

Abstract
Alzheimer’s disease (AD) is a common, genetically complex, fatal neurodegenerative disorder of late life. Although several genes are known to play a role in early-onset AD, identification of the genetic basis of late onset AD (LOAD) has been challenging, with only the APOE gene known to have a high contribution to both AD risk and age-at-onset. Here we present the first genome-scan analysis of the complete, well-characterized University of Washington LOAD sample of 119 pedigrees, using age-at-onset as the trait of interest. The analysis approach used allows for a multilocus trait model while at the same time accommodating age censoring, effects of APOE as a known genetic covariate, and full pedigree and marker information. The results provide strong evidence for linkage of loci contributing to age-at-onset to genomic regions on chromosome 6q16.3, and to 19q13.42 in the region of the APOE locus. There was evidence for interaction between APOE and the locus on chromosome 6q and suggestive evidence for linkage to chromosomes 11p13, 15q12-14, and 19p13.12. These results provide the first independent confirmation of an AD age-at-onset locus on chromosome 6 and suggest that further efforts towards identifying the underlying causal locus or loci are warranted.
Keywords: linkage analysis, MCMC, oligogenic, dementia, age-censored
INTRODUCTION
Alzheimer disease (AD: MIM 104300) is the most common cause of dementia and seventh leading cause of death in the US (Alzheimer’s Association 2010). AD is typically classified into early onset AD (EOAD, age-at-onset (AAO) < 60 yrs) and late onset AD (LOAD, AAO ≥ 60 yrs). The more common LOAD evolves slowly from mildly impaired memory to severe cognitive loss. The annual incidence rate of LOAD increases from ~1% at ages 65–70 yrs to 6–8% at ages ≥85 yrs (Mayeux 2003), with 10–30% of individuals affected by age 85 yrs (Hebert et al. 2003; Rocca et al. 1991). There are few effective treatments. A few drugs alleviate symptoms in the early or states of the disease, and many more are in various stages of clinical trials. However, there is still no cure, effective prevention, or delay of age-at-onset, any of which would provide substantial benefit. Therefore, research into the underlying causes and onset age of AD is key to developing better diagnostic and treatment tools.
A substantial fraction of AD is clearly attributable to genetic factors. Family history of dementia is a key risk factor (Lautenschlager et al. 1996; Mayeux et al. 1991), with high heritability of 58–79% (Bergem et al. 1997; Gatz et al. 1997; Gatz et al. 2006; Tanzi and Bertram 2005). A number of rare mutations in three autosomal genes - amyloid precursor protein (APP [MIM 104760]) and the two presenilins (PSEN1 [MIM 104311] and PSEN2 [MIM 600759]) -are causal in EOAD (Bertram and Tanzi 2008; Goate et al. 1991; Levy-Lahad et al. 1995; Sherrington et al. 1995). To date, only one gene (APOE [MIM 107741]) has been confirmed to have substantial genotype-specific differences in risk for LOAD (Corder et al. 1993) with meta-analysis odds ratios of 3.68 for allele ε4 vs. allele ε3 (Bertram et al. 2007). In contrast, although ~9 additional loci have been implicated, and confirmed in European-ancestry samples by genomewide association studies (GWAS) (Harold et al. 2009; Lambert et al. 2009; Naj et al. 2011; Schellenberg and Montine 2012; Seshadri et al. 2010; Wijsman et al. 2011), all have small effects. Also, confirmation with many of the same SNPs was not obtained in African American samples (Logue et al. 2011), suggesting that these SNPs are non-causal and tag unidentified causal variants. Even the most convincing of these SNPs, near bridging integrator 1 (BIN1, [MIM 601248]), has an odds ratio of only 1.17 for the risk allele (Bertram et al. 2007) – far less than that of APOE. APOE is not only associated with risk of AD, but also with AAO, accounting for only 4 – 20% of the variance in AAO (Bennett et al. 1995; Daw et al. 2000; Slooter et al. 1998; Tunstall et al. 2000). There is evidence for several other loci contributing to variance in AAO (Daw et al. 2000), suggesting that it would be of interest to identify such genes (Hagnell et al. 1992; Miech et al. 2002).
Previous genome scans for LOAD focused primarily on two traits: the risk of AD, and to a lesser extent, AAO. Use of AD risk in pedigree-based linkage scans identified several regions with suggestive or significant evidence of linkage. From these, a meta-analysis highlighted several regions, on chromosomes (chr) 1, 3, 6, 7, 8, 17, and 19 (Butler et al. 2009). Using AAO as the trait, a small number of regions have also provided consistent but modest evidence for linkage with LOAD across multiple pedigree-based samples (Choi et al. 2011; Dickson et al. 2008; Holmans et al. 2005; Lee et al. 2008; Li et al. 2002; Wijsman et al. 2004). Several of these studies include the NIMH AD sample, and are therefore not independent. Only one smaller GWAS study has been published for AAO that implicated a new locus (Kamboh et al. 2011), but without confirmation to date. These combined results from both pedigree and population-based samples, therefore, are all consistent with a model for LOAD risk that includes multiple rare or uncommon variants in multiple genes, with genetic heterogeneity both within and between samples.
The potential power of AAO as a trait makes it of interest for linkage analysis of AD. Several models based on censored AAO in pedigree data have been proposed. These include a semi-parametric survival model combined with multivariate random effects (Li 2002; Pankratz et al. 2005; Scurrah et al. 2000), and use of residuals from a survival analysis as input for variance-component (VC) linkage analysis (Dickson et al. 2008; Li et al. 2002). Although the semi-parametric model makes use of right-censored traits such as AAO with inclusion of familial dependence, the computational complexity and model assumptions limit use. Similarly, the VC approach is based on use of residuals obtained by ignoring the pedigree information (Dickson et al. 2008; Li et al. 2002). Lack of full incorporation of genetic covariates such as APOE because of inability to impute genotypes into unsampled subjects, more sensitivity of VC methods than other methods to pedigree ascertainment (Forrest and Feingold 2000; Kleensang et al. 2010; Williams et al. 1997), and inflexibility to group analysis also limit the use of this method.
Markov chain Monte Carlo (MCMC) provides a computationally tractable approach for analysis that circumvents some of the limitations of these other methods. Bayesian reversible-jump MCMC oligogenic linkage analysis provides a tractable approach for analysis of pedigree-based censored AAO data (Daw et al. 1999; Heath 1997). This approach provides high efficiency for three reasons: (1) it uses a multilocus (oligogenic) trait; (2) it allows a multilocus marker model in pedigrees of virtually any size, together with multilocus genotype data; and (3) it incorporates age censoring, allowing efficient use of all available age data. One advantage that distinguishes this approach from other analysis options is that inclusion of APOE as a genetic covariate allows the model to impute, and therefore use, APOE genotypes even in subjects missing genotype data. This provides larger samples with covariate-adjusted data for analysis of AAO. For example, use of this approach was able to identify AAO effects of APOE in the presence of a PSEN2 mutation in a modest-sized sample (Wijsman et al. 2005) and contributed to identification of regions with AAO modifier loci in the presence of a PSEN2 mutation and APOE adjustment (Marchani et al. 2010).
Here we present the results of a genome scan of LOAD AAO on the University of Washington (UW) dataset using such MCMC-based oligogenic analysis. Although a previous analysis examined five chromosomes in an earlier smaller subset of this sample (Wijsman et al. 2004), this is the first complete scan of this entire sample of well-documented, autopsy-verified LOAD families, and therefore increases the number of family-based LOAD samples on which a genome scan has been performed. This data set supports evidence for AAO loci on chromosomes 6, 11, 15, and 19, with the strongest evidence for a non-APOE locus on chromosome 6.
METHODS
Sample and Phenotypes
Families in this study were identified and recruited primarily by the University of Washington (76 families). Additional families were obtained from Oregon Health Sciences University (19 families), the University of Minnesota (3 families) and from the National Cell Repository for Alzheimer’s Disease (18 families). Finally, 3 additional families were multiply ascertained as described under quality control (below). Families were included in the analysis if the family’s mean age-at-onset was ≥60, and no family members had identified mutations in known early-onset Alzheimer’s disease genes or genes associated with related dementias: APP, PSEN1, PSEN2, or MAPT. Twenty-six of the families used here were also submitted to the NIA LOAD sample with members of 14 of these represented in the recent GWAS of the NIA LOAD sample (Wijsman et al. 2011). After completion of the analyses and as we finalized this report, we learned that one small 13-member family was also part of a late-onset family under study elsewhere in which a potential mutation in PSEN1 had been identified (Kauwe et al. 2007); this family was not removed from the analyses since the oligogenic model used for analysis accommodates multiple trait loci. The study was approved by the University of Washington institutional review board.
The study included 1323 individuals in 119 families (Table I). Family sizes ranged from 4–53 individuals, in 2–6 generations. Of these individuals, 541 had a diagnosis of Alzheimer’s Disease (AD affected), 659 were free of AD (unaffected), and 123 had unknown disease status. For purposes of our analysis here an individual was considered to have missing phenotype data when either age or AD diagnostic status was missing. AAO was determined as that age at which family members and/or colleagues first noticed a persistent change in memory or behavior. After accounting for diagnosis and age information, phenotype data were available for 981 individuals, of whom 479 were affected by AD with a mean (standard deviation, sd) AAO of 71.4 (8.0) years, and 502 were unaffected with a mean (sd) censoring age of 68.7 (15.3) years. Of the 119 families, 104 families had neuropathological documentation by CERAD and Reagan criteria and/or Braak staging (Consensus Report 1998; McKhann et al. 1984; Mirra et al. 1991) for at least one individual affected by AD, with 1–8 autopsies per family and a total of 246 autopsies across all families.
TABLE I.
Sample Characteristics
| Alzheimer’s Disease Status | ||||
|---|---|---|---|---|
| Affected | Unaffected | Unknown | Total | |
| Subjects | 541 | 659 | 123 | 1323 |
| Female | 333 (61.6%) | 327 (49.6%) | 60 (48.8%) | 720 (54.5%) |
| Age info | 479 (88.5%) | 502 (76.2%) | N/A | 981 (74.1%) |
| Age, yrs * | 71.4 (8.0) | 68.7 (15.3) | N/A | 70.0 (12.3) |
| Autopsies | 238 | 8 | 0 | 246 |
| > 90% genotyped | 289 (53.4%) | 207 (31.4%) | 0 | 496 (37.5%) |
| Genotypes missing | 168 (31.3%) | 386 (58.6%) | 123 (100%) | 677 (51.2%) |
| APOE genotyped | 403 (74.5%) | 355 (53.9%) | 0 | 758 (57.3%) |
| APOE E2 AF ** | 0.024 (0.005) | 0.059 (0.009) | N/A | 0.040 (0.005) |
| APOE E4 AF ** | 0.503 (0.018) | 0.344 (0.018) | N/A | 0.428 (0.013) |
Shown in mean (sd) format
AF stands for allele frequency; mean (sd) is shown
Genotypes
A total of 401 autosomal multiallelic microsatellite markers (ABI Prism Linkage Mapping Set, Version 2.5, http://home.appliedbiosystems.com/) were genotyped across the genome, with methods described elsewhere (Wijsman et al. 2004). Average spacing between markers was about 10 cM and the mean (range) of heterozygosity was 0.79 (0.53 – 0.92). Genotype data were available for 609 individuals, 346 of whom were affected and 263 were unaffected (Table I). Of these 609 individuals, 113 had more than 10% missing genotypes, and represented samples with little available DNA. A total of 677 individuals in older generations were unavailable for sampling and thus were completely missing genotype data, including 168 affected, 386 unaffected, and 123 with unknown disease status. In addition, for some individuals in each pedigree, APOE genotypes were available, for a total of 758 individuals, of whom 403 were affected and 355 were unaffected. APOE genotypes were available for 71.2% of all subjects with age and affection status. APOE genotyping methods were previously described (Hixson and Vernier 1990; Levy-Lahad et al. 1995).
Marker positions were obtained from the Rutger’s map (Matise et al. 2007). We converted these positions based on the Haldane map function (Haldane 1919), and used a sex-averaged map for analysis. Marker allele frequencies were estimated from the observed data by direct allele counting, and were provided to the analysis program.
Prior to analysis, the data were subjected to a variety of quality control checks. The program RELPAIR (http://csg.sph.umich.edu/boehnke/relpair.php) (Epstein et al. 2000), used with a 1% genotyping error assumption, was used to identify sample duplicates and incorrect relationships, which were corrected prior to further analysis. After pedigree structure corrections were made, the program Loki (version 2.4.7, http://www.stat.washington.edu/thompson/Genepi/pangaea.shtml) (Daw et al. 1999; Heath 1997) was used to identify remaining Mendelian inconsistent genotypes; these genotypes were removed for all subjects in families with such inconsistencies. Markers with excessive numbers of Mendelian inconsistencies were also removed for all families. Markers were also evaluated for evidence of temporal drift in allele-size calling, and corrected, when necessary.
Statistical methods
Analysis Overview
We used an oligogenic trait model as our primary analysis approach, implemented in a Bayesian reversible-jump (Green 1995) MCMC framework (Daw et al. 1999; Heath 1997). Key features of this model are the allowance for multiple underlying trait loci (e.g., an oligogenic trait model) and ability to incorporate full multipoint marker information on all pedigrees without need for pedigree simplification. This approach improves power to detect evidence of linkage, while at the same time providing location resolution within the implicated region (Heath et al. 1997). After data cleaning, oligogenic segregation analyses followed by joint linkage and oligogenic segregation analyses were conducted, both without and with APOE as a covariate. Analysis without APOE as a covariate was the most similar to analyses by others of AD treated as a discrete trait. Inclusion of APOE as a covariate allowed for removal of genetic variation attributable to APOE, thereby increasing power to detect additional, weaker, QTLs which were hidden by the APOE effect. Also, inclusion of APOE as a covariate was expected to remove from the oligogenic model the somewhat more complicated sub-models that tend to occur for trait loci with more than 2 alleles (Rosenthal and Wijsman 2010), as occurs for the triallelic APOE locus. Oligogenic segregation analysis was performed without use of marker information in order to provide a description of the underlying QTL models consistent with the distribution of AAO within the pedigrees. This serves as a useful indication and estimation of QTL models that may be identified in further linkage analysis. Joint linkage and segregation analysis makes use of additional information from linked markers to estimate the location of QTLs in the genome, to provide a measure of strength of evidence for linkage, and to provide effect sizes of such localized QTLs. The statistical significance of identified linkage signals were then estimated by resampling simulations.
Statistical Model
Age-at-onset (AAO) was our response variable, treated as a censored quantitative trait with affected subjects having observed AAO, and unaffected subjects having censored AAO. AAO was assumed to have an additive relationship with quantitative trait locus (QTL) effects, with these effects acting independently of each other. All QTLs were assumed to be diallelic with allele frequency Pa for the allele associated with the baseline homozygous genotype, and with additive effects across QTLs, although genotypes within a QTL could include dominance. Explicitly, the model can be expressed as: where Y is age-at-onset, μ is the overall mean AAO of a baseline genotype, X is the incidence matrix for covariate effects, β is the vector of covariate effects, Qi is the incidence matrix for the effect of QTL i, αi is the vector of additive and dominant effects for QTL i, k is the number of QTLs, and e is the residual effect. The number of QTLs was assumed to be Poisson distributed, and the residual effect was assumed to have a Normal distribution with mean 0, as is typical in linear modeling. In the analyses here, the covariate effects, when included, were the individual APOE genotypes. All effects in this model are assumed to be additive. The posterior distributions of model parameters were estimated conditional on prior distributions and the data. More details of the Bayesian MCMC approach and theory are described elsewhere (Daw et al. 1999; Heath 1997). Additional information is also available for readers with less familiarity with the underlying statistical methods (Wijsman 2002; Wijsman and Yu 2004). All analysis runs were conducted with Loki ver. 2.4.7.
APOE was included as a genetic covariate for some analyses since it is well established as influencing AAO based on simple survival analysis methods (Corder et al. 1993) or the more sophisticated oligogenic models also used here (Daw et al. 2000). Missing APOE information was imputed using standard methods based on a Mendelian inheritance model (Elston and Stewart 1971; Lander and Green 1987), based on pedigree information plus APOE genotypes observed on some members of each pedigree together with genotypes at one flanking, highly-polymorphic marker (heterozygosity ~80%) situated ~3 cM on each side of APOE. Imputed genotypes were used in a multiple-imputation framework (Heath 1997; Thompson and Basu 2003). This is considered a gold-standard for accommodating missing data, and allows the analysis to incorporate both maximum available information as well to fully allow for the inherent uncertainty introduced by using variables with missing data (Rubin 1996; Schafer 1999). Unlike population-based approaches, pedigree-based genotype imputation does not rely on linkage disequilibrium among markers, and instead uses the information present in the joint inheritance of linked loci in pedigrees, coupled with observed genotype data on a subset of subjects. Evaluation of the accuracy of imputed genotypes has been carried out in situations where underlying true, but masked, genotypes are available for measurement of accuracy, and indicates accuracy of >98% in the context of flanking genome scan STR marker data (Cheung et al. 2010).
Oligogenic segregation analysis
Oligogenic segregation analysis was conducted marker-free to identify QTL models that explain the observed data, with and without adjustment for APOE genotype, as described above. Parameters of interest included covariate (APOE) effects (if included), the number of QTLs, and the mean trait value for each genotype for each QTL. The prior distribution on the number of QTLs in the model was Poisson with mean 4, based on previous results (Daw et al. 2000), with a range of 0–17. The prior distribution of the variance in genotypic effects (τβ) was Normal with mean 0 and variance of 512. The sensitivity of results to the choice of τβ was evaluated by performing a set of sensitivity analyses with τβ ranging from 200 to 1000. Results were insensitive to this choice of τβ over this range in both segregation and linkage analysis (not shown). The analysis was based on 10,000,000 MCMC sweeps, with a burn-in of 10,000 sweeps and a thinning interval of 10 sweeps, where one sweep defines one round of updating of all parameter values in the model. Inference was based on the posterior distribution of the model parameters. Given allele labels of ‘A’ and ‘a’ for each diallelic QTL, the homozygote effect for each QTL is defined as the difference between the mean trait value for subjects with trait genotypes AA vs. the baseline aa genotype, μAA - μaa, with the heterozygote effect similarly defined as μAa - μaa. The parameters of interest were the homozygote and heterozygote effect for each QTL in the model, which were summarized by the mean homozygote and heterozygote effects.
Joint linkage-segregation analysis
Joint multipoint linkage-segregation analysis was conducted with genome scan markers to localize the QTLs in the genome. Parameters of interest included all those also of interest in the oligogenic segregation analysis, plus the location of each QTL. The prior distributions specified and run conditions were the same as in the oligogenic segregation analysis, except that 500,000 sweeps with a burn-in of 1,000 sweeps were used. Analysis runs were conducted genomewide. The Bayes’ Factor (BF) (Kass and Raftery 1995) was used to quantify the evidence of linkage, where the BF was defined as the ratio of the posterior to prior odds of a parameter value indicating that the location of a QTL was in a particular interval. In this analysis, BFs were computed in 2 cM bins across each chromosome. To further refine information about linkage signals on chrs 6, 11, 15, and 19, additional longer runs with 10,000,000 sweeps were carried out for chr 6, and runs with 2,000,000 sweeps were carried out for chrs 11, 15, and 19. Each such run was repeated three times, with the same prior distributions as for the other analysis. Single-marker analysis, which is not sensitive to map assumptions, was used to investigate the possibility of spurious positive linkage signals that can result from map misspecification in multipoint analysis (Daw et al. 2000). In each of these analysis runs, each single marker from one of the four chromosomes was included, with the same analysis conditions as in the genomewide scan used for analysis. No evidence of spurious positive signals was observed: all regions with maximum BF in multipoint analysis also produced a maximum BF for the individual markers in the same region in single marker analysis.
Measurement of evidence for linkage
Simulations were conducted to obtain empirical p-values for the strongest linkage signal identified on chr 6. One thousand simulations with the program SimSuite (http://faculty.washington.edu/wijsman/software.shtml) were conducted under the null hypothesis of no linkage, as described above, but with a re-sampled set of marker genotypes in each simulation (Igo and Wijsman 2008). The runs consisted of 200,000 sweeps, with a burn-in of 1,000 sweeps and a thinning interval of 10 sweeps. Other settings were the same as described above. The relatively small number of simulations and sweeps was dictated by the computational intensity of these analyses. We carried out these analyses over a 75 cM window surrounding the position of interest to get a region-specific empirical p-value. One analysis run with the true genotype data was conducted and used as the positive control. The maximum BF (BFmax) was obtained from each simulation and the positive control, and the empirical p-value was defined as the proportion of the BFmax’s from the simulations that exceed the BFmax from the positive control.
RESULTS
Oligogenic segregation analysis
In the absence of APOE as a covariate, the analyses indicated the presence of several QTLs in the oligogenic model (Figure 1A, B). The most distinct QTL model (model 1, left) described an essentially recessive model for elevated AAO, with a protective homozygote (AA) for the minor allele that had a mean AAO that was 25.9 years later than that of the more common (aa) baseline homozygote. This model was slightly under-dominant with a mean heterozygote (Aa) effect of −6.3 years relative to that of the aa baseline homozygote. Previous studies indicate that this under-dominance is likely to be an artifact resulting from the additive model assumption coupled with ascertainment of pedigrees with multiple living cases (Ma et al. 2007). As such, this model should therefore not to be over-interpreted. Under these same analysis conditions, there are additional QTLs that are discernible, representing generally dominant or over-dominant models for later AAO, but with multiple modes and overlapping distributions that make interpretation difficult.
Figure 1.

Posterior distribution of QTL model genotype effects in the absence (A, B) and presence (C, D) of APOE as a covariate. Panels A and C depict 3-dimensional histograms of the posterior density of parameter estimates for the heterozygous vs. homozygous effects (see text in Methods). Panels B and D depict contour plots of the same information.
When APOE was included as a covariate, the individual QTL models within the oligogenic model were more distinct (Figure 1C, D, Table II). This is consistent with expectations for simplification of the oligogenic model with removal of the APOE effect. Model 1 was similar to that obtained in the absence of adjustment for APOE, but was more compact, i.e., the bivariate credible intervals (Bayesian confidence intervals) for the homozygote and heterozygote genotype effects were smaller than they were without APOE adjustment. With APOE adjustment, the additional indistinct QTL models obtained in the absence of APOE adjustment (Figure 1A) coalesced and spread apart into a much stronger and compact model 2 and a diffuse model 3 (Figure 1C). Model 2 was essentially dominant for the major allele for elevated AAO (or recessive for the minor allele for reduced AAO) since individuals with the heterozygous genotype have an average AAO that is 21.3 years higher than that of individuals with the baseline aa homozygous genotype, and individuals with the AA homozygous genotype having a similar increased average AAO of 23.9 years (Table II). The most diffuse model 3 (center bottom) had both low homozygote and heterozygote effects, hence is of less interest and is not further discussed or summarized in Table II.
TABLE II.
Estimated Parameters for QTL Models with APOE Adjustment
| Analysis | Chromosome | Region (cM) | Model | Pa * | Heterozygote effect | Homozygote effect |
|---|---|---|---|---|---|---|
| Segregation Analysis | NA | NA | 1 | 0.69 (0.12) | −7.08 (2.90) | 26.36 (9.20) |
| NA | NA | 2 | 0.17 (0.11) | 21.34 (4.54) | 23.85 (4.13) | |
| Joint linkage and segregation Analysis | 6q16.3 | 100 – 125 | 1 | 0.69 (0.07) | −6.93 (2.17) | 27.71 (8.53) |
| 11p13 | 25 – 90 | 2 | 0.21 (0.11) | 17.89 (4.71) | 21.91 (4.69) | |
| 15q12 | 0 – 23 | 2 | 0.2 (0.08) | 18.53 (3.53) | 22.09 (4.14) | |
| 15q14 | 23 – 70 | 2 | 0.18 (0.05) | 21.27 (3.46) | 23.47 (3.25) | |
| 19p13.12 | 15 – 60 | 2 | 0.17 (0.08) | 19.55 (3.68) | 25.26 (4.14) | |
| 19q13.42 | 65 – 80 | 2 | 0.17 (0.10) | 21.09 (3.80) | 24.04 (4.01) |
Allele frequency of allele a. Posterior mean and sd are shown; same format for heterozygote effect and homozygote effect
Joint linkage-segregation analysis
The initial genome scan analysis identified the strongest genome wide evidence for linkage on chr 6q. In the initial genome scan with 500,000 sweeps, the maximum evidence for linkage, BFmax = 18, was obtained at around 109 cM, flanked by markers D6S1671 and D6S434, whether or not APOE was included as a covariate (Figure 2). For the convenience of the reader, the online supplementary table contains both meiotic and sequence-based map positions for these, and other markers of interest reported here. Longer analysis runs with 10,000,000 sweeps showed results that were similar to those obtained from the shorter runs, but with stronger evidence of linkage without APOE adjustment (BFmax = 30) than with APOE adjustment (BFmax = 18) (Figure 3A). Empirical p-values were 0.023, 0.006, and 0.004 for BFmax of 10, 18, and 30, respectively.
Figure 2.
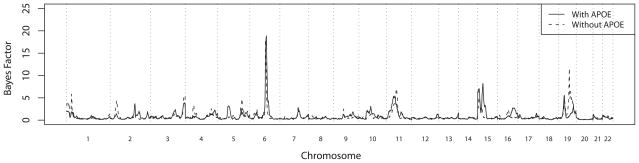
Results of genome scan of age-at-onset with (solid line) and without (dashed line) adjusting for APOE as a covariate, measured with Bayes Factors (see text in Methods). Vertical dotted lines show boundaries between chromosomes.
Figure 3.

Detailed depiction of results for QTL models that have location parameter values anywhere on chromosome 6, based on a long MCMC run. Parameters are as described in Figure 1. Linkage analysis results (panel A) with (solid line) and without (dashed line) adjusting for APOE as a covariate, and contour plots of posterior distribution of QTL model parameter densities from the same analysis runs with (Panel B) and without (Panel C) adjustment for APOE as a covariate. The majority of QTL models with parameter values for QTL location on chromosome 6 fall near the region with the strongest evidence for linkage, and thus describe the QTL models with strongest support in this region.
The QTL models for all of chr 6 were concentrated on model 1, whether or not APOE was included as a covariate (Figure 3B, C). The QTL model for the more localized region where the linkage signal occurred showed a very similar pattern, but was even more concentrated on model 1 (results not shown). After accounting for the genetic variance in AAO contributed by APOE, this QTL explains 45.9% of the total genetic variance, and 40.4% of the total variance that remains. The genetic variance for this QTL is very similar to that of APOE,
The QTL models for additional chromosomes with evidence for linkage were slightly more complicated, as expected given the complex QTL models identified in oligogenic segregation analysis. Not surprisingly, in the absence of APOE adjustment, there was also strong evidence for linkage to chr 19q at 75cM (BFmax = 14; Figure 2), with this evidence similar in strength to that obtained for chromosome 6. This location on chr 19 is consistent with the location of APOE (72 cM). This signal was much weaker (BFmax = 4; Figure 2) in the presence of APOE adjustment, as is expected if the evidence for linkage to this region is derived from the effects of APOE. Other regions with more moderate evidence for linkage were found on chr 11p (61cM) with or without APOE adjustment (BFmax = 8 and 7, respectively), chr 15q (15 cM and 35 cM) with APOE adjustment (BFmax = 7.5 and 9 respectively), and chr 19p (35 cM) with or without APOE adjustment (BFmax = 4.5 and 6, respectively; Figure 2). Long runs with 2,000,000 sweeps confirmed the results obtained from the initial shorter runs (Figure 4), with relative insensitivity of the evidence for linkage to inclusion of APOE as a covariate. These additional linkage signals were not overwhelming, but they consistently appeared in multiple analysis runs and were insensitive to the choice of parameters of prior distributions (not shown).
Figure 4.

Detailed depiction of results of QTL models with location parameters on chromosomes 11, 15 and 19. Parameters are as described in Figure 1. Panels A, D and G depict linkage analysis results and posterior QTL model parameters from the same analysis runs for chromosomes 11, 15, and 19, respectively, with (solid line) and without (dashed line) adjusting for APOE as a covariate. Panels B, E and H depict contour plots of posterior distribution of QTL model parameters densities with adjustment for APOE as a covariate. Panels C, F, and I depict contour plots of posterior distributions of QTL model parameter densities without adjustment for APOE as a covariate. In all cases, the posterior distributions of QTL model parameter densities are based on the QTLs with a location parameter on the depicted chromosome.
We did further analysis for each of these signals. The QTL models for chr 11 and 15 showed a similar pattern: without APOE adjustment, the QTL models consisted of model 2 and the less distinct model 3; when APOE was used as a covariate, the models shifted from the ambiguous model 3 to a clear model 2 (Figure 4B, C, E, F). This corresponds to the stronger evidence of linkage on chr 11 and 15 with APOE adjustment. Results for chr 19 went in the opposite direction: when APOE was not a covariate, the model concentrated on model 2, with a small part in model 3, giving stronger evidence of linkage; when APOE was a covariate, the model shifted to the ambiguous model 3, and the linkage evidence became weaker (Figure 4H, I), as is consistent with APOE as the causal locus in this region.
DISCUSSION
Our genome-wide scan identified strong evidence of linkage of loci affecting AAO to two chromosomes: 6q16.3 and 19q13.42. The strongest evidence for linkage, to chr 6q, overlaps a previously reported region for an AAO locus in samples that included the NIMH AD dataset (Choi et al. 2011; Dickson et al. 2008; Holmans et al. 2005) (Table III), and therefore provides the first independent confirmation in a different sample of an AAO locus in this region. The region on chr 19q13.42 is explained by the well-established APOE locus, and was noted in an earlier analysis of a subset of this sample (Wijsman et al. 2004) as well as in several previous genome scans of AAO in other samples (Choi et al. 2011; Dickson et al. 2008; Holmans et al. 2005; Li et al. 2002), all of which included at least part of the NIMH AD sample. The families used in the current sample provided a dataset that is suitable for identifying loci contributing to AAO because of the large sample size, the high proportion of large families, the ascertainment on a large number of subjects with confirmed autopsies, and the relatively complete age information in both affected subjects and unaffected subjects. The MCMC-based multipoint analysis with an oligogenic trait model was also able to incorporate a multilocus mode-of-inheritance, age censoring, and APOE covariate effects without requiring pedigree simplification, thus making full use of the available data. The NIMH AD sample analyzed previously for AAO had characteristics similar to ours, including a relatively consistent measure of AAO across collection sites. This may have facilitated detection of evidence for linkage to AAO, which is still a novel endpoint compared to use of AD risk in most genome scans of AD. These results suggest that there still may be important lessons to be learned from the study of other AD-related phenotypes and endophenotypes.
Table III.
Linkage analyses on chromosome 6 across studies that used AAO as the trait.
| Study | Sample | Chromosome 6 Linkage signal | ||||
|---|---|---|---|---|---|---|
| Marker | Map Position** | Physical Position# | Evidence | APOE covar. | ||
| {Holmans, 2005 #1478} | NIMH | Not provided | 70 cM | NA | MLS = 1.22 | No |
| Not provided | 70 cM | NA | MLS = 2.44 | Yes | ||
| Not provided | 100 cM | NA | MLS ≈ 1.45## | Yes | ||
| {Dickson, 2008 #3449} | NIMH | D6S1017 | 67 cM | 41785151 | MLS = 2.3 | No |
| D6S1053 | 85 cM | 64648238 | MLS = 2.2 | No | ||
| D6S1021 | 112 cM | 104780614 | MLS = 1.6 | No | ||
| {Choi, 2011 #4668} | NIMH | D6S1017 | 67 cM | 41785151 | BFmax = 42.9 | No |
| NIMH C51* | D6S1021 | 112 cM | 104780614 | BFmax = 4.8 | No | |
| NIMH C50* | D6S1021 | 112 cM | 104780614 | BFmax = 7.8 | Yes | |
| Current | UW | D6S1671 D6S434 |
109–111 cM | 100662132 102542632 |
BFmax = 30 | No |
| Current | UW | D6S1671 D6S434 |
109–111 cM | 100662132 102542632 |
BFmax = 18 | Yes |
Sub-sample: NIMH center 50 (C50), center 51 (C51)
Map positions (cM) are based on the Haldane map function based on on the Rutger’s map{Matise, 2007 #3904}, see methods. The exception is Holmans (2005) study, for which positions are as originally supplied, since no information about the map function used was provided by the authors.
Human Genome Sequence Build 36
“≈”: Values were not reported and were therefore estimated from a figure in the paper.
The main region identified in our sample on chr 6 overlaps a region reported previously for AAO in the independent NIMH AD dataset analyzed alone (Choi et al. 2011; Dickson et al. 2008) or together with other samples (Holmans et al. 2005) (Table III). This region of overlap is not at the position with the strongest previously-reported linkage statistic in the NIMH AD sample, but represents a secondary location at ~110 cM, with evidence for linkage to this position driven by a subset of the sites contributing to the NIMH AD sample (Choi et al. 2011). This locus also differs from that on chromosome 6 reported in a recent GWAS of AD risk (Naj et al. 2010), and other AD risk loci implicated by GWAS with evidence of replication across samples are on other chromosomes (Schellenberg and Montine 2012). Note that although our results show a stronger BF at chr 6q16 than at APOE, this does not imply a stronger effect size for AAO in the general population. This is because the strength of a linkage signal is affected by the trait allele frequency in the sample, with common trait alleles leading to weaker linkage signals (Risch and Merikangas 1996). Our sample has an inflated APOE ε4 allele frequency compared to the general population because of ascertainment of multiplex AD families. This leads to an elevation of the frequency of ε4 homozygotes, which were approximately 8-fold more frequent in this sample than in a sample from the general population. Such homozygosity obscures the information needed to determine the recombinant status of transmitted gametes near APOE, thus reducing evidence for linkage. This effect on linkage information near APOE as a function of sample ascertainment was also noted previously in analysis of the data from the individual centers contributing to the NIMH AD sample (Choi et al. 2011).
Glutamate receptor ionotropic kainate 2 (GRIK2) is an excellent candidate gene in this region on chromosome 6, which otherwise contains relatively few genes. Glutamate receptors serve as excitatory neurotransmitter receptors in the mammalian brain (Lerma 2006; Vincent and Mulle 2009). Aberrant glutamaterigic activity has been proposed to explain some pathogenic aspects of the AD disease process (Lipton 2007), and is supported by demonstration of altered glutamate transport in AD cases vs. controls (Jacob et al. 2007; Masliah et al. 1996; Woltjer et al. 2010). GRIK2 has already been implicated in several studies as a gene affecting AAO in Huntington’s disease (Li et al. 2006; MacDonald et al. 1999; Rubinsztein et al. 1997), which represents another neurodegenerative disease. Also, transgenic mice carrying the N141I PSEN2 EOAD mutation have been reported to have altered resistance to excitotoxic treatment with kainite compared to their wild-type littermates (Schulte et al. 2009), further supporting a possible role in modulating AD risk and/or AAO.
Adjustment for APOE affected the evidence for linkage to chr 6 in this sample, suggesting possible interaction with APOE. Similar results were obtained previously, also in the independent NIMH AD sample (Choi et al. 2011; Holmans et al. 2005), supporting such interaction. However, the oligogenic model used here for analysis of AAO assumes additive effects across loci, and cannot be used for a test of an interactive (epistatic) model. With an additive model, in the absence of a true interaction, adjustment for APOE serves only as a precision variable, and would be expected to have little effect on evidence for linkage. In the presence of an unmodeled interaction, however, the estimated effect of the chr 6 QTL could be inflated, possibly increasing evidence for linkage. Further work is needed to investigate the possibility of mechanistic interaction that may be driving evidence for a statistical interaction.
The moderate evidence for linkage on several additional chromosomes is also consistent with results reported in other studies. The region implicated on chr 11p13 overlaps the signal observed in an early-onset sample screened for AAO modifier loci (Marchani et al. 2010). This region contains brain-derived neurotrophic factor (BDNF), which has been reported to be associated with LOAD (Kunugi et al. 2001; Riemenschneider et al. 2002; Ventriglia et al. 2002) although current meta analysis results are less convincing (Bertram et al. 2007). This region also contains the glutamate transporter gene GLT-1, which is the predominant glutamate transporter in the cerebral cortex and hippocampus (Lehre and Danbolt 1998). The signal on chr 19p13.12 overlaps a strong signal observed in analysis of this chromosome using an earlier subset version of the University of Washington data set (Wijsman et al. 2004) and has also been reported in a study of dementia in an Amish sample (Hahs et al. 2006). There are a number of strong candidate loci in this region, some of which have been implicated in association or mechanistic studies, such as peptidylprolyl cis/trans isomerase NIMA-interacting 1 (PIN1) (Lu et al. 1999; Pastorino et al. 2006; Segat et al. 2007) and the low density lipoprotein receptor (LDLR) (Carter 2007; Gopalraj et al. 2005). The region implicated in the current sample on chr 15q also overlaps a previously reported signal from a combined analysis of the NIMH AD sample combined with additional families (Holmans et al. 2005), although evidence for linkage in both cases is relatively modest.
The results obtained here are typical of genome scan studies in both Alzheimer’s disease, and in complex traits in general. It should come as no surprise that only a subset of loci implicated here overlap with those implicated in other samples. The presence of partial, but incomplete overlap of such regions is a typical outcome of any study of a complex trait, as has been well-understood for almost two decades (Suarez et al. 1994). The fact that several regions replicate across subsets of the small number of available independent samples is highly encouraging, including the locus identified here on chromosome 6. Identification of the causal variants responsible for this signal, and other, promising linkage signals therefore awaits future application of newer sequencing technologies. A challenge will be to identify families that contribute to the linkage signals as prime candidates for sequencing. The method used here is not suitable for single family analysis given the available family sizes, the need for large data sets when using this method, and the current implementation of the software. This is a challenge for not only the current study, but many other studies of complex traits, and awaits development of appropriate statistical methods along with development of the software needed for use in practical settings with complex traits.
Supplementary Material
Acknowledgments
We thank the patients and their families, whose help and participation made this work possible. We thank Leslie Leong and Hiep Nguyen for technical assistance; Adele Sadovnick, Leonard Heston, and Haydeh Payami for family material; and David Cook for discussions. Supported by National Institutes of Health (NIH) grants P50 AG005136, T32 AG000258, and K99 AG040184; Veterans Affairs Research Funds; and The Metropolitan Life Foundation. Some samples used in this study were obtained from the National Cell Repository for Alzheimer’s Disease (NCRAD), supported by NIH U24 AG021886.
Footnotes
The authors have no conflicts of interest to declare.
References
- Alzheimer’s Association. 2010 Alzheimer’s Disease Facts and Figures. Alzheimer’s and Dementia; 2010. p. 6. [DOI] [PubMed] [Google Scholar]
- Bennett C, Crawford F, Osborne A, Diaz P, Hoyne J, Lopez R, Roques P, Duara R, Rossor M, Mullan M. Evidence That the Apoe Locus Influences Rate of Disease Progression in Late-Onset Familial Alzheimer’s Disease but Is Not Causative. American Journal of Medical Genetics. 1995;60(1):1–6. doi: 10.1002/ajmg.1320600102. [DOI] [PubMed] [Google Scholar]
- Bergem ALM, Engedal K, Kringlen E. The role of heredity in late-onset Alzheimer disease and vascular dementia- A twin study. Archives of General Psychiatry. 1997;54(3):264–270. doi: 10.1001/archpsyc.1997.01830150090013. [DOI] [PubMed] [Google Scholar]
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nature Genetics. 2007;39(1):17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nature Reviews Neuroscience. 2008;9(10):768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- Butler AW, Ng MYM, Hamshere ML, Forabosco P, Wroe R, Al-Chalabi A, Lewis CM, Powell JF. Meta-analysis of linkage studies for Alzheimer’s disease-A web resource. Neurobiology of Aging. 2009;30(7):1037–1047. doi: 10.1016/j.neurobiolaging.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Carter CJ. Convergence of genes implicated in Alzheimer’s disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochemistry International. 2007;50(1):12–38. doi: 10.1016/j.neuint.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Cheung CYK, Thompson EA, Wijsman EM. In Silico Genotype Imputation on Large Pedigrees. Genetic Epidemiology. 2010;34:919. [Google Scholar]
- Choi Y, Marchani EE, Bird TD, Steinbart EJ, Blacker D, Wijsman EM. Genome Scan of Age-at-Onset in the NIMH Alzheimer Disease Sample Uncovers Multiple Loci, Along With Evidence of Both Genetic and Sample Heterogeneity. American Journal of Medical Genetics Part B-Neuropsychiatric Genetics. 2011;156B(7):785–798. doi: 10.1002/ajmg.b.31220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consensus Report. Consensus Report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer’s Disease. Neurobiology of Aging. 1998;19(2):109–116. [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Daw EW, Heath SC, Wijsman EM. Multipoint oligogenic analysis of age-at-onset data with applications to Alzheimer’s disease pedigrees. American Journal of Human Genetics. 1999;64(3):839–851. doi: 10.1086/302276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw EW, Payami H, Nemens EJ, Nochlin D, Bird TD, Schellenberg GD, Wijsman EM. The number of trait loci in late-onset Alzheimer disease. American Journal of Human Genetics. 2000;66:196–204. doi: 10.1086/302710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MR, Li J, Wiener HW, Perry RT, Blacker D, Bassett SS, Go RCP. A genomic scan for age at onset of Alzheimer’s disease in 437 families from the NIMH genetic initiative. American Journal of Medical Genetics Part B-Neuropsychiatric Genetics. 2008;147B(6):784–792. doi: 10.1002/ajmg.b.30689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Human Heredity. 1971;21:523–542. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- Epstein MP, Duren WL, Boehnke M. Improved inference of relationship for pairs of individuals. American Journal of Human Genetics. 2000;67(5):1219–31. doi: 10.1016/s0002-9297(07)62952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest WF, Feingold E. Composite statistics for QTL mapping with moderately discordant sibling pairs. American Journal of Human Genetics. 2000;66:1642–1660. doi: 10.1086/302897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, Pedersen NL, Berg S, Johansson B, Johansson K, Mortimer JA, Posner SF, Viitanen M, Winblad B, Ahlbom A. Heritability for Alzheimer’s disease: The study of dementia in Swedish twins. Journals of Gerontology Series a-Biological Sciences and Medical Sciences. 1997;52(2):M117–M125. doi: 10.1093/gerona/52a.2.m117. [DOI] [PubMed] [Google Scholar]
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Archives of General Psychiatry. 2006;63(2):168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Gopalraj RK, Zhu H, Kelly JF, Mendiondo M, Pulliam JF, Bennett DA, Estus S. Genetic association of low density lipoprotein receptor and Alzheimer’s disease. Neurobiology of Aging. 2005;26(1):1–7. doi: 10.1016/j.neurobiolaging.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Green PJ. Reversible jump Markov chain Monte Carlo computation and Bayesian model determination. Biometrika. 1995;82:711–732. [Google Scholar]
- Hagnell O, Ojesjo L, Rorsman B. Incidence of Dementia in the Lundby-Study. Neuroepidemiology. 1992;11:61–66. doi: 10.1159/000110981. [DOI] [PubMed] [Google Scholar]
- Hahs DW, McCauley JL, Crunk AE, McFarland LL, Gaskell PC, Jiang L, Slifer SH, Vance JM, Scott WK, Welsh-Bohmer KA, Johnson SR, Jackson CE, Pericak-Vance MA, Haines JL. A genome-wide linkage analysis of dementia in the Amish. American Journal of Medical Genetics Part B-Neuropsychiatric Genetics. 2006;141B(2):160–166. doi: 10.1002/ajmg.b.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. The combination of linkage values, and the calculation of distance between the loci of linked factors. Journal of Genetics. 1919;8:299–309. [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics. 2009;62:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath SC. Markov Chain Monte Carlo Segregation and linkage analysis for oligogenic models. American Journal of Human Genetics. 1997;61:748–760. doi: 10.1086/515506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath SC, Snow GL, Thompson EA, Tseng C, Wijsman EM. MCMC Segregation and linkage analysis. Genetic Epidemiology. 1997;14:1011–1016. doi: 10.1002/(SICI)1098-2272(1997)14:6<1011::AID-GEPI75>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population - Prevalence estimates using the 2000 census. Archives of Neurology. 2003;60(8):1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. Journal of Lipid Research. 1990;31(3):545–8. [PubMed] [Google Scholar]
- Holmans P, Hamshere M, Hollingworth P, Rice F, Tunstall N, Jones S, Moore P, DeVrieze FW, Myers A, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Williams N, Norton N, Abraham R, Kehoe P, Williams H, Rudrasingham V, O’Donovan M, Jones L, Hardy J, Goate A, Lovestone S, Owen M, Williams J. Genome screen for loci influencing age at onset and rate of decline in late onset Alzheimer’s disease. American Journal of Medical Genetics Part B-Neuropsychiatric Genetics. 2005;135B(1):24–32. doi: 10.1002/ajmg.b.30114. [DOI] [PubMed] [Google Scholar]
- Igo RP, Wijsman EM. Empirical significance values for linkage analysis: Trait simulation using posterior model distributions from MCMC oligogenic segregation analysis. Genetic Epidemiology. 2008;32(2):119–131. doi: 10.1002/gepi.20267. [DOI] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grunblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. Journal of Alzheimers Disease. 2007;11(1):97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- Kamboh MI, Barmada MM, Demirci FY, Minster RL, Carrasquillo MM, Pankratz VS, Younkin SG, Saykin AJ, Sweet RA, Feingold E, Dekosky ST, Lopez OL The Alzheimer’s Disease Neuroimaging Initiative. Genome-wide association analysis of age-at-onset in Alzheimer’s disease. Molecular Psychiatry. 2011 doi: 10.1038/mp.2011.135. ((in press)) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RE, Raftery AE. Bayes Factors. Journal of the American Statistical Association. 1995;90(430):773–795. [Google Scholar]
- Kauwe JSK, Jacquart S, Chakraverty S, Wang J, Mayo K, Fagan AM, Holtzman DM, Morris JC, Goate AM. Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Annals of Neurology. 2007;61(5):446–453. doi: 10.1002/ana.21099. [DOI] [PubMed] [Google Scholar]
- Kleensang A, Franke D, Alcais A, Abel L, Muller-Myhsok B, Ziegler A. An Extensive Comparison of Quantitative Trait Loci Mapping Methods. Human Heredity. 2010;69(3):202–211. doi: 10.1159/000289596. [DOI] [PubMed] [Google Scholar]
- Kunugi H, Ueki A, Otsuka M, Isse K, Hirasawa H, Kato N, Nabika T, Kobayashi S, Nanko S. A novel polymorphism of the brain-derived neurotrophic factor (BDNF) gene associated with late-onset Alzheimer’s disease. Molecular Psychiatry. 2001;6(1):83–86. doi: 10.1038/sj.mp.4000792. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lander ES, Green PJ. Construction of multilocus genetic maps in humans. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:2363–2367. doi: 10.1073/pnas.84.8.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenschlager NT, Cupples LA, Rao VS, Auerbach SA, Becker R, Burke J, Chui H, Duara R, Foley EJ, Glatt SL, Green RC, Jones R, Karlinsky H, Kukull WA, Kurz A, Larson EB, Martelli K, Sadovnick AD, Volicer L, Waring SC, Growdon JH, Farrer LA. Risk of dementia among relatives of Alzheimer’s disease patients in the MIRAGE study: What is in store for the oldest old? Neurology. 1996;46(3):641–650. doi: 10.1212/wnl.46.3.641. [DOI] [PubMed] [Google Scholar]
- Lee JH, Barral S, Cheng R, Chacon I, Santana V, Williamson J, Lantigua R, Medrano M, Jimenez-Velazquez IZ, Stern Y, Tycko B, Rogaeva E, Wakutani Y, Kawarai T, St George-Hyslop P, Mayeux R. Age-at-onset linkage analysis in Caribbean Hispanics with familial late-onset Alzheimer’s disease. Neurogenetics. 2008;9(1):51–60. doi: 10.1007/s10048-007-0103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult rat brain. Journal of Neuroscience. 1998;18(21):8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J. Kainate receptor physiology. Current Opinion in Pharmacology. 2006;6(1):89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995;269(5226):970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- Li HZ. An additive genetic gamma frailty model for linkage analysis of diseases with variable age of onset using nuclear families. Lifetime Data Analysis. 2002;8(4):315–334. doi: 10.1023/a:1020500720254. [DOI] [PubMed] [Google Scholar]
- Li JL, Hayden MR, Warby SC, Durr A, Morrison PJ, Nance M, Ross CA, Margolis RL, Rosenblatt A, Squitieri F, Frati L, Gomez-Tortosa E, Garcia CA, Suchowersky O, Klimek ML, Trent RJA, McCusker E, Novelletto A, Frontali M, Paulsen JS, Jones R, Ashizawa T, Lazzarini A, Wheeler VC, Prakash R, Xu G, Djousse L, Mysore JS, Gillis T, Hakky M, Cupples LA, Saint-Hilaire MH, Cha JHJ, Hersch SM, Penney JB, Harrison MB, Perlman SL, Zanko A, Abramson RK, Lechich AJ, Duckett A, Marder K, Conneally PM, Gusella JF, MacDonald ME, Myers RH. Genome-wide significance for a modifier of age at neurological onset in Huntington’s Disease at 6q23–24: the HD MAPS study. BMC Medical Genetics. 2006:7. doi: 10.1186/1471-2350-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YJ, Scott WK, Hedges DJ, Zhang FY, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen AA, Goetz CG, Mastaglia F, Stajich JM, Gibson RA, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer KA, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA. Age at onset in two common neurodegenerative diseases is genetically controlled. American Journal of Human Genetics. 2002;70(4):985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. Pathologically activated therapeutics for neuroprotection. Nature Reviews Neuroscience. 2007;8(11):803. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- Logue MW, Schu M, Vardarajan BN, Buros J, Green RC, Go RCP, Griffith P, Obisesan TO, Shatz R, Borenstein A, Cupples LA, Lunetta KL, Fallin MD, Baldwin CT, Farrer LA, Grp MS. A Comprehensive Genetic Association Study of Alzheimer Disease in African Americans. Archives of Neurology. 2011;68(12):1569–1579. doi: 10.1001/archneurol.2011.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Wulf G, Zhou X, Davies P, Lu K. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- Ma JZ, Amos CI, Daw EW. Ascertainment correction for Markov chain Monte Carlo segregation and linkage analysis of a quantitative trait. Genetic Epidemiology. 2007;31(6):594–604. doi: 10.1002/gepi.20231. [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Vonsattel MP, Shrinidhi J, Couropmitree NN, Cupples LA, Bird ED, Gusella JF, Myers RH. Evidence for the GluR6 gene associated with younger onset age of Huntington’s disease. Neurology. 1999;53(6):1330–1332. doi: 10.1212/wnl.53.6.1330. [DOI] [PubMed] [Google Scholar]
- Marchani EE, Bird TD, Steinbart EJ, Rosenthal E, Yu CE, Schellenberg GD, Wijsman EM. Evidence for three loci modifying age-at-onset of Alzheimer’s disease in early-onset PSEN2 families. American Journal of Medical Genetics Part B-Neuropsychiatric Genetics. 2010;153B:1031–1041. doi: 10.1002/ajmg.b.31072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Annals of Neurology. 1996;40(5):759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- Matise TC, Chen F, Chen WW, De la Vega FM, Hansen M, He CS, Hyland FCL, Kennedy GC, Kong XY, Murray SS, Ziegle JS, Stewart WCL, Buyske S. A second-generation combined linkage-physical map of the human genome. Genome Research. 2007;17(12):1783–1786. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux R. Epidemiology of neurodegeneration. Annual Review of Neuroscience. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Sano M, Chen J, Tatemichi T, Stern Y. Risk of Dementia in 1st-Degree Relatives of Patients with Alzheimers-Disease and Related Disorders. Archives of Neurology. 1991;48(3):269–273. doi: 10.1001/archneur.1991.00530150037014. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Miech RA, Breitner JCS, Zandi PP, Khachaturian AS, Anthony JC, Mayer L. Incidence of AD may decline in the early 90s for men, later for women - The Cache County study. Neurology. 2002;58(2):209–218. doi: 10.1212/wnl.58.2.209. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, Van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Naj AC, Beecham GW, Martin ER, Gallins PJ, Powell EH, Konidari I, Whitehead PL, Cai GQ, Haroutunian V, Scott WK, Vance JM, Slifer MA, Gwirtsman HE, Gilbert JR, Haines JL, Buxbaum JD, Pericak-Vance MA. Dementia Revealed: Novel Chromosome 6 Locus for Late-Onset Alzheimer Disease Provides Genetic Evidence for Folate-Pathway Abnormalities. Plos Genetics. 2010;6(9) doi: 10.1371/journal.pgen.1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JSK, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature Genetics. 2011;43(5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz VS, de Andrade M, Therneau TM. Random-effects Cox proportional hazards model: general variance components methods for time-to-event data. Genetic Epidemiology. 2005;28(2):97–109. doi: 10.1002/gepi.20043. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li XJ, Xia WM, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440(7083):528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Riemenschneider M, Schwarz S, Wagenpfeil S, Diehl J, Muller U, Forstl H, Kurz A. A polymorphism of the brain-derived neurotrophic factor (BDNF) is associated with Alzheimer’s disease in patients lacking the Apolipoprotein E epsilon 4 allele. Molecular Psychiatry. 2002;7(7):782–785. doi: 10.1038/sj.mp.4001073. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas KR. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Rocca WA, Hofman A, Brayne C, Breteler MMB, Clarke M, Copeland JRM, Dartigues JF, Engedal K, Hagnell O, Heeren TJ, Jonker C, Lindesay J, Lobo A, Mann AH, Molsa PK, Morgan K, Oconnor DW, Droux AD, Sulkava R, Kay DWK, Amaducci L. Frequency and Distribution of Alzheimers-Disease in Europe - a Collaborative Study of 1980–1990 Prevalence Findings. Annals of Neurology. 1991;30(3):381–390. doi: 10.1002/ana.410300310. [DOI] [PubMed] [Google Scholar]
- Rosenthal EA, Wijsman EM. Joint linkage and segregation analysis under multiallelic trait inheritance: Simplifying interpretations for complex traits. Genetic Epidemiology. 2010;34:344–353. doi: 10.1002/gepi.20490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin DB. Multiple imputation after 18+ years. Journal of the American Statistical Association. 1996;91(434):473–489. [Google Scholar]
- Rubinsztein DC, Leggo J, Chiano M, Dodge A, Norbury G, Rosser E, Craufurd D. Genotypes at the GluR6 kainate receptor locus are associated with variation in the age of onset of Huntington disease. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):3872–3876. doi: 10.1073/pnas.94.8.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer JL. Multiple imputation: a primer. Statistical Methods in Medical Research. 1999;8(1):3–15. doi: 10.1177/096228029900800102. [DOI] [PubMed] [Google Scholar]
- Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathologica. 2012 doi: 10.1007/s00401-012-0996-2. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte EC, Slawik H, Schule R, Gunther T, Hull M. Alterations in excitotoxicity and prostaglandin metabolism in a transgenic mouse model of Alzheimer’s disease. Neurochemistry International. 2009;55(7):689–696. doi: 10.1016/j.neuint.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Scurrah KJ, Palmer LJ, Burton PR. Variance components analysis for pedigree-based censored survival data using generalized linear mixed models (GLMMs) and Gibbs sampling in BUGS. Genetic Epidemiology. 2000;19(2):127–148. doi: 10.1002/1098-2272(200009)19:2<127::AID-GEPI2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, Clerici A, Arosio B, Crovella S. PIN1 promoter polymorphisms are associated with Alzheimer’s disease. Neurobiology of Aging. 2007;28(1):69–74. doi: 10.1016/j.neurobiolaging.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EMC, Ramirez-Lorca R, Debette S, Longstreth WT, Janssens A, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du YC, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MMB. Genome-wide Analysis of Genetic Loci Associated With Alzheimer Disease. Journal of the American Medical Association. 2010;303(18):1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Slooter AJ, Cruts M, Kalmijn S, Hofman A, Breteler MM, Van Broeckhoven C, van Duijn CM. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Archives of Neurology. 1998;55(7):964–968. doi: 10.1001/archneur.55.7.964. [DOI] [PubMed] [Google Scholar]
- Suarez BK, Hampe CL, van Eerdewegh P. Problems of replicating linkage claims in psychiatry. In: Gerson ES, Cloninger CR, editors. Genetic approaches to mental disorders. Washington, DC: American Psychiatric Press; 1994. pp. 23–46. [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell. 2005;120(4):545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Thompson E, Basu S. Genome sharing in large pedigrees: Multiple imputation of ibd for linkage detection. Human Heredity. 2003;56(1–3):119–125. doi: 10.1159/000073739. [DOI] [PubMed] [Google Scholar]
- Tunstall N, Owen MJ, Williams J, Rice F, Carty S, Lillystone S, Fraser L, Kehoe P, Neill D, Rudrasingham V, Sham P, Lovestone S. Familial influence on variation in age of onset and behavioural phenotype in Alzheimer’s disease. British Journal of Psychiatry. 2000;176:156–159. doi: 10.1192/bjp.176.2.156. [DOI] [PubMed] [Google Scholar]
- Ventriglia M, Chiavetto LB, Benussi L, Binetti G, Zanetti O, Riva MA, Gennarelli M. Association between the BDNF 196 A/G polymorphism and sporadic Alzheimer’s disease. Molecular Psychiatry. 2002;7(2):136–137. doi: 10.1038/sj.mp.4000952. [DOI] [PubMed] [Google Scholar]
- Vincent P, Mulle C. Kainate Receptors in Epilepsy and Excitotoxicity. Neuroscience. 2009;158(1):309–323. doi: 10.1016/j.neuroscience.2008.02.066. [DOI] [PubMed] [Google Scholar]
- Wijsman EM. Joint linkage and segregation analysis using Markov chain Monte Carlo methods. In: Camp N, Cox A, editors. Quantitative trait loci: methods and protocols. Totowa: Humana Press; 2002. pp. 139–161. [DOI] [PubMed] [Google Scholar]
- Wijsman EM, Daw EW, Yu CE, Payami H, Steinbart EJ, Nochlin D, Conlon EM, Bird TD, Schellenberg GD. Evidence for a novel late-onset Alzheimer’s disease locus on chromosome 19p13.2. American Journal of Human Genetics. 2004;75:398–409. doi: 10.1086/423393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsman EM, Daw EW, Yu X, Steinbart EJ, Nochlin D, Bird TD, Schellenberg G. APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. American Journal of Medical Genetics Part B-Neuropsychiatric Genetics. 2005;132B:14–20. doi: 10.1002/ajmg.b.30087. [DOI] [PubMed] [Google Scholar]
- Wijsman EM, Pankratz ND, Choi Y, Rothstein JH, Faber KM, Cheng R, Lee JH, Bird TD, Bennett DA, Diaz-Arrastia R, Goate AM, Farlow MR, Ghetti B, Sweet RA, Foroud TM, Mayeux R. Genome wide association of familial late onset Alzheimer’s disease replicates BIN1 and CLU, and nominates CUGBP2 in interaction with APOE. PLoS Genetics. 2011;7(2):e1001308. doi: 10.1371/journal.pgen.1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsman EM, Yu D. Joint Oligogenic Segregation and Linkage Analysis using Bayesian Markov chain Monte Carlo Methods. Molecular Biotechnology. 2004;28:205–226. doi: 10.1385/MB:28:3:205. [DOI] [PubMed] [Google Scholar]
- Williams JT, Duggirala R, Blangero J. Statistical properties of a variance components method for quantitative trait linkage analysis in nuclear families and extended pedigrees. Genetic Epidemiology. 1997;14(6):1065–1070. doi: 10.1002/(SICI)1098-2272(1997)14:6<1065::AID-GEPI84>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Woltjer RL, Duerson K, Fullmer JM, Mookherjee P, Ryan AM, Montine TJ, Kaye JA, Quinn JF, Silbert L, Erten-Lyons D, Leverenz JB, Bird TD, Pow DV, Tanaka K, Watson GS, Cook DG. Aberrant Detergent-Insoluble Excitatory Amino Acid Transporter 2 Accumulates in Alzheimer Disease. Journal of Neuropathology and Experimental Neurology. 2010;69(7):667–676. doi: 10.1097/NEN.0b013e3181e24adb. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.