

Abstract
How to identify a set of genes that are relevant to a key biological process is an important issue in current molecular biology. In this paper, we propose a novel method to discover differentially expressed genes based on robust principal component analysis (RPCA). In our method, we treat the differentially and non-differentially expressed genes as perturbation signals S and low-rank matrix A, respectively. Perturbation signals S can be recovered from the gene expression data by using RPCA. To discover the differentially expressed genes associated with special biological progresses or functions, the scheme is given as follows. Firstly, the matrix D of expression data is decomposed into two adding matrices A and S by using RPCA. Secondly, the differentially expressed genes are identified based on matrix S. Finally, the differentially expressed genes are evaluated by the tools based on Gene Ontology. A larger number of experiments on hypothetical and real gene expression data are also provided and the experimental results show that our method is efficient and effective.
Background
One of the challenges in current molecular biology is how to find the genes associated with key cellular processes. Up to date, using microarray technology, these genes associated with a special biological process have been detected more comprehensively than ever before.
DNA microarray technology has enabled high-throughput genome-wide measurements of gene transcript levels [1,2], which is promising in providing insight into biological processes involved in gene regulation [3]. It allows researchers to measure the expression levels of thousands of genes simultaneously in a microarray experiment. Gene expression data usually contain thousands of genes (sometimes more than 10,000 genes), and yet only a small number of samples (usually less than 100 samples). Gene expression is believed to be regulated by a small number of factors (compared to the total number of genes), which act together to maintain the steady-state abundance of specific mRNAs. Some of these factors could represent the binding of one (or more) transcription factor(s) (TFs) to the promoter region(s) of the gene [4]. So, it can be assumed that the genes associated with a biological process are influenced only by a small subset of TFs [5]. Although the expression levels of thousands of genes are measured simultaneously, only a small number of genes are relevant to a special biological process. Therefore, it is important how to find a set of genes that are relevant to a biological process.
Various methods have been proposed for identifying differentially expressed genes from gene expression data. These methods can be roughly divided into two categories: univariate feature selection (UFS) and multivariate feature selection (MFS). The commonest scheme of UFS is utilized as follows. First, a score for each gene is independently calculated. Then the genes with high scores were selected [6]. The main virtues of UFS are simple, interpretable and fast. However, UFS has some drawbacks. For example, if each gene is independently selected from gene expression data, a large part of the mutual information contained in the data will be lost.
To overcome the drawbacks of UFS, the methods of MFS use all the features simultaneously to select the genes. So far, many mathematical methods for MFS, such as principal component analysis (PCA), independent component analysis (ICA), nonnegative matrix factorization (NMF), lasso logistic regression (LLR) and penalized matrix decomposition (PMD), have been devised to analyze gene expression data. For example, Lee et al. applied PCA to analyze gene expression data [7]. Liu et al. proposed a method of weighting principal components by singular values to select characteristic genes [8]. Probabilistic PCA was used to analyze gene expression data by Nyamundanda et al. [9]. Huang et al. used ICA to analyze gene expression data [10]. NMF was used to select the gene by Zheng et al. [11]. Liu et al. used LLR to select characteristic gene using gene expression data [12]. In [13], Witten et al. proposed penalized matrix decomposition (PMD), which was used to extract plant core genes by Liu et al. [14]. However, the brittleness of these methods with respect to grossly corrupted observations often puts its validity in jeopardy.
Recently, a new method for matrix recovery, namely robust PCA, has been introduced in the field of signal processing [15]. The problem of matrix recovery can be described as follows, assume that all the data points are stacked as column vectors of a matrix , and the matrix (approximately) have low rank:
| (1) |
where has low-rank and is a small perturbation matrix. The robust PCA proposed by Candes et al. can recover a low-rank matrix from highly corrupted measurements [15]. Here, the entries in can have arbitrary large magnitude, and their support is assumed to be sparse but unknown.
Although the method has been successfully applied to model background from surveillance video and to remove shadows from face images [15], it's validity for gene expression data analysis is still need to be studied. The gene expression data all lie near some low-dimensional subspace [16], so it is natural to treat these genes data of non-differential expression as approximately low rank. As mentioned above, only a small number of genes are relevant to a biological process, so these genes with differential expression can be treated as sparse perturbation signals.
In this paper, based on robust PCA, a novel method is proposed for identifying differentially expressed genes. The differentially and non-differentially expressed genes are treated as perturbation signals and low-rank matrix . Firstly, the matrix of expression data is decomposed into two adding matrices and by using RPCA. Secondly, the differentially expressed genes are discovered according to the matrix . Finally, the differentially expressed genes are evaluated by the tools based on Gene Ontology. The main contributions of our work are as follows: firstly, it proposes, for the first time, the idea and method based on RPCA for discovery of differentially expressed genes; secondly, it provides a larger number of experiments of gene selection.
Methods
The definition of Robust PCA (RPCA)
This subsection simply introduces robust PCA (RPCA) proposed by Candes et al. [15]. Let denote the nuclear norm of the matrix , that is, the sum of its singular values, and let denote the -norm of . Supposing that denotes the observation matrix given by Eq.(1), RPCA solves the following optimization problem:
| (2) |
where is a positive regulation parameter. Due to the ability to exactly recover underlying low-rank structure in the data, even in the presence of large errors or outliers, this optimization is referred to as Robust Principal Component Analysis (RPCA).
For the RPCA problem Eq.(2), a Lagrange multiplier Y is introduced to remove the equality constraint. According to [17], the augmented Lagrange multiplier method on the Lagrangian function can be applied:
| (3) |
where is a positive scalar and denotes the Frobenius norm. Lin et al. gave a method for solving the RPCA problem, which is referred to as the inexact ALM (IALM) method [17]. The details of this algorithm can be seen in [17].
The RPCA model of gene expression data
Considering the matrix of gene expression data with size , each row of represents the transcriptional responses of a gene in all the samples, and each column of represents the expression levels of all the genes in one sample. Without loss of generality, , so it is a classical small-sample-size problem.
Our goal of using RPCA to model the microarray data is to identify these significant genes. As mentioned in Introduction, it is reasonable to view the significant genes as sparse signals, so the differential ones are viewed as the sparse perturbation signals and the non-differential ones as the low-rank matrix . Consequently, the genes of differential expression can be identified according to the perturbation signals . The RPCA model of microarray data is shown in Figure 1. The white and yellow blocks denote zero and near-zero in Figure 1. Red and blue blocks denote the perturbation signals. As shown in Figure 1, the matrix of differentially expressed genes (red or blue block) can be recovered from the matrix of gene expression data.
Figure 1.
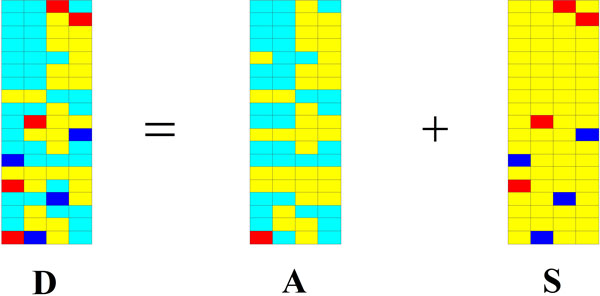
The RPCA model of microarray data. The white and yellow blocks denote zero and near-zero in this figure. Red and blue blocks denote the perturbation signals.
Suppose the matrix decomposition has been done by using RPCA. By choosing the appropriate parameter , the sparse perturbation matrix can be obtained, i.e., most of entries in are zero or near-zero (as white and yellow blocks shown in Figure 1). The genes corresponding to non-zero entries can be considered as ones of differential expression.
Identification of differentially expressed genes
After observation matrix has been decomposed by using RPCA, sparse perturbation matrix can be obtained. Therefore the differentially expressed genes can be identified according to sparse matrix .
Denote the perturbation vector associated with -th sample as:
| (4) |
Then the sparse matrix can be expressed as follows:
| (5) |
So the sparse matrix can be denoted as:
| (6) |
The differentially expressed genes can be classified into two categories: up-and down-regulated ones [18], which are reflected by the positive and negative entries in the sparse matrix . Here, to discover the differentially expressed genes, only the absolute value of entries in need to be considered. Then the following two steps are executed: firstly, the absolute values of entries in the sparse matrix are find out; secondly, to get the evaluating vector , the matrix is summed by rows. Mathematically, it can be expressed as follows:
| (7) |
Consequently, to obtain the new evaluating vector , which is sorted in descending order. Without loss of generality, suppose that the first entries in are non-zero, that is,
| (8) |
Generally, the larger the element in is, the more differential the gene is. So, the genes associated with only the first () entries in are picked out as differentially expressed ones.
Results and discussion
This section gives the experimental results. Firstly, in the first subsection, hypothetical data are exploited to clarify how to set the parameter. Secondly, in the second subsection, our method is compared with the following methods on the real gene expression data of plants responding to abiotic stresses: (a) PMD method using the left singular vectors to identify the differentially expressed genes (proposed by Witten et al. [13]); (b) SPCA method using all the PCs of SPCA (proposed by Journée et al. [19]) to identify the differentially expressed genes. Finally, in the third subsection, the three methods are compared on the real gene expression data of colon tumor.
Experimental results on hypothetical data
Matrices randomly generated will be used for the simulation experiments. The true solution is denoted by the ordered pairs , which are generated by using the method in [17]. The rank-r matrix is generated as , where and are independent and matrices, respectively. Elements of and are i.i.d. Gaussian random variables with zero mean and unit variance. is a sparse matrix whose support is chosen uniformly at random, and whose non-zero entries are i.i.d. uniformly in the space . denotes the sparse degree of matrix , which is defined as the number of non-zero entries divided by the number of all the entries. The matrix is the input data to the RPCA. To evaluate the identification performance of RPCA, denotes the recognition accuracy of matrix , which is defined as follows.
| (9) |
where correct identified entries mean that the identified entries in approximately equal to the ones in .
In [17,20], a fixed regulation parameter is used, where . In order to clarify how to set , the following two different cases are considered: first, ; second, , the small-size-sample problem.
Results while m=n
In this experiment, let , , . Table 1 lists the recognition results with different . As Table 1 listed, when , the recognition accuracy can be achieved above 90%. When , the matrix can be completely identified, i.e. .
Table 1.
The recognition accuracy with different
| n | 500 | 1000 | 2000 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rank/n | 0.05 | 0.05 | 0.10 | 0.10 | 0.05 | 0.05 | 0.10 | 0.10 | 0.05 | 0.05 | 0.10 | 0.10 |
| 0.05 | 0.10 | 0.05 | 0.10 | 0.05 | 0.10 | 0.05 | 0.10 | 0.05 | 0.10 | 0.05 | 0.10 | |
| 0.1 | 1.00 | 0.30 | 0.96 | 0.02 | 1.00 | 0.64 | 1.00 | 0.07 | 1.00 | 0.71 | 1.00 | 0.08 |
| 0.2 | 1.00 | 1.00 | 1.00 | 0.92 | 1.00 | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.3 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.4 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.5 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.6 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.7 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.8 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.9 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 1.0 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
Results while m>n
In this experiment, let ,, and increase from 10 to 100 with an interval 10. Table 2, 3, 4, 5 list the results. As tables 2 and 3 listed with , when , the recognition accuracy can be achieved above 90%. As tables 4 and 5 listed with , when , the recognition accuracy can be achieved above 90%. In words, to achieve the recognition accuracy above 90%, must be equal to or larger than three times of (). As tables 2, 3, 4, 5 listed, by rows, the larger the number of column is, the higher the recognition accuracy can be achieved.
Table 2.
The recognition accuracy with and
| n | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 | |
| 0.1 | 1.00 | 0.30 | 0.96 | 0.02 | 1.00 | 0.64 | 1.00 | 0.07 | 1.00 | 0.71 |
| 0.2 | 1.00 | 1.00 | 1.00 | 0.92 | 1.00 | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 |
| 0.3 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.4 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.5 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.6 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.7 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.8 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.9 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 1.0 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
Table 3.
The recognition accuracy with and
| n | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 | |
| 0.1 | 0.01 | 0.02 | 0.07 | 0.15 | 0.24 | 0.36 | 0.43 | 0.51 | 0.59 | 0.66 |
| 0.2 | 0.24 | 0.84 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.3 | 0.50 | 0.95 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.4 | 0.61 | 0.97 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.5 | 0.62 | 0.96 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.6 | 0.64 | 0.94 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.7 | 0.64 | 0.93 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.8 | 0.65 | 0.91 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.9 | 0.66 | 0.89 | 0.98 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 1.0 | 0.67 | 0.86 | 0.97 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
Table 4.
The recognition accuracy with and
| n | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 | |
| 0.1 | 0.00 | 0.06 | 0.50 | 0.92 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.2 | 0.06 | 0.61 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.3 | 0.15 | 0.77 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.4 | 0.27 | 0.74 | 0.98 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.5 | 0.40 | 0.67 | 0.96 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.6 | 0.50 | 0.63 | 0.93 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.7 | 0.59 | 0.60 | 0.88 | 0.98 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.8 | 0.66 | 0.59 | 0.82 | 0.97 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.9 | 0.71 | 0.61 | 0.76 | 0.94 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 1.0 | 0.75 | 0.65 | 0.72 | 0.90 | 0.98 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
Table 5.
The recognition accuracy with and
| n | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 | |
| 0.1 | 0.01 | 0.01 | 0.00 | 0.01 | 0.01 | 0.01 | 0.02 | 0.04 | 0.07 | 0.09 |
| 0.2 | 0.22 | 0.16 | 0.50 | 0.89 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.3 | 0.51 | 0.43 | 0.89 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.4 | 0.62 | 0.56 | 0.93 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.5 | 0.64 | 0.59 | 0.92 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.6 | 0.64 | 0.58 | 0.88 | 0.98 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.7 | 0.65 | 0.58 | 0.83 | 0.96 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.8 | 0.65 | 0.59 | 0.79 | 0.94 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.9 | 0.67 | 0.61 | 0.73 | 0.91 | 0.98 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 1.0 | 0.68 | 0.65 | 0.70 | 0.86 | 0.96 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 |
Now, we investigate how different influences the recovery accuracy . For example, when , Figure 2 shows the recognition accuracy with different . As shown in Figure 2, when , the recognition of matrix can reach highest accuracy. With increasing, the recovery accuracy drops. For example, when , s3 and s4 are degraded to 90%.
Figure 2.

The recognition accuracy of matrix with different . s1 denotes the recognition accuracy series with and . s2 denotes the recognition accuracy series with and . s3 denotes the recognition accuracy series with and . s4 denotes the recognition accuracy series with and .
From these experiments, a conclusion can be drawn that when the optimal empirical value of is given as: , where the size of data matrix is , the highest identification accuracy can be obtained.
Experimental results on gene expression data of plants responding to abiotic stresses
Along with other two state-of-the-art methods, namely PMD and SPCA, used as comparison, three methods, including RPCA, are used to discover the differentially expressed genes responding to abiotic stresses based on real gene expression data.
Data source
The raw data were downloaded from NASCArrays [http://affy.arabidopsis.info/] [21], which include two classes: roots and shoots in each stress. The reference numbers are: control, NASCArrays-137; cold stress, NASCArrays-138; osmotic stress, NASCArrays-139; salt stress, NASCArrays-140; drought stress, NASCArrays-141; UV-B light stress, NASCArrays-144; heat stress, NASCArrays-146. Table 6 lists the sample number of each stress type. There are 22810 genes in each sample. The data are adjusted for background of optical noise using the GC-RMA software by Wu et al. [22] and normalized using quartile normalization. The results of GC-RMA are gathered in a matrix for further processed.
Table 6.
The sample number of each stress type in the raw data
| Stress Type | cold | drought | salt | UV-B | heat | osmotic | control |
|---|---|---|---|---|---|---|---|
| Number of Samples | 6 | 7 | 6 | 7 | 8 | 6 | 8 |
Selection of the parameters
In this paper, for PMD method, the -norm of is taken as the penalty function, i.e. . Because of , let , where . For simplicity, let , that is, only one factor is used. The results with -norm () and -norm (, i.e. the number of nonzero coefficients, or cardinality) penalty in SPCA are similar, which is also shown in [19], so -norm penalty and the parameter are taken in SPCA. For a fair comparison, 500 genes are roughly selected by these methods via choosing appropriate parameters and of the two methods, PMD and SPCA, which are listed in Table 7 for different data set. As the first subsection of experiments mentioned, while , RPCA gives the optimization results. Then, according to methods section, the first 500 genes are selected.
Table 7.
The values of and on different data set
| Stress | shoot | shoot | root | root |
|---|---|---|---|---|
| PMD | SPCA | PMD | SPCA | |
| drought | 0.0928 | 0.4224 | 0.0999 | 0.4065 |
| salt | 0.0924 | 0.4920 | 0.1057 | 0.5261 |
| UV-B | 0.1036 | 0.4505 | 0.0966 | 0.4329 |
| cold | 0.1026 | 0.4660 | 0.0983 | 0.4726 |
| heat | 0.0765 | 0.3770 | 0.0931 | 0.3710 |
| osmotic | 0.1049 | 0.5139 | 0.0946 | 0.5338 |
Gene ontology (GO) analysis
Recently, many tools have been developed for the functional analysis of large lists of genes [23,24]. Most of them focus on the evaluation of Gene Ontology (GO) annotations. GOTermFinder is a web-based tool that finds the significant GO terms shared among a list of genes, helping us discover what these genes may have in common. The analysis of GOTermFinder provides significant information for the biological interpretation of high-throughput experiments.
In this subsection, the genes identified by these methods, RPCA, PMD and SPCA, are sent to GOTermFinder [24], which is publicly available at http://go.princeton.edu/cgi-bin/GOTermFinder. Its threshold parameters are set as following: minimum number of gene products = 2 and maximum P-value = 0.01. Here, the key results are shown. Table 8 lists the terms of Response to abiotic stimulus (GO:0009628), whose background frequency in TAIR set is 1539/29556 (5.2%). Response to abiotic stimulus is the ancestor term of all the abiotic stresses. In GOTermFinder, a p-value is calculated using the hyper-geometric distribution, its details can be seen in [24]. Sample frequency denotes the number of genes hit in the selected genes, such as 107/500 denotes 107 genes associated with the GO term in 500 ones selected by these methods. As listed in Table 8, all the three experimented methods, PMD, SPCA and RPCA, can extract the significant genes with very lower P-value, as well as very higher sample frequency. In Table 8, the superior results are in bold type. In the twelve items, there is only one of them (cold on root) that PMD is equal to our method. In other items, our method is superior to SPCA and PMD.
Table 8.
Response to abiotic stimulus (GO:0009628)
|
Stress type |
PMD | SPCA | RPCA | ||||
|---|---|---|---|---|---|---|---|
| P-value | Sample frequency | P-value | Sample frequency | P-value | Sample frequency | ||
| drought | s | 3.91E-34 | 107/500 (21.4%) | 7.5E-21 | 87/500 (17.4%) | 1.09E-45 | 122/500 (24.4%) |
| drought | r | 1.78E-10 | 68/500 (13.6%) | 4.14E-08 | 63/500 (12.6%) | 1.03E-27 | 98/500 (19.6%) |
| salt | s | 9.93E-39 | 113/500 (22.6%) | 9.83E-33 | 105/500 (21.0%) | 1.35E-55 | 134/500 (26.8%) |
| salt | r | 1.36E-15 | 78/500 (15.6%) | 6.18E-12 | 71/500 (14.2%) | 1.65E-22 | 90/500 (18.0%) |
| UV-B | s | 1.76E-13 | 74/500 (14.8%) | 7.84E-23 | 90/500 (18.0%) | 5.9E-41 | 116/500 (23.2%) |
| UV-B | r | 5.3E-10 | 67/500 (13.4%) | 8.00 E-4 | 52/500 (10.4%) | 4.73E-29 | 100/500 (20.0%) |
| cold | s | 5.82E-35 | 106/500 (21.6%) | 1.17E-19 | 85/500 (17.0%) | 2.13E-46 | 123/500 (24.6%) |
| cold | r | 2.74E-23 | 91/500 (18.2%) | 4.1E-19 | 84/500 (16.8%) | 4.02E-23 | 91/500 (18.2%) |
| heat | s | 1.44E-24 | 93/500 (18.6%) | 4.64E-22 | 89/500 (17.8%) | 7.46E-55 | 133/500 (26.6%) |
| heat | r | 1.41E-15 | 78/500 (15.6%) | 1.35E-08 | 64/500 (12.8%) | 1.07E-34 | 108/500 (21.6%) |
| osmotic | s | 6.55E-38 | 112/500 (22.4%) | 2.02E-18 | 83/500 (16.6%) | 6.83E-54 | 132/500 (26.4%) |
| osmotic | r | 1.4E-14 | 76/500 (15.2%) | 2.87E-17 | 81/500 (16.2%) | 9.98E-35 | 108/500 (21.6%) |
In this table, 's' denotes the shoot samples; 'r' denotes the root samples.
Figure 3 shows the sample frequency of response to abiotic stimulus (GO:0009628) given by the three methods. From Figure 3(a), RPCA method outperforms others in all the data sets of shoot samples with six different stresses. Figure 3(b) shows that only in cold-stress data set of root samples, PMD is equal to our method and they are superior to SPCA. In other data sets, our method is superior to the others.
Figure 3.
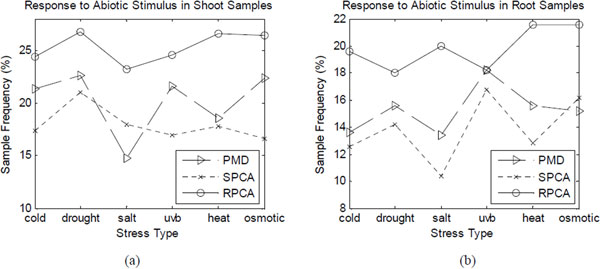
The sample frequency of response to abiotic stimulus.
The characteristic terms are listed in Table 9, in which the superior results are in bold type. As listed in Table 9, PMD method outperforms SPCA and our method in three items, such as drought in shoot, salt in root and cold in root, among the whole items. However, it shows that, on one of the twelve items (osmotic in shoot), our method has the same competitive result as PMD, while both methods are superior to SPCA. In other eight items, our method excels PMD and SPCA methods. In addition, on all the characteristic items, our method has superiority over SPCA.
Table 9.
Characteristic terms selected from GO by algorithms
| Stress type | GO Terms | Background frequency | Sample frequency | |||
|---|---|---|---|---|---|---|
| PMD | SPCA | RPCA | ||||
| drought | s | GO:0009414 response to water deprivation | 207/29887 (0.7%) | 47/500 (9.4%) | 23/500 (4.6%) | 34/500 (6.8%) |
| drought | r | GO:0009415 response to water deprivation | 207/29887 (0.7%) | 26/500 (5.2%) | 24/500 (4.8%) | 30/500 (6.0%) |
| salt | s | GO:0009651 response to salt stress | 395/29887 (1.3%) | 41/500 (8.2%) | 28/500 (5.6%) | 48/500 (9.8%) |
| salt | r | GO:0009651 response to salt stress | 395/29887 (1.3%) | 33/500 (6.6%) | 22/500 (4.4%) | 31/500 (6.2%) |
| UV-B | s | GO:0009416Response to light stimulus | 557/29887 (1.9%) | 23/500 (4.6%) | 30/500 (6.0%) | 42/500 (8.4%) |
| UV-B | r | GO:0009416Response to light stimulus | 557/29887 (1.9%) | 24/500 (4.8%) | none | 36/500 (7.2%) |
| cold | s | GO:0009409 response to cold | 276/29887 (0.9%) | 44/500 (8.8%) | 34/500 (6.8%) | 58/500 (11.6%) |
| cold | r | GO:0009410 response to cold | 276/29887 (0.9%) | 43/500 (8.6%) | 33/500 (6.6%) | 38/500 (7.6%) |
| heat | s | GO:0009408 response to heat | 140/29887 (0.5%) | 45/500 (9.0%) | 30/500 (6.0%) | 47/500 (9.4%) |
| heat | r | GO:0009409 response to heat | 140/29887 (0.5%) | 43/500 (8.6%) | 28/500 (5.6%) | 48/500 (9.6%) |
| osmotic | s | GO:0006970 response to osmotic stress | 474/29887 (1.6%) | 55/500 (11.0%) | 29/500 (5.8%) | 55/500 (11.0%) |
| osmotic | r | GO:0006970 response to osmotic stress | 474/29887 (1.6%) | 39/500 (7.8%) | 27/500 (5.4%) | 41/500 (8.2%) |
In this table, 's' denotes the shoot samples; 'r' denotes the root samples; 'none' denotes that the algorithm cannot give the GO terms.
From the results of experiments, it can be concluded that our method is efficient and effective.
Experimental results on colon data
The three methods, SPCA, PMD and RPCA, are compared on colon cancer data set [25]. Colon cancer is the fourth most common cancer for males and females and the second most frequent cause of death.
Data source
The raw data were downloaded from http://genomics-pubs.princeton.edu/oncology/affydata/I2000.html, which include gene expression levels for 2000 gene and contain 40 tumor and 22 normal tissue samples.
Selection of the parameters
In this subsection, for PMD method, the -norm of is taken as the penalty function, i.e. . Let , where . For SPCA method, let , that is, only one factor is used. -norm penalty and the parameter are taken in SPCA. For a fair comparison, 100 genes are roughly selected by these methods via choosing appropriate parameters. PMD and SPCA use the parameters and on colon data set, respectively. As the first subsection of experiments mentioned, while , RPCA gives the optimization results. Then, according to Methods section,the first 100 genes are selected using our method.
Gene ontology (GO) analysis
The genes identified by these methods, RPCA, PMD and SPCA, are evaluated by using AmiGO [26]. Its threshold parameters are set as following: minimum number of gene products = 2 and maximum P-value = 0.1. A number of lines of evidence suggest that immune, stimulus and tumor have affinity, so Table 10 lists the key results: the terms of Response to stimulus (GO:0050896) and Immune system process (GO:0002376). As listed in Table 10, RPCA outperforms its competitive methods with higher sample frequency.
Table 10.
Characteristic terms selected from GO on colon data
| GO Term | Response to stimulus | Immune system process |
|---|---|---|
| Accession No. | GO:0050896 | GO:0002376 |
| Background frequency | 32294/155706 (20.7%) | 7011/155706 (4.5%) |
| P-value(RPCA) | 1.76E-10 | 5.74E-09 |
| Sample frequency (RPCA) | 38/57 (66.7%) | 19/57 (33.3%) |
| P-value(SPCA) | 8.71E-06 | 2.95E-04 |
| Sample frequency (SPCA) | 32/57 (56.1%) | 14/57 (24.6%) |
| P-value(PMD) | 7.93E-04 | 8.27E-01 |
| Sample frequency (PMD) | 27/51 (52.9%) | 9/51 (17.6%) |
Function analysis
Table 11 lists the top 30 genes selected by using RPCA. To further study the biology functions of the selected genes, we also make the network analysis of the top 100 genes selected by our algorithm using the GeneMANIA tool [27] on the Web sitehttp://genemania.org/. The result is listed in Table 12. From the table it can be seen that there are 215 genes of this chip participating in the cytokine-mediated signalling pathway, in which there are 21 genes discovered by our method. This pathway has the lowest p-value. It is considered as the most probable pathway with these top 100 genes. Recent findings also indicate that cytokine receptors can regulate immune cell functions by transcription-independent mechanisms [28]. Some other pathways with the most significance are also listed in Table 12.
Table 11.
The top 30 genes of colon data selected by RPCA
| Gene No. | Sequence | Gene Name |
|---|---|---|
| M27190 | gene | Homo sapiens secretary pancreatic stone protein (PSP-S) mRNA, complete cds. |
| R89823 | 3' UTR | INORGANIC PYROPHOSPHATASE (Bos taurus) |
| M87789 | gene | IG GAMMA-1 CHAIN C REGION (HUMAN). |
| T48904 | 3' UTR | HEAT SHOCK 27 KD PROTEIN (HUMAN). |
| M26383 | gene | Human monocyte-derived neutrophil-activating protein (MONAP) mRNA, complete cds. |
| J00231 | gene | Human Ig gamma3 heavy chain disease OMM protein mRNA. |
| X02761 | gene | Human mRNA for fibronectin (FN precursor). |
| R80612 | 3' UTR | PHOSPHOLIPASE A2, MEMBRANE ASSOCIATED PRECURSOR (HUMAN). |
| M31994 | gene | Human cytosolic aldehyde dehydrogenase (ALDH1) gene, exon 13. |
| T47377 | 3' UTR | S-100P PROTEIN (HUMAN). |
| X02492 | gene | INTERFERON-INDUCED PROTEIN 6-16 PRECURSOR (HUMAN); contains L1 repetitive element. |
| M94132 | gene | Human mucin 2 (MUC2) mRNA sequence. |
| X67325 | gene | H.sapiens p27 mRNA. |
| D28137 | gene | Human mRNA for BST-2, complete cds. |
| L05144 | gene | PHOSPHOENOLPYRUVATE CARBOXYKINASE, CYTOSOLIC (HUMAN); contains Alu repetitive element; contains element PTR5 repetitive element. |
| X02874 | gene | Human mRNA for (2'-5') oligo A synthetase E (1,6 kb RNA). |
| T55117 | 3' UTR | ALPHA-1-ANTITRYPSIN PRECURSOR (HUMAN). |
| M19045 | gene | Human lysozyme mRNA, complete cds. |
| Y00711 | gene | L-LACTATE DEHYDROGENASE H CHAIN (HUMAN);. |
| X60489 | gene | Human mRNA for elongation factor-1-beta. |
| T57780 | 3' UTR | IG LAMBDA CHAIN C REGIONS (HUMAN). |
| T60778 | 3' UTR | MATRIX GLA-PROTEIN PRECURSOR (Rattus norvegicus). |
| H58397 | 3' UTR | TRANS-1, 2-DIHYDROBENZENE-1, 2-DIOL DEHYDROGENASE (HUMAN). |
| L08044 | gene | Human intestinal trefoil factor mRNA, complete cds. |
| M18216 | gene | Human nonspecific cross reacting antigen mRNA, complete cds. |
| K03474 | gene | Human Mullerian inhibiting substance gene, complete cds. |
| L33930 | gene | Homo sapiens CD24 signal transducer mRNA, complete cds and 3' region. |
| T48014 | 3' UTR | HEMOGLOBIN ALPHA CHAIN (HUMAN). |
| H73908 | 3' UTR | METALLOTHIONEIN-IA (Bos taurus) |
| R70030 | 3' UTR | IG MU CHAIN C REGION (HUMAN). |
Table 12.
Pathway analysis of the top 100 genes selected by RPCA on colon data
| rank | Go annotation | Q-value | Genes in network | Genes in genome |
|---|---|---|---|---|
| 1 | cytokine-mediated signalling pathway | 2.27E-20 | 21 | 215 |
| 2 | cellular response to cytokine stimulus | 1.70E-19 | 21 | 244 |
| 3 | response to cytokine stimulus | 2.62E-18 | 21 | 283 |
| 4 | type I interferon-mediated signalling pathway | 1.61E-17 | 14 | 71 |
| 5 | cellular response to type I interferon | 1.61E-17 | 14 | 71 |
| 6 | response to type I interferon | 1.67E-17 | 14 | 72 |
| 7 | interferon-gamma-mediated signalling pathway | 2.60E-08 | 9 | 77 |
| 8 | cellular response to interferon-gamma | 3.64E-08 | 9 | 81 |
| 9 | response to interferon-gamma | 1.04E-07 | 9 | 92 |
| 10 | response to other organism | 3.69E-05 | 10 | 243 |
Conclusion
In this paper, a novel RPCA-based method of discovering differentially expressed genes was proposed. It combined RPCA and sparsity of gene differential expression to provide an efficient and effective approach for gene identification. Our method mainly included the following two steps: firstly, the matrix of differential expression was discovered from gene expression data matrix by using robust PCA; secondly, the differentially expressed genes were discovered according to matrix . The experimental results on real gene data showed that our method outperformed the other state-of-the-art methods. In future, we will focus on the biological meaning of the differentially expressed genes.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Jin-Xing Liu, Email: sdcavell@126.com.
Yu-Tian Wang, Email: wytsd75@163.com.
Chun-Hou Zheng, Email: zhengch99@126.com.
Wen Sha, Email: wensha@ahu.edu.cn.
Jian-Xun Mi, Email: mijianxun@gmail.com.
Yong Xu, Email: laterfall2@yahoo.com.cn.
Acknowledgements
This work was supported by fund for China Postdoctoral Science Foundation Funded Project, No. 2012M510091; Program for New Century Excellent Talents in University (No.NCET-08-0156), NSFC under grant No. 61071179, 61272339, 61202276 and 61203376, and the Key Project of Anhui Educational Committee, under Grant No. KJ2012A005; the Foundation of Qufu Normal University under grant no. XJ200947.
Declarations
The publication costs for this article were funded by fund for China Postdoctoral Science Foundation Funded Project, No. 2012M510091.
This article has been published as part of BMC Bioinformatics Volume 14 Supplement 8, 2013: Proceedings of the 2012 International Conference on Intelligent Computing (ICIC 2012). The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/14/S8.
References
- Wang B, Wong H, Huang DS. Inferring protein-protein interacting sites using residue conservation and evolutionary information. Protein and peptide letters. 2006;13(10):999. doi: 10.2174/092986606778777498. [DOI] [PubMed] [Google Scholar]
- Huang DS, Zhao XM, Huang GB, Cheung YM. Classifying protein sequences using hydropathy blocks. Pattern recognition. 2006;39(12):2293–2300. doi: 10.1016/j.patcog.2005.11.012. [DOI] [Google Scholar]
- Wang L, Li PCH. Microfluidic DNA microarray analysis: A review. Analytica chimica acta. 2011;687(1):12–27. doi: 10.1016/j.aca.2010.11.056. [DOI] [PubMed] [Google Scholar]
- Liao JC, Boscolo R, Yang YL, Tran LM, Sabatti C, Roychowdhury VP. Network component analysis: reconstruction of regulatory signals in biological systems. Proceedings of the National Academy of Sciences. 2003;100(26):15522–15527. doi: 10.1073/pnas.2136632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueck D, Morris QD, Frey BJ. Multi-way clustering of microarray data using probabilistic sparse matrix factorization. Bioinformatics. 2005;21(suppl 1):i144–i151. doi: 10.1093/bioinformatics/bti1041. [DOI] [PubMed] [Google Scholar]
- Dudoit S, Shaffer JP, Boldrick JC. Multiple hypothesis testing in microarray experiments. Statistical Science. 2003;18(1):71–103. doi: 10.1214/ss/1056397487. [DOI] [Google Scholar]
- Lee D, Lee W, Lee Y, Pawitan Y. Super-sparse principal component analyses for high-throughput genomic data. BMC bioinformatics. 2010;11(1):296. doi: 10.1186/1471-2105-11-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Xu Y, Zheng CH, Wang Y, Yang JY. Characteristic Gene Selection via Weighting Principal Components by Singular Values. Plos One. 2012;7(7):e38873. doi: 10.1371/journal.pone.0038873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyamundanda G, Brennan L, Gormley IC. Probabilistic Principal Component Analysis for Metabolomic Data. BMC bioinformatics. 2010;11(1):571. doi: 10.1186/1471-2105-11-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DS, Zheng CH. Independent component analysis-based penalized discriminant method for tumor classification using gene expression data. Bioinformatics. 2006;22(15):1855–1862. doi: 10.1093/bioinformatics/btl190. [DOI] [PubMed] [Google Scholar]
- Zheng CH, Huang DS, Zhang L, Kong XZ. Tumor clustering using nonnegative matrix factorization with gene selection. Information Technology in Biomedicine, IEEE Transactions on. 2009;13(4):599–607. doi: 10.1109/TITB.2009.2018115. [DOI] [PubMed] [Google Scholar]
- Liu J, Zheng C, Xu Y. Advanced Computational Intelligence (IWACI), 2011 Fourth International Workshop on. IEEE; 2011. Lasso logistic regression based approach for extracting plants coregenes responding to abiotic stresses; pp. 461–464. [Google Scholar]
- Witten DM, Tibshirani R, Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009;10(3):515–534. doi: 10.1093/biostatistics/kxp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Zheng CH, Xu Y. Extracting plants core genes responding to abiotic stresses by penalized matrix decomposition. Comput Biol Med. 2012;42(5):582–589. doi: 10.1016/j.compbiomed.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Candes EJ, Li X, Ma Y, Wright J. Robust principal component analysis? Arxiv preprint ArXiv:09123599. 2009.
- Eckart C, Young G. The approximation of one matrix by another of lower rank. Psychometrika. 1936;1(3):211–218. doi: 10.1007/BF02288367. [DOI] [Google Scholar]
- Lin Z, Chen M, Wu L, Ma Y. The augmented Lagrange multiplier method for exact recovery of corrupted low-rank matrices. 2010. http://Arxivorg/abs/10095055v2
- Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D'Angelo C, Bornberg-Bauer E, Kudla J, Harter K. The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. The Plant Journal. 2007;50(2):347–363. doi: 10.1111/j.1365-313X.2007.03052.x. [DOI] [PubMed] [Google Scholar]
- Journée M, Nesterov Y, Richtarik P, Sepulchre R. Generalized power method for sparse principal component analysis. The Journal of Machine Learning Research. 2010;11:517–553. [Google Scholar]
- Candes EJ, Li X, Ma Y, Wright J. Robust Principal Component Analysis? Journal of the ACM. 2011;58(3):11. [Google Scholar]
- Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S. NASCArrays: a repository for microarray data generated by NASC's transcriptomics service. Nucleic Acids Res. 2004;32:D575–D577. doi: 10.1093/nar/gkh133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. Journal of the American Statistical Association. 2004;99(468):909–917. doi: 10.1198/016214504000000683. [DOI] [Google Scholar]
- Sartor MA, Mahavisno V, Keshamouni VG, Cavalcoli J, Wright Z, Karnovsky A, Kuick R, Jagadish H, Mirel B, Weymouth T. ConceptGen: a gene set enrichment and gene set relation mapping tool. Bioinformatics. 2010;26(4):456–463. doi: 10.1093/bioinformatics/btp683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EI, Weng SA, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder-open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20(18):3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. P Natl Acad Sci USA. 1999;96(12):6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu SQ, Marshall B, Lewis S. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25(2):288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome biology. 2008;9(Suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezbradica JS, Medzhitov R. Integration of cytokine and heterologous receptor signaling pathways. Nature immunology. 2009;10(4):33–339. doi: 10.1038/ni.1713. [DOI] [PubMed] [Google Scholar]