76.4 %, as determined by elemental analysis and FTIR spectroscopy), monomethoxy-poly(ethylene glycol) (m-PEG, Mn 5,000 Da), 1-hydroxybenzotrizole (HOBt) and succinic anhydride were purchased from Aldrich (USA). 4-Dimethylaminopyridine (DMAP) was obtained from Sigma (St Louis, MO, USA). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC.HCL) was provided by Fluka Chemical Corp. (Milwaukee, WI, USA). Sodium alginate (low viscosity; 250 cps for a 2% solution at 25°C), triethyl amine, phthalic anhydride, dioxane and dimethyl formamide (DMF) were obtained from Sigma-Aldrich, SIAL (St Louis, MO, USA). Phosphate buffer saline (PBS pH 7.4), absolute ethanol and all other reagents were of analytical grade and used as received.
2.2. Methods
2.2.1. Preparation of PEG/NPHCs graft copolymer
The copolymer of PEG grafted onto NPHCs was prepared by a modified method reported in our earlier study (El-Sherbiny, et al., 2010a) and described briefly as follow:
Firstly, monohydroxy-terminated monomethoxy-poly(ethylene glycol) macromer (m-PEG) was converted into carboxyl-capped precursor by reacting with succinic anhydride. Briefly, m-PEG (100 g, 20 mmol), succinic anhydride (2.4 g, 24 mmol), DMAP (2.44 g, 20 mmol) and triethylamine (2.02 g, 20 mmol) were dissolved in dry dioxane (350 ml). The reaction mixture was then stirred at room temperature for 48 h under a dry nitrogen atmosphere. The dioxane was evaporated under vacuum and the residue was taken up in CCl4, filtered and precipitated by diethyl ether to produce a white powder of m-PEG-COOH (yield: 98%). Secondly, NPHCs was synthesized by reaction of 10 g Cs with 44.8 g phthalic anhydride (5 mol equivalent to pyranose rings) in presence of 200 ml of DMF at 130°C under dry nitrogen atmosphere for 8 h. The reaction mixture was then left to reach room temperature before pouring it onto ice-water. The precipitated product was collected by filtration, washed with methanol, and then dried at 40°C under vacuum to give a pale brown product (NPHCs). PEG-g-NPHCs copolymer was then prepared by stirring of 37.9 g of m-PEG-COOH with 5.0 g of dried NPHCs (0.4 mol equivalent to m-PEG-COOH) in 75 ml of DMF. Then, HOBt (3.4 g, 3 mol equivalent to m-PEG-COOH) was added as a catalyst and the reaction mixture was stirred at room temperature until the solution became clear. Then, EDC.HCl (4.25 g, 3 mol equivalent to m-PEG-COOH) was added and the reaction was continued overnight under stirring at room temperature. The mixture was then dialyzed against distilled water and washed with ethanol to remove impurities and unreacted macromere and to obtain the white product of PEG-g-NPHCs (5.47g).
m-PEG-COOH: FTIR (υmax, cm−1) 3496 (OH stretching), 2882 (C–H stretching), 1733 (C=O of carboxylic group), and 1102 (C-O-C stretching); EA, (C231H460O117), Anal. calculated (%): C, 54.35 and H, 9.02, found (%): C, 56.8 and H, 9.19.
NPHCs: FTIR (υmax, cm−1) 3281(OH stretching, NH bending and intermolecular H-bonding), 2961 (C–H stretching), 1775 and 1698 (C=O anhydride), 1395 (C=C, phthaloyl), 1058 (C-O, pyranose) and 732 (aromatic ring of phthaloyl); EA, found (%): C, 60.31; H, 4.83; and N, 4.92.
PEG-g-NPHCs: FTIR (υmax, cm−1) 3423 (OH stretching, NH bending and intermolecular H-bonding), 2879 (C–H stretching), 1736 (C=O ester and anhydride), 1703 (C=O anhydride), 1096 (C-O-C stretching) and 723 (aromatic ring of phthaloyl). EA, found (%): C, 56.16; H, 4.69; and N, 5.15.
2.2.2. Characterization
The elemental analysis for m-PEG-COOH, Cs, NPHCs and PEG-g-NPHCs was carried out using Costech ECS4010 elemental analyzer coupled to a Thermo-Finnigan Delta Plus isotope ratio mass spectrometer. FTIR characterization of the synthesized polymers was performed using Nicolet 6700 FTIR spectrometer and the differential scanning calorimetry (DSC) was carried out using DSC 2920 (Modulated DSC, TA Instruments) in a nitrogen atmosphere from −40°C to 400°C at a heating rate of 10°C/min. Samples of weight 10–12 mg were placed in aluminum sample pans and sealed. An empty aluminum pan of approximately equal weight was used as a reference. All peaks were determined and the areas were converted into enthalpy values. The crystallography patterns of powdered polymer samples were investigated by X-ray diffraction (XRD) using a Scintag Pad V diffractometer with Data-Scan 4 software (MDI, Inc.) for system automation and data collection. Cu-K-α radiation (40 kV, 35 mA) was used with a Bicron Scintillation detector (with a pyrolitic graphite curved crystal monochromator). Data were analyzed with Jade Software (Ver 9.0 from MDI, Inc.). The samples were run between 2θ values of 5° and 45° at a continuous-scan data collection rate of 0.5 deg/min.
2.2.3. Preparation of the semi-IPN hydrogel microspheres
The semi-IPN hydrogel matrices were developed via spray drying of PEG-g-NPHCs NPs dispersed in sodium alginate solution followed by ionotropic gelation using divalent cations (Ca2+). In a typical procedure, homogenous solutions of the synthesized copolymer, PEG-g-NPHCs (1% w/v) and sodium alginate (3% w/v) were prepared using 0.06% acetic acid and distilled water as solvents, respectively. The self-assembled PEG-g-NPHCs NPs were prepared by sonication of the copolymer solution (1% w/v) using a probe type sonicator (Misonix ultrasonic processor, S-4000, Misonix Inc, CT, USA) at 60 W for 2 min. The sonication step was repeated three times until the desired size was obtained. The sonication process was performed in an ice-water bath with applying pulse function (pulse on 5 s & pulse off 5 s) in order to avoid the heat built-up of the polymer solution during the sonication. Then, a predetermined volume of PEG-g-NPHCs NPs suspension was added dropwise with homogenization (15,000 rpm) to a calculated volume of 3% aqueous alginate solution (Table 1). The polymer mixture was then completed with distilled water until obtaining same final concentration (1.5% w/v) in all the prepared formulations. Then, the homogenized polymer mixtures were spray-dried with a 0.7 mm two-fluid pressurized atomizer at a feed rate of 25% (6 ml/min) in a Büchi Mini spray dryer B-290 (Büchi, Switzerland). The atomizing air flow rate was 500–600 NL/h. The inlet temperature was adjusted at 125°C and the outlet temperature was varying between 60 and 65°C. The obtained microspheres powder was collected and the spray drying yield (%) was calculated. Then, the semi-IPN hydrogel microspheres were obtained via soaking a part of the collected powder in 10 ml of 0.1 M CaCl2 solution for 10 min with slow stirring. The crosslinked powder was then filtered through 0.2 μm filter paper followed by washing with 10 ml of distilled water and freeze dried. The obtained hydrogel microspheres were stored in dry conditions until further investigation.
Table 1.
The composition of the developed semi-IPN hydrogel microspheres and some of their characteristics
| Sample Code | Alg (W%) | PEG-g-NPHCs (W%) | Moisture Content (%)
|
1 E% ± SD |
2 VMD, (μm)
|
3 Span
|
Bulk Density
|
4 da
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plain | BSA-Loaded | Plain | BSA-Loaded | Plain | BSA-Loaded | Plain | BSA-Loaded | Plain | BSA-Loaded | ||||
| SIPN0 | 100 | 00 | 10.42±1.74 | 7.52±0.39 | 78.72±2.78 | 2.18±0.12 | 2.80±0.21 | 2.88±0.06 | 2.45±0.07 | 0.219 | 0.705 | 1.02±0.09 | 1.52±0.13 |
| SIPN1 | 80 | 20 | 10.15±1.04 | 6.50±0.36 | 90.32±0.90 | 2.56±0.10 | 3.99±0.10 | 2.52±0.03 | 1.74±0.09 | 0.251 | 0.361 | 1.28±0.12 | 2.40±0.68 |
| SIPN2 | 60 | 40 | 8.70±1.33 | 5.76±0.44 | 85.21±1.93 | 2.24±0.02 | 3.44±0.03 | 2.42±0.01 | 1.75±0.11 | 0.297 | 0.308 | 1.22±0.17 | 1.91±0.51 |
| SIPN3 | 50 | 50 | 8.29±1.49 | 5.47±0.34 | 49.60±2.21 | 2.86±0.08 | 4.36±0.02 | 2.43±0.02 | 1.81±0.32 | 0.340 | 0.365 | 1.67±0.06 | 2.63±0.79 |
E%: Entrapment Efficiency %;
Volume median diameter, VMD;
Span= (D90-D10)/D50,
da, aerodynamic diameter.
2.2.4. Determination of particle size
The size of the developed PEG-g-NPHCs self-assembled NPs was estimated using Dynamic Light Scattering, DLS. Copolymer solutions of different concentrations (0.1, 0.2 and 1%) were sonicated three times for 2 min each at a power of 60 watt. Then, the size of the resulting NPs was estimated using Wyatt Technology Corporation Dyna Pro-titan DLS. The size of the hydrogel microspheres and particle size distribution were determined using laser diffraction (SYMPATEC, Sympatec Gmbt, System Partikl-Technik, Germany with a He-Ne laser beam 5 mW max at 632.8 nm) using WINDOX 5.1.2 software. The measurements were carried out in the range 0.5–175 μm for the suspension of the microspheres in acetone. Volume median diameters (VMD, μm) were calculated from the particle size distribution curves for both plain and drug-loaded microspheres. VMD is the particle diameter at which half the particles volume is contributed by particles larger than the VMD and half by particles smaller than the VMD. Moreover, the particle densities were approximated from tapped density experiments then the aerodynamic diameters of the microspheres were calculated using the following relationship:
| (1) |
where, da is the microsphere aerodynamic diameter (μm), dg is the geometric diameter (VMD, μm), ρp is the microspheres density (g/cc), ρ0 is the standard density (1 g/cc) and χ is the dynamic shape factor (χ =1, assuming that the particles developed in this study are spherical).
2.2.5. In-vitro aerosolization study
An in-vitro dry powder aerosolization study was carried out for the BSA-loaded SIPN2 microspheres, as example, using a Next Generation Impactor (NGI, MSP Corporation, MN, USA). In a typical procedure, a size 3CS capsule (Capsugel®, MA, USA) was packed with 10–15 mg of hydrogel microspheres powder. The capsule was then placed in a handheld, dry-powder, breath-activated inhaler device (Handihaler®). The capsule was punctured prior to inhalation and a pump (Copley HCP5, Nottingham, UK) was actuated to simulate an inspiration (air flow rate of 60±5% L/min for duration of 6 s). Three capsules were emptied, one in each run. The powder emitted from each capsule was deposited on different stages of the NGI. Each stage was containing a specific volume (5–15 ml) of sodium acetate buffer, pH 3.4 as a collecting solvent. Moreover, the powders deposited on the inhaler, capsule and the adapter were also collected in appropriate volumes of the collecting solvent. The amount of BSA-loaded-powder deposited on each stage was then determined by Bradford method (Bradford, 1976) with measuring the absorbance at λmax of 595 nm using Infinite M-200 fluorophotometer (TECAN, USA). The emitted fraction (EF%) that is corresponding the percent of total loaded powder mass exiting the capsule was determined gravimetrically and it can be expressed as follow:
| (2) |
where mfull and mempty are the weights (mg) of the capsule before and after simulating the inhalation and mpowder is the initial weight (mg) of the powder in the capsule.
Also, the analysis of drug deposition in the device, capsule, adapter, throat and NGI stages allows determination of different deposition parameters (Respirable fraction, RF% and the fine particle fraction, FPF%) as shown in the following relationships:
| (3) |
2.2.6. Scanning electron microscopy analysis
The morphology of the developed drug-free and drug-loaded hydrogel microspheres was investigated by SEM (Hitachi S-800 field emission scanning electron microscope operated in secondary electron mode with a Robinson backscatter detector and with a Hitachi PCI system for digital image capture). Dry microspheres were mounted on aluminum stubs with double-sided conducting carbon tapes and coated with a 50/50 mixture of Au/Pd to minimize surface charging. The samples were scanned at an accelerating voltage of 20 KV.
2.2.7. Determination of moisture content
The moisture percentages of the developed microspheres were determined using HR83 Halogen Moisture Analyzer (Mettler-Toledo GmbH, Switzerland). The samples weights were in the range of 120–200 mg and the applied drying temperature was 120°C for 2 min. The moisture content was obtained as a weight loss (%) and calculated as the average ±SD from three independent measurements.
2.2.8. Dynamic swelling study
The swelling behavior of drug (BSA)-loaded hydrogel microspheres in PBS, pH 7.4 was studied by determining the increase in both volume median diameter (VMD, μm) and medium diameter (X50, μm) of the microspheres with time using laser diffractometer (SYMPATEC, Sympatec Gmbt, System Partikl-Technik, Germany).
2.2.9. In-vitro biodegradation study
A biodegradation study of the microspheres was carried out in presence of lysozyme. Typically, lysozyme was dissolved in PBS (pH 7.4) to prepare a solution with a concentration of 2 mg/ml (Hirano, et al., 1989). Predetermined weights of microspheres (20–25 mg) were transferred to microphage tubes and incubated with 1.0 ml of lysozyme solution in a shaking (100 rpm) water bath at 37°C for 1 h until the equilibrium swelling of these microspheres almost attained. Then, the samples were centrifuged for 3 min at a speed of 14,500 rpm and the supernatant was removed. The weights of the swollen microspheres (Wo) were determined. Then 1 ml of fresh lysozyme solution was added to the swollen microspheres. At certain intervals, starting from determination of Wo, the steps of centrifugation and weighing were repeated and the final weights (Wt) of microspheres at these intervals were determined. The percent weight remaining (Wr%) of the samples due to enzymatic degradation were determined according to the following equation:
| (4) |
where Wo is the weight of sample after 1 h swelling in lysozyme solution and Wt is the weight of the sample after incubation with lysozyme for a given time, t.
2.2.10. Entrapment of a model drug and determination of entrapment efficiency
Semi-IPN hydrogel microspheres loaded with bovine serum albumin (BSA), as a model for protein drugs, were prepared in the same manner described earlier in section 2.2.3. Predetermined volumes of aqueous BSA solution (10% w/v) were added, with stirring, to the PEG-g-NPHCs solution before sonication. The initial percentage of added BSA was 40% w/w based on the total weight of polymer mixture (PEG-g-NPHCs and alginate) before the spray drying. After collecting the spray dried powder, a certain amount of it was then, soaked in 10 ml of 0.1 M CaCl2 solution for 10 min with stirring then filtered through 0.2 μm filter paper, washed with 10 ml of distilled water and freeze dried.
Both the washings (10 ml) and the CaCl2 solution (10 ml) were collected and filtered. Then, their content of BSA was estimated using the Bradford assay (Bradford, 1976) from the absorption at λmax 595 nm with the aid of UV-Vis spectrophotometry, (Infinite M 200 fluorophotometer, TECAN, USA). The difference between the amount of BSA initially added to the microspheres and that estimated in the 20 ml (washings plus CaCl2) was taken as a measure of the amount of BSA entrapped. The mean values for three replicate determinations and their mean ±SD values were reported. The entrapment efficiency (E%) of BSA was calculated as follows:
| (5) |
where mi and mr are the amounts (mg) of the BSA initially uploaded and remained in the microspheres respectively.
2.2.11. In-vitro release studies
The in-vitro release profiles of the uploaded BSA were determined by transferring a certain amount (40–50 mg) of BSA-loaded microspheres to a vial containing 2 ml of PBS, pH 7.4. Then, all samples were maintained at 37°C in an incubator with shaking (100 rpm). At predetermined intervals, 100 μl aliquot was withdrawn and analyzed by the Bradford method (Bradford, 1976) at λmax 595 nm using a UV-Vis spectrophotometry. The withdrawn sample was replaced with an equal volume of fresh buffer, to keep the volume of the release medium constant. It has been found prior the studies that no absorbance interference from the plain microspheres under the same conditions. The amounts of BSA released (mg) from hydrogel microspheres were then calculated using a standard curve of BSA in PBS, pH 7.4. The results were calculated in terms of cumulative release (%, w/w) relative to the actual initially entrapped weight of BSA in the microspheres. The data points represent mean ±SD from three independent experiments.
2.2.12. Statistical analysis
The results were analyzed and expressed as mean ±SD. Effects of various parameters on the properties of the developed semi-IPN hydrogel microspheres were statistically analyzed by one-way ANOVA using Excel (Microsoft Office 2007). Differences were considered significant at the level of p < 0.05.
3. Results and discussion
3.1. Synthesis of PEG-g-NPHCs copolymers
The copolymer of m-PEG grafted onto NPHCs was successfully synthesized through a modified method reported in our earlier study (El-Sherbiny, et al., 2010a). The copolymer was prepared through a systematic sequence (Scheme 1) started with converting of m-PEG into carboxyl-capped m-PEG precursor (m-PEG-COOH) upon reaction with succinic anhydride. The prepared m-PEG-COOH was obtained in a very good yield then characterized using FTIR and EA. The free NH2 groups of Cs were masked via phthaloylation process upon reaction with phthalic anhydride in DMF to produce phthaloyl Cs (NPHCs). The synthesis of NPHCs was confirmed by FTIR through appearance of bands at 1395 and 732 cm−1 which correspond C=C and the aromatic ring of the phthaloyl groups respectively. Then the PEG-g-NPHCs copolymer was synthesized upon reaction of m-PEG-COOH with NPHCs in DMF. The grafting % of PEG-g-NPHCs was found to be 9.3 %. The obtained PEG-g-NPHCs copolymer was characterized by EA and FTIR. The copolymer synthesis was also confirmed by studying its thermal characteristics in comparison with that of starting materials, PEG-COOH and NPHCs. For instance, the DSC thermogram of PEG-COOH showed an endothermic peak at about 65°C which corresponds to its melting process. This endothermic peak appeared also in the thermogram of the copolymer, PEG-g-NPHCs at 58°C due to the melting of the grafted PEG side chains. The PEG-g-NPHCs showed also an exotherm at 231°C which was ascribed to the crystallization of the graft copolymer (El-Sherbiny, et al., 2010a). In addition, the XRD patterns of the prepared PEG-g-NPHCs, as compared to that of PEG-COOH and NPHCs, (Figure 1) represent another experimental proof on the synthesis of the copolymer. From the figure, the diffractogram of NPHCs reflects an amorphous nature of it with a broad hump over a range of approximately 18° and centered at 2 value of 23°. In case of m-PEG-COOH and PEG-g-NPHCs, they showed development of very similar crystalline patterns with a pronounced greater peak intensity and indicative of more crystalline material in case of m-PEG-COOH.
Scheme 1.
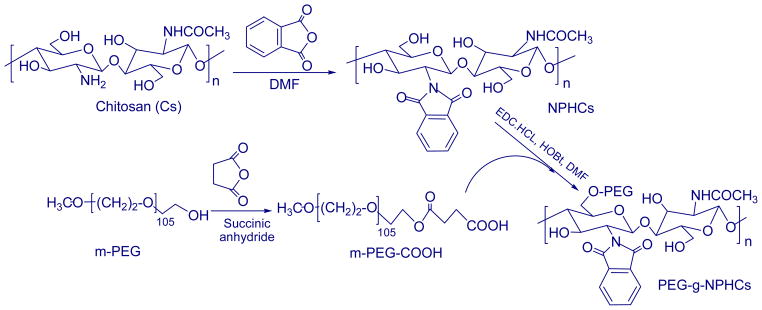
Synthesis of PEG-g-NPHCs copolymer
Figure 1.
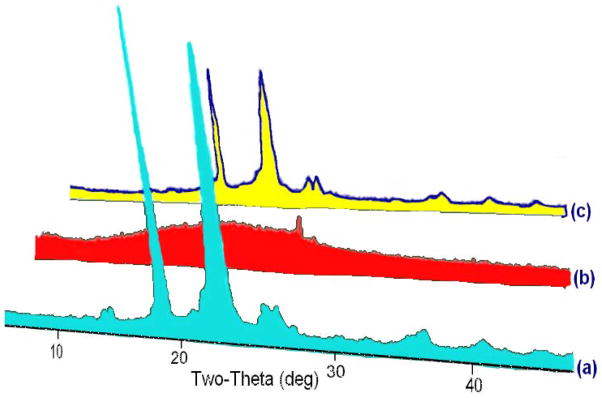
A 3D overlay of the XRD patterns of (a) m-PEG-COOH, (b) NPHCs and (c) PEG-g-NPHCs copolymer
3.2. Preparation of semi-IPN hydrogel microspheres
One of the most commonly used strategies to enhance the physicochemical characteristics of a polymer upon using it in preparation of a hydrogel is the incorporation of this polymer in an interpenetrating polymeric network (IPN). In these IPNs, the polymer is incorporated with either hydrophilic polymer(s) or with hydrophilic monomer(s) treated to bring about in situ copolymerization in the presence of a suitable crosslinking agent. In general, an IPN can be defined as a combination of two or more chemically distinct polymers held together ideally and solely by their permanent mutual entanglements. The IPNs have two basic characteristics: first, one or more of the polymers must be synthesized and/or crosslinked, in the immediate presence of the other(s), and second, the combination produces an advanced multicomponent polymeric system, with new desired property profiles (Klempner, 1994; Kim, et al., 2004). Generally, when only one polymer undergoes crosslinking, the resulting hydrogel matrix is called semi-IPN. In this study, the synthesized PEG-g-NPHCs was self-assembled into NPs. Then respirable/swellable semi-IPN hydrogel microspheres were developed via incorporating these self-assembled PEG-g-NPHCs NPs within a calcium-crosslinked spray dried alginate matrix (Scheme 2). The main purpose was to investigate these nano-/microspheres biodegradable semi-IPN hydrogel matrices as new carrier systems for sustained pulmonary drug delivery that combines the benefits of both NPs and the swellable hydrogel microspheres suggested in our previous studies (El-Sherbiny, et al., 2010a; El-Sherbiny and Smyth, 2010) while avoiding their shortcomings. As shown in Table 1, three different formulations (SIPN1-SIPN3) were prepared in addition to a control formulation based on alginate alone (SIPN0).
Scheme 2.

A schematic illustration for preparation of the semi-IPN hydrogel microspheres
3.3. Particle size
Designing of a suitable respirable drug carrier system with adequate aerodynamic characteristics represents a critical issue in inhalation therapy. These carriers should be small enough to reach the deep lung, but not so small that they fail to deposit and are exhaled again. The size of the developed PEG-g-NPHCs self-assembled NPs was found to be 206.4±34.8 nm (for 1% solution) as determined by DLS. A trial was also made to investigate the effect of PEG-g-NPHCs concentration onto size of the resulting NPs. It was found (as shown in Figure 2) that, increasing the concentration of copolymer solution from 0.1% to 0.2% has significantly increased the NPs size from 170.0±36.5 nm to 203.5±20.7 nm, respectively. This increase in size continued upon further increasing the concentration to 1% (206.4±34.8 nm). However, the increase in the size of NPs was found to be statistically non significant beyond the copolymer concentration of 0.2%. In case of microparticles, there are different types of diameters depending on the method of their determination. Volume median diameter, VMD is very common upon using laser diffraction technique and it represents the cumulative diameter of 50% of microparticles and it can provide an appropriate estimation of particle size for dry powder inhalation purposes. From the size data in Table 1, the VMDs of the developed plain and BSA-loaded microspheres are in the range 2.18±0.12 to 2.86±0.08 and 2.80±0.21 to 4.36±0.02 μm, respectively. It was noted that uploading BSA in the developed microspheres increases their sizes significantly. It seems also from the obtained results that increasing the percentage of alginate in the microspheres tends, mostly, to decrease the size. This can be attributed to increasing the crosslinking extent. However, this effect of alginate content onto particle size was found to be statistically insignificant. Moreover, as both aerosolization performance and lung deposition efficiencies are determined by aerodynamic diameters (da), the densities of the developed microspheres were also determined because aerodynamic diameters are related to both density and geometric diameters (VMD) as described earlier in equation 1. As apparent in Table 1, the bulk densities of the microspheres are relatively low (in the range 0.22–0.34 and 0.31–0.71 g/cc for plain and BSA-loaded microspheres, respectively). Therefore, the aerodynamic diameters of the microspheres are likely to be significantly less than the geometric diameters (in the range 1.02±0.09 to 1.67±0.06 and 1.52±0.13 to 2.63±0.79 μm for plain and BSA-loaded microspheres, respectively). This explains the relatively high respirability (high RF values) of the developed microspheres as demonstrated in the aerosol dispersion study (section 3.9). The cumulative size and density distributions of the developed semi-IPN microspheres are shown in Figure 3.
Figure 2.

Dependence of the size (R, nm) of the synthesized self-assembled PEG-g-NPHCs nanoparticles on the concentration of copolymer solution (%), as determined by DLS
Figure 3.

The cumulative size and density distributions of the developed semi-IPN microspheres
3.4. Scanning electron microscopy
Figure 4 shows the scanning electron micrographs of some plain and BSA-loaded semi-IPN hydrogel microspheres. The developed hydrogel particles have, in general, spherical shapes with different extents of regularity and surface smoothness. For instance, the drug-free spray dried microspheres (Figure 4a) showed a regular spherical nature with highly smooth, dense and integrated surfaces. This regular spheres turned to show a relatively distorted spheres with a kind of surface roughness upon crosslinking of these drug-free spray dried microspheres with Ca2+ (Figure 4b). In case of the BSA-loaded Ca2+-crosslinked spray dried microspheres (Figure 4c), the shape seems more regular than the BSA-free Ca2+-crosslinked spray dried microspheres and their surfaces appear also relatively smoother and integrated.
Figure 4.

Surface morphology of the developed semi-IPN hydrogel microspheres: (a) Drug-free spray dried microspheres; (b) Drug-free Ca2+-crosslinked spray dried microspheres and (c) BSA-loaded Ca2+-crosslinked spray dried microspheres
3.5. Moisture contents
Table 1 illustrates the moisture contents (%) of the plain and BSA-loaded hydrogel microspheres developed in this study. As noted from the results, the moisture percentages fall in the ranges of (8.29±1.49 to 10.42±1.74) and (5.47±0.34 to 7.52±0.39) for plain and BSA-loaded microspheres, respectively. From the table, plain microspheres tend to have higher percentages of moisture as compared to BSA-loaded microspheres. Although the differences in moisture contents of different formulations are not statistically significant, it seems that moisture percentages decrease with increasing the PEG-g-NPHCs content in the microspheres.
3.6. Dynamic swelling study
The swelling patterns of the BSA-loaded hydrogel microspheres in PBS, pH 7.4 are shown in Figure 5. Swelling behavior was estimated via determining the increase in both volume median diameter (VMD, μm) and medium diameters (X50, μm) of the microspheres with time using laser diffractometer. As appeared from the figure, the developed formulations showed a fast initial swelling within the first 2 min. For instance, the VMD of SIPN1, SIPN2 and SIPN3 microspheres has increased from about 4 μm when dry to 23.6, 26.0 and 48.1 μm, respectively. This swelling continues regularly with time to reach 84.1, 66.3 and 84.2 μm at 20 min for SIPN1, SIPN2 and SIPN3, respectively. It can be noted also from Figure 5 that, increasing the percent of alginate upon moving from SIPN3 (50% alginate) to SIPN2 (60% alginate) and SIPN1 (80% alginate) tends to decrease the swelled sizes of the microspheres. This behavior may be attributed to increasing the crosslinking extent of the semi-IPN hydrogel microspheres upon increasing the alginate percent due to the interaction with CaCl2. This speculation can be confirmed also by comparing the swelling profiles of the microspheres formulations SIPN1-SIPN3 with that of the control, SIPN0 (100% alginate) which attained only a VMD value of 31.7 μm after 20 min. The same conclusion was also obtained from the X50 (μm) values of swelled microspheres (Figure 5b).
Figure 5.

Volume median diameter (VMD, μm) and medium diameter (X50, μm) of swelled BSA-loaded semi-IPN hydrogel microspheres
3.7. In-vitro biodegradation study
The in-vitro enzymatic degradation study of the developed microspheres was carried out in PBS, pH 7.4 in presence of 2 mg/ml lysozyme as has been the protocol in previous studies (Hirano, et al., 1989). In this study, the percent weight remaining of the microspheres as a function of time was determined and taken as a measure of degradation. Figure 6 shows the enzymatic degradation profiles of the developed microspheres. As shown in the figure, degradation of the three prepared formulations was started to occur within the first 2 h. It seems also that, in general, increasing the percent of PEG-g-NPHCs copolymer in the microspheres tends to reduce the degradation rate (high remaining weights). For instance, at 6 h, the remaining weights of SIPN1 (20% PEG-g-NPHCs), SIPN2 (40% PEG-g-NPHCs) and SIPN3 (50% PEG-g-NPHCs) are 65.9±2.5, 74.3±0.2 and 82.4±3.3, respectively. This effect of copolymer percent became relatively non significant beyond 144 h. The retarding effect of the PEG-g-NPHCs for the enzymatic degradation of microspheres is in agreement with the results obtained by our group in an earlier study (El-Sherbiny, et al., 2010a) with other formulations containing PEG-g-NPHCs. This influence of PEG-g-NPHCs can be attributed to both hydrophobicity and the steric hindrance arises from their bulky phthaloyl groups. These two effects tend to impede the diffusion of enzyme through the microspheres matrices and consequently retard their degradation. The SIPN1-SIPN3 microspheres showed better biodegradation rates as compared to that of the control formulation, SIPN0 which is attributed to the high crosslinking extent in SIPN0.
Figure 6.

In-vitro degradation profiles of the developed semi-IPN hydrogel microspheres as investigated in PBS, pH 7.4 at 37°C in presence of lysozyme
3.8. In-vitro cumulative release studies
The entrapment efficiency (E%) of the model protein, BSA in the developed hydrogel microspheres were estimated as shown in Table 1. As noted from the table, the E% values are in the range 49.60±2.21 to 90.32±0.90. The partial loss of the entrapped drug takes place particularly during both the crosslinking in CaCl2 solution and washing of the microspheres after crosslinking. Moreover, it seems that the E% tends to decrease, in most cases, with decreasing the alginate content in the developed microspheres. Decreasing the alginate content leads to a reduction in the extent of ionotropic crosslinking and consequently facilitates loss of more drug during washing.
The cumulative release profiles of BSA from the developed hydrogel microspheres in PBS, pH 7.4 at 37°C is shown in Figure 7. As apparent from the figure, the SIPN1-SIPN3 microspheres showed enhanced release profiles of BSA than that of the control formulation (SIPN0). The BSA release from the microspheres showed three different phases. Initially, there was a burst release within the first 2 h. This burst release can be attributed to the fast initial swelling of the hydrogel microspheres (see Figure 5). Then, the microspheres showed a slow sustained release of BSA up to 4 days. The drug released in this stage may be the drug that is physically entrapped in the microspheres. In the final phase, a slower release was observed which may be attributed to the release from the NPs upon starting degradation of the microspheres (see Figure 6). However this BSA release observed in the last phase is not a quantitative reflection of the extent of biodegradation. Moreover, the figure shows that increasing the alginate content in the microspheres tends to decrease the percentage of BSA released which can be attributed to increasing the crosslinking extent and consequently decreasing the swelling (Figure 5).
Figure 7.

The cumulative release profiles of the model protein, BSA from the developed semi-IPN hydrogel microspheres in PBS, pH 7.4 at 37°C
3.9. In-vitro aerosolization study
The powder aerosolization characteristics of the formulation SIPN2-BSA, as an example for the semi-IPN hydrogel microspheres developed in this study, were assessed in-vitro using a dry-powder, breath-activated inhaler device (Handihaler® DPI) with aid of NGI at air flow rate of 60±5% L/min. The percentage of drug deposition on each stage of the NGI, adapter, capsule and the device was calculated and plotted as in Figure 8. The average emitted fraction, EF (%) of the investigated powder (SIPN2-BSA) was found to be relatively high (97.36%). It was found also that, for the formulation SIPN2-BSA, the average respirable fraction, RF (%) of the powder is 30.69% and its fine particle fraction, FPF% (cut-off diameters, da < 4.46 μm) is 31.52%. This FPF value represents the fraction of the dry powder that would likely deposit in the deep lung. In conclusion, the data obtained from this preliminary powder aerosolization study implies that the semi-IPN hydrogel microspheres developed in this contribution are very suitable for inhalation and drug delivery to the deep lung.
Figure 8.

Dry powder aerosolization study using NGI with a flow rate; 60 L/min and using a Handihaler® as inhaler device
Conclusion
In this contribution, PEG-g-NPHCs self-assembled nanoparticles were synthesized then used in a combination with sodium alginate to prepare respirable semi-IPN hydrogel microspheres as novel biodegradable carriers for pulmonary sustained drug delivery. The developed nano-/microspheres carrier systems were obtained via spray drying technique followed by ionotropic crosslinking in mild aqueous medium. The carrier systems were designed in such a way to display appropriate morphological and aerodynamic characteristics. The preliminary in-vitro evaluation of these carriers showed that they can be used as good potential carriers for pulmonary sustained drug delivery.
Acknowledgments
This research was supported by National Institutes of Health, National Institute of Biomedical Imaging and Bioengineering (REB006892A), Oxnard Foundation, PhRMA Foundation and CF Foundation.
Footnotes
Declaration of Interest
The authors report no declarations of interest.
References
- Ahsan F, Rivas IP, Khan MA, Suárez-Torres AI. Targeting to macrophages: role of physicochemical properties of particulate carriers–liposomes and microspheres on the phagocytosis by macrophages. J Control Release. 2002;2(79):29–40. doi: 10.1016/s0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cascone MG, Pot PM, Lazzeri L, Zhu Z. Release of dexamethasone from PLGA nanoparticles entrapped into dextran/poly(vinyl alcohol) hydrogels. J Mater Sci, Mater Med. 2002;13:265–269. doi: 10.1023/a:1014006800514. [DOI] [PubMed] [Google Scholar]
- El-Sherbiny IM, Abdel-Bary EM, Harding DRK. Swelling characteristics and in vitro drug release study with pH-and thermally sensitive hydrogels based on modified chitosan. J Appl Polym Sci. 2006;102:977–985. [Google Scholar]
- El-Sherbiny IM, McGill S, Smyth HDC. Swellable microparticles as potential carriers for pulmonary sustained drug delivery. J Pharm Sci. 2010a;99(5):2343–2356. doi: 10.1002/jps.22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sherbiny IM, Abdel-Bary EM, Harding DRK. Preparation and in vitro evaluation of new pH-sensitive hydrogel beads for oral delivery of protein drugs. J Appl Polym Sci. 2010b;115 (5):2828–2837. [Google Scholar]
- El-Sherbiny IM. Enhanced pH-responsive carrier system based on alginate and chemically modified carboxymethyl chitosan for oral delivery of protein drugs: Preparation and in-vitro assessment. Carbohydr Polym. 2010 doi: 10.1016/j.carbpol.2010.01.034. in press. [DOI] [Google Scholar]
- El-Sherbiny IM, Smyth HDC. Novel cryomilled physically crosslinked biodegradable hydrogel microparticles as carriers for inhalation therapy. J Microencapsulation. 2010 doi: 10.3109/02652041003739840. in press. [DOI] [PubMed] [Google Scholar]
- Grenha A, Grainger CI, Dailey LA, Seijo B, Martin GP, Remunan-Lopez C, Forbes B. Chitosan nanoparticles are compatible with respiratory epithelial cells in vitro. Eur J Pharm Sci. 2007;31:73–84. doi: 10.1016/j.ejps.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Heyder J, Rudolf G. Mathematical models of particle deposition in the human respiratory tract. J Aerosol Sci. 1984;15:697–707. [Google Scholar]
- Hirano S, Tsuchida H, Nagao N. N-Acetylation in chitosan and the rate of its enzymic hydrolysis. Biomater. 1989;10:574–6. doi: 10.1016/0142-9612(89)90066-5. [DOI] [PubMed] [Google Scholar]
- Kabbaj M, Phillips NC. Anticancer activity of mycobacterial DNA: Effect of formulation as chitosan nanoparticles. J Drug Target. 2001;9:317–328. doi: 10.3109/10611860108998768. [DOI] [PubMed] [Google Scholar]
- Kawaguchi H, Koiwai N, Ohtsuka Y, Miyamoto M, Sasakawa S. Phagocytosis of latex particles by leukocytes. I Dependence of phagocytosis on the size and surface potential of particles. Biomater. 1986;7:61–66. doi: 10.1016/0142-9612(86)90091-8. [DOI] [PubMed] [Google Scholar]
- Kim DH, Martin DC. Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomater. 2006;27:3031–3037. doi: 10.1016/j.biomaterials.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Yoon SG, Kim NY, Kim SI. Swelling characterization of the semi-interpenetrating polymer network hydrogels composed of chitosan and poly(diallyldimethylammonium chloride) J Appl Polym Sci. 2004;91:2876–2880. [Google Scholar]
- Klempner D. Advances in interpenetrating polymer networks. In: Frisch KC, editor. Vol. 4. Technomic Publishing Co; Lancaster, PA: 1994. p. 312. [Google Scholar]
- Krenis LJ, Strauss B. Effect of size and concentration of latex particles on respiration of human blood leucocytes. Proc Soc exp Biol (NY) 1961;107:748–750. doi: 10.3181/00379727-107-26743. [DOI] [PubMed] [Google Scholar]
- Majeti NV, Kumar R. A review of chitin and chitosan applications. React Funct Polym. 2000;46:1–27. [Google Scholar]
- Makino K, Yamamoto N, Higuchi K, Harada N, Ohshima H, Terada H. Phagocytic uptake of polystyrene microspheres by alveolar macrophages: effects of the size and surface properties of the microspheres. Colloids Surf B, Biointerfaces. 2003;27:33–39. [Google Scholar]
- Maoa S, Shuaia X, Ungera F, Wittmara M, Xiec X, Kissel T. Synthesis, characterization and cytotoxicity of poly(ethylene glycol)-graft-trimethyl chitosan block copolymers. Biomater. 2005;26:6343–6356. doi: 10.1016/j.biomaterials.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Martonen T, Smyth HDC, Isaccs K, Burton R. Issues in drug delivery: dry powder inhaler performance and lung deposition. Respir Care. 2005;50:1228–1252. [PubMed] [Google Scholar]
- Murata Y, Kodama Y, Isobe T, Kofuji K, Kawashima S. Drug release profile from calcium-induced alginate–phosphate composite gel beads. Int J Polym Sci. 2009:1–4. [Google Scholar]
- Oberdorster G. Pulmonary effects of inhaled ultrafine particles. Int Arch Occup Environ Health. 2001;74:1–8. doi: 10.1007/s004200000185. [DOI] [PubMed] [Google Scholar]
- Ohya Y, Cai R, Nishizawa H, Hara K, Ouchi T. Preparation of PEG-grafted chitosan nanoparticles as peptide drug carriers. STP Pharma Sci. 2000;10:77–82. [Google Scholar]
- Opanasopit P, Ngawhirunpat T, Chaidedgumjorn A, Rojanarata T, Apirakaramwong A, Phongying S, et al. Incorporation of camptothecin into N-phthaloyl chitosan-g-mPEG self-assembly micellar system. Eur J Pharm Biopharm. 2006;64:269–76. doi: 10.1016/j.ejpb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Opanasopit P, Ngawhirunpat T, Rojanarata T, Choochottiros C, Chirachanchai S. N-Phthaloyl chitosan-g-mPEG design for all-trans retinoic acid-loaded polymeric micelles. Eur J Pharm Sci. 2007;30:424–31. doi: 10.1016/j.ejps.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Parka JH, Kwon S, Lee M, Chung H, Kim JH, Kim YS, Park RW, Kim IS, Seo SB, Kwon IC, Jeong SY. Self-assembled nanoparticles based on glycol chitosan bearing hydrophobic moieties as carriers for doxorubicin: In vivo biodistribution and anti-tumor activity. Biomater. 2006;27:119–126. doi: 10.1016/j.biomaterials.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Prego C, Torres D, Fernandez-Megia E, Novoa-Carballal R, Quiñoá E, Alonso MJ. Chitosan–PEG nanocapsules as new carriers for oral peptide delivery: Effect of chitosan pegylation degree. J Control Release. 2006;111:299–308. doi: 10.1016/j.jconrel.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Rudt S, Muller RH. In vitro phagocytosis assay of nano- and microparticles by chemiluminescence. I Effect of analytical parameters, particle size and particle concentration. J Control Release. 1992;22:263–271. [Google Scholar]
- Smyth HDC, Hickey AJ. Carriers in drug powder delivery: implications for inhalation system design. Am J Drug Deliv. 2005;3:117–132. [Google Scholar]
- Tsapis N, Bennett D, Jackson B, Weitz DA, Edwards DA. Trojan particles: Large porous carriers of nanoparticles for drug delivery. PNAS. 2002;99:12001–12005. doi: 10.1073/pnas.182233999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Zhang C, Ping Q, Yu L. A series of novel chitosan derivatives: synthesis, characterization and micellar solubilization of paclitaxel. Carbohydr Polym. 2007;68:781–92. [Google Scholar]
- Zahoor A, Sharma S, Khuller GK. Inhalable alginate nanoparticles as antitubercular drug carriers against experimental tuberculosis. Int J Antimicrob Ag. 2005;26:298–303. doi: 10.1016/j.ijantimicag.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang H, Wu Z, Wang Z, Niu H, Li C. Nasal absorption enhancement of insulin using PEG-grafted chitosan nanoparticles. Eur J Pharm Biopharm. 2008;68:526–34. doi: 10.1016/j.ejpb.2007.08.009. [DOI] [PubMed] [Google Scholar]