

Abstract
Background
Fungal polyketides include commercially important pharmaceuticals and food additives, e.g. the cholesterol-lowering statins and the red and orange monascus pigments. Presently, production relies on isolation of the compounds from the natural producers, and systems for heterologous production in easily fermentable and genetically engineerable organisms, such as Saccharomyces cerevisiae and Escherichia coli are desirable. Rubrofusarin is an orange polyketide pigment that is a common intermediate in many different fungal biosynthetic pathways.
Results
In this study, we established a biosynthetic pathway for rubrofusarin in S. cerevisiae. First, the Fusarium graminearum gene encoding polyketide synthase 12 (PKS12) was heterologously co-expressed with the Aspergillus fumigatus gene encoding phosphopantetheinyl transferase (npgA) resulting in production of YWA1. This aromatic heptaketide intermediate was converted into nor-rubrofusarin upon expression of the dehydratase gene aurZ from the aurofusarin gene cluster of F. graminearum. Final conversion into rubrofusarin was achieved by expression of the O-methyltransferase encoding gene aurJ, also obtained from the aurofusarin gene cluster, resulting in a titer of 1.1 mg/L. Reduced levels of rubrofusarin were detected when expressing PKS12, npgA, and aurJ alone, presumably due to spontaneous conversion of YWA1 to nor-rubrofusarin. However, the co-expression of aurZ resulted in an approx. six-fold increase in rubrofusarin production.
Conclusions
The reconstructed pathway for rubrofusarin in S. cerevisiae allows the production of a core scaffold molecule with a branch-point role in several fungal polyketide pathways, thus paving the way for production of further natural pigments and bioactive molecules. Furthermore, the reconstruction verifies the suggested pathway, and as such, it is the first example of utilizing a synthetic biological “bottom up” approach for the validation of a complex fungal polyketide pathway.
Keywords: Iterative polyketide synthase, Pigment, Heterologous production, YWA1, Nor-rubrofusarin, Aurofusarin
Background
Polyketides exhibit large structural diversity and have attracted considerable interest from the pharmaceutical industry as they form a chemical class rich in bioactive molecules. The biological activities of fungal polyketides span from potent carcinogenic toxins (aflatoxin) over immunosuppressants (dalesconols), anti-tumor compounds (aspergiolide A) to block-buster cholesterol-lowering drugs (statins) [1-3]. In addition, many fungal polyketides also display vibrant colors due to their polyaromatic structures, which have made them attractive for the textile and food industries as natural dyes [4,5].
Fungal polyketides of commercial interest are often produced in nature by species that are not easily cultured in standard liquid fermentation setups, due to their filamentous growth, and formation of dense mycelium clumps resulting in increased viscosity and reduced oxygen solubility. The transfer of such biosynthetic pathways to standard industrial cell factories, such as Saccharomyces cerevisiae, could offer an attractive alternative as it would allow easier fermentation and process optimization via the existing metabolic engineering strategies. However, only a few examples of successful heterologous expression of genes encoding fungal polyketide synthases in yeast have been reported in literature. These include the 6-MSA synthase gene from Penicillium patulum resulting in 6-methylsalicylic acid (6-MSA) production [6-8], rdc1/rdc5 from Pochonia chlamydosporia[9] resulting in (R)-monocillin II production, and hpm3/hpm8 resulting in hypothemycin production [10]. Most recently the individual expression of three PKSs from Chaetomium globosum and one from Coprinopsis cinerea was reported in an effort to characterize orphan PKSs [11]. These results show that yeast heterologous expression can provide an avenue for production of industrially relevant polyketides and for characterizing orphan polyketide synthase genes from non-model organisms. However, common for these examples is that the studies only aimed to produce the first stable intermediate in the respective biosynthetic pathway and have not included the heterologous expression of any tailoring enzymes responsible for modification of the synthase product. To test the feasibility of using yeast for production of complex polyketides we aimed to reconstruct a fungal polyketide pigment biosynthetic pathway, which leads to the formation of rubrofusarin (Figure 1). Rubrofusarin is an orange-brown pigment produced by a wide range of different fungal species, such as Fusarium graminearum, Aspergillus niger, Aspergillus parasiticus and Ustilaginoidea virens[12-14]. The biological activity of rubrofusarin is reported to include inhibition of human DNA topoisomerase II-α [15] and general antibiotic effects on Mycobacterium tuberculosis and various filamentous fungi [16]. Interestingly, the fate of this polyketide core scaffold differs in the particular fungal species due to their unique repertoires of tailoring enzymes, resulting in production of such different end products as rubrofusarin B, aurofusarin, nigerone, nigerasperone A, chaetochromin, ustilaginoidin and parasperone A [12,17].
Figure 1.
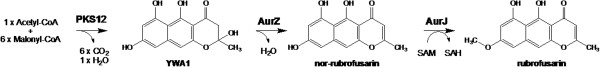
The rubrofusarin biosynthetic pathway. Biosynthetic pathway of rubrofusarin in F. graminearum established by targeted gene deletion experiments [12,18].
Currently, the best described biosynthetic pathway, which includes rubrofusarin as an intermediate, is the aurofusarin pathway in F. graminearum. In this species, the first stable pathway intermediate on the path to rubrofusarin is the yellow naphthopyrone YWA1, which is synthesized by the condensation of one acetyl-CoA and six malonyl-CoA molecules, catalyzed by the type I iterative non-reducing PKS12 [18]. Similar reactions have been reported for Aspergillus nidulans[19]. The formed YWA1 is dehydrated by AurZ to form the orange pigment nor-rubrofusarin, which is then O-methylated by AurJ to yield rubrofusarin (Figure 1) [12,18]. Polyketide biosynthetic pathways are typically encoded by gene clusters in fungi and mapping of the aurofusarin pathway in F. graminearum has therefore primarily been based on targeted deletion of the genes surrounding PKS12[12,18,20]. Thus, the possibility exists that genes located elsewhere in the genome might be directly involved in the formation of rubrofusarin. Hence, an appropriate bottom-up proof of the sole involvement of PKS12, aurZ and aurJ in rubrofusarin formation would be their heterologous expression in a phylogenetically distant organism.
Heterologous production in S. cerevisiae of other important compound classes with a complex biosynthetic origin, e.g. terpenoids [21] and plant type III polyketides [22], has already been well established. To provide a proof-of-concept that this approach is also a viable option for polyketide pathways with a fungal origin, and to prove the proposed biosynthetic pathway, we here present the stepwise construction of a rubrofusarin pathway in the heterologous host S. cerevisiae.
Results and discussion
Rubrofusarin has previously been described to have antifungal properties, and to avoid problems with toxic effects of intermediates or the end-product during strain construction, the open reading frame (ORF) encoding the wild type PKS12 was placed under control of the inducible CUP1 promoter to allow control of expression onset and strength. Codon optimized ORFs for aurZ, aurJ and npgA were expressed using unique constitutive promoters and terminators (Table 1). The genes were inserted individually into different plasmids, derived from pRS413-6 [23], each carrying unique auxotrophic selection marker genes along with the unique promoter and terminator elements. This plasmid-based approach was chosen to allow for easy construction of any desired combination of genes in S. cerevisiae to study the sequential order of individual catalytic steps in the biosynthetic pathway (Table 1).
Table 1.
S. cerevisiae strains used in the study
| Strain | Plasmids carried | Relevant genotype |
|---|---|---|
|
S. cerevisiae control |
pRS416 (URA3) |
- |
| pRS413 (HIS3) | ||
| pRS414 (TRP1) | ||
| pRS415 (LEU2) | ||
|
S. cerevisiae PKS12/npgA |
pRS416::pCUP1-PKS12-tADH1, |
PKS12, npgA |
| pRS413::pPYK1-npgA-tTEF1 | ||
| pRS414 | ||
| pRS415 | ||
|
S. cerevisiae PKS12/npgA/aurZ |
pRS416::pCUP1-PKS12-tADH1, |
PKS12, npgA, aurZ |
| pRS413::pPYK1-npgA-tTEF1 | ||
| pRS414::pTEF1-aurZ-tENO2 | ||
| pRS415 | ||
|
S. cerevisiae PKS12/npgA/aurJ |
pRS416::pCUP1-PKS12-tADH1, |
PKS12, npgA, aurJ |
| pRS413::pPYK1-npgA-tTEF1 | ||
| pRS415::pGPD1-aurJ-tCYC1 | ||
| pRS414 | ||
| S. cerevisiae PKS12/npgA/aurZ/aurJ | pRS416::pCUP1-PKS12-tADH1, |
PKS12, npgA, aurZ, aurJ |
| pRS413::pPYK1-npgA-tTEF1 | ||
| pRS414::pTEF1-aurZ-tENO2 | ||
| pRS415::pGPD1-aurJ-tCYC1 |
S. cerevisiae strains that were analyzed for production of rubrofusarin and precursors. Final plasmids transformed into S. cerevisiae strains showing the vector backbone in which the constructed promoter (p) – ORF – terminator (t) gene cassettes were inserted. The auxotrophic marker gene is also shown. All plasmids were maintained by the low copy number sequences ARS/CEN.
Heterologous production of YWA1
Expression of PKS12 and npgA in S. cerevisiae resulted in the production of a yellow pigment which was not seen in the control strain carrying empty plasmids. In contrast to plates without added copper, yellow-colored pigment was visually observed after approx. 24 hours of cultivation on synthetic complete (SC) agar plates supplemented with copper. Following incubation for an additional 2-3 days, a yellow pigment became visible in the medium surrounding the S. cerevisiae colonies, indicating that the produced pigment was escaping from the cells. In SC liquid cultures a yellow color became visually observable after 2-3 days of cultivation (Figure 2). Chemical analysis, using LC-MS, of the growth medium from the induced PKS12/npgA strain revealed a new peak with a retention time (RT) of 5.3 min and a [M + H]+ of 277.07 m/z. Based on the exact mass, the compound was identified as the pigment YWA1 (Figure 3). The intensity of the peak was, however, relatively weak suggesting either a low production level, poor extraction or ionization of YWA1, or rapid degradation as was previously reported by Fujii and colleagues working with YWA1 in A. oryzae[24]. This compound is also known from Aspergillus fumigatus in which it is converted to 1,8-dihydroxynaphthalene (DHN) in multiple steps, including a chain-shortening reaction catalyzed by Ayg1 [25], and in A. nidulans where it is the precursor for the green conidial pigment [19].
Figure 2.

Phenotype of the constructed S. cerevisiae strains. Cultures of S. cerevisiae strains, carrying the genes shown, were grown in A) SC medium with 300 μM CuSO4 and without uracil, histidine, tryptophan and leucine. Cells were grown for 4 days at 30°C, 160 rpm shaking, and collected by centrifugation. B) SC agar plates with 500 μM CuSO4 and without uracil, histidine, tryptophan and leucine. Grown for 4 days at 30°C. The strains accumulated the indicated rubrofusarin pathway products.
Figure 3.
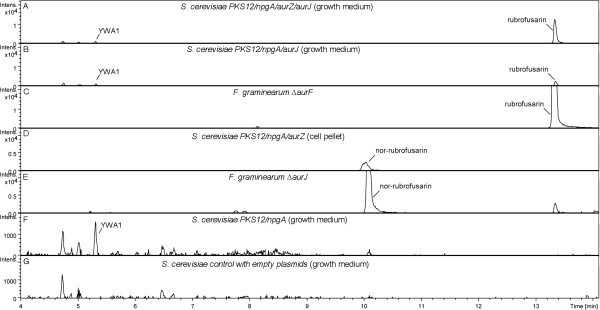
Chemical analysis of strains. Extracted ion chromatograms (EIC) for the analyzed strains. EIC for A) S. cerevisiae PKS12/npgA/aurZ/aurJ, growth medium (GM), B) S. cerevisiae PKS12/npgA/aurJ, (GM), C) rubrofusarin-accumulating reference F. graminearum ΔaurF, D) S. cerevisiae PKS12/npgA/aurZ, cell pellet, E) nor-rubrofusarin-accumulating reference F. graminearum ΔaurJ, F) S. cerevisiae PKS12/npgA (GM), G) Control S. cerevisiae strain with empty plasmids (GM). Note that the scales on the Y-axes are not identical between the different chromatograms.
Heterologous production of nor-rubrofusarin
Introduction of the dehydratase-encoding aurZ gene in the YWA1-producing strain resulted in colonies that visually appeared more intensely orange after 3 days of cultivation as compared to the parent YWA1-producing PKS12/npgA strain (Figure 2). The yellow pigment observed in the plate medium surrounding S. cerevisiae colonies of the PKS12/npgA strain was no longer visible, suggesting conversion of YWA1 into another orange pigment, which did not readily escape the cells (Figure 2).
Chemical analysis revealed a single new peak with a RT of 10.0 min and a [M + H]+ = 259.06 m/z which displayed a distinct UV absorption spectrum (see UV spectrum in Additional file 1). Based on a comparison to nor-rubrofusarin extracted from a nor-rubrofusarin-accumulating F. graminearum ΔaurJ reference strain, and by comparison to a previously published UV spectrum, the compound was identified as nor-rubrofusarin (Figure 3) [18]. Nor-rubrofusarin could be detected in both the cell pellet and growth medium, while YWA1 was still only detectable in the growth medium, further indicating that the observed change in colour is linked to the compounds of the identified peaks (Figure 4). A low level of nor-rubrofusarin was also detected in the cell pellet of the PKS12/npgA parent strain likely resulting from spontaneous conversion of YWA1. Spontaneous dehydration of YWA1 and YWA1-like compounds has previously been observed with purified compounds [18,26].
Figure 4.

Detection of intermediates inside and outside of the cells. Detection of the rubrofusarin pathway intermediates in the constructed S. cerevisiae strains, shown by mean areas of the integrated peaks from extracted ion chromatograms for A) YWA1, B) Nor-rubrofusarin, and shown by mean titer (mg/L) calculated for C) Rubrofusarin. Measurements performed for the analyzed cell pellet (C) and growth medium (GM) of the constructed S. cerevisiae strains (see Table 1). Error bars indicate the std. deviation of each integrated peak (n = 3). Note that the mean areas are not comparable between different compounds.
Heterologous production of rubrofusarin
Introduction of aurJ, encoding an O-methyltransferase, into the S. cerevisiae PKS12/npgA/aurZ strain changed the visual phenotype to appear less orange during growth on plates (strain PKS12/npgA/aurZ/aurJ, Figure 2). Chemical analysis showed that nor-rubrofusarin was no longer accumulating. Instead, a new compound accumulated which had a RT of 13.3 min, [M + H]+ of 273.07 m/z and a unique UV absorption spectrum (see UV spectrum in Additional file 1). The compound was identified as rubrofusarin by comparison to rubrofusarin extracted from a rubrofusarin-accumulating F. graminearum ΔaurF reference strain (Figure 3) and by comparison to the previously published UV spectrum for rubrofusarin [18]. The rubrofusarin could be detected both in the extracted cell pellet and growth medium. YWA1 was still measurable in the growth medium, however at a reduced level (Figure 4A).
Surprisingly, introduction of aurJ in the PKS12/npgA strain resulted in colonies that were more orange than the PKS12/npgA strain which solely accumulates YWA1 (Figure 2). Chemical analysis showed the production of rubrofusarin, evidenced by the appearance of a peak at the same RT and [M + H]+ = 273.07 m/z as in the PKS12/npgA/aurZ/aurJ strain, but at an approx. six-fold lower level. These results are contrary to what has previously been observed in F. graminearum ΔaurZ mutants which only accumulate YWA1 even though they still express aurJ[18]. However, as we observed in the PKS12/npgA strain, low levels of YWA1 may undergo spontaneous conversion to nor-rubrofusarin, and subsequent methylation of this by AurJ may then explain the formation of rubrofusarin without the presence of AurZ. The difference in rubrofusarin concentration observed between the PKS12/npgA/aurZ/aurJ and PKS12/npgA/aurJ strains shows that AurZ is responsible for catalyzing the specific dehydration of YWA1 to yield nor-rubrofusarin, as previously suggested [18]. It is noteworthy that no methylated YWA1 was observed in the PKS12/npgA/aurJ strain at [M + H]+ = 291.08 m/z, suggesting that formation of rubrofusarin only proceeds through the nor-rubrofusarin intermediate, as previously proposed by Frandsen et al [12].
The presented heterologous production of rubrofusarin supports the biosynthetic pathway suggested by Frandsen et al. 2011 i.e. that PKS12, AurZ, and AurJ are stepwise responsible for formation of YWA1, nor-rubrofusarin, and rubrofusarin, respectively. In addition, it rules out the possibility that further, as yet unidentified enzymes are required for completion of the pathway.
The accumulated rubrofusarin from the strains PKS12/npgA/aurZ/aurJ and PKS12/npgA/aurJ was quantified by HPLC. Integration of rubrofusarin peaks from strain culture extracts showed a titer of 1.1 mg/L rubrofusarin produced by the PKS12/npgA/aurZ/aurJ strain, while 0.19 mg/L rubrofusarin was measured in the strain extract, when AurZ was excluded from the pathway.
In the present study no optimization strategies for production were employed. However, several simple optimization strategies would be applicable, such as increasing the gene copy number, tuning the individual expression level of pathway enzymes, and increasing the acetyl-CoA and malonyl-CoA precursor pools in the cell. In addition, optimizing the fermentation conditions is likely to substantially increase the titer. Production of cell pigmentation would provide a simple and convenient read-out for successful optimization.
The structurally simpler polyketide 6-MSA has previously been heterologously produced in S. cerevisiae reaching a titer of 1.7 g/L [6]. In another study, the 6-MSA titer was increased 60% by increasing the malonyl-CoA supply, via overexpression of the acetyl-CoA carboxylase ACC1, reaching a final 554 mg/L with their particular strain background [8]. These results suggest that the potential for rubrofusarin production in yeast may be significantly higher than what we detected in this pathway reconstruction study.
S. cerevisiae as a heterologous host for fungal natural or synthetic polyketide pathways
Simple eukaryotic hosts, such as the unicellular yeast S. cerevisiae, hold great potential for expression and study of heterologous biosynthetic pathways, including those of entire gene clusters typical of secondary metabolism of filamentous fungi. In synthetic biology terms, S. cerevisiae represents a more “minimal host” than filamentous fungi, due to the absence of any significant endogenous secondary metabolites [27], which could otherwise result in pleiotropic effects, complicating product formation and the ensuing characterization of the heterologous pathways. Hence, a yeast-based system for heterologous expression of fungal polyketide biosynthetic pathways is very well-suited for efficient characterization of the continuously increasing pool of orphan fungal polyketide gene clusters being revealed by genome sequencing projects [28,29].
Reconstruction of the rubrofusarin pathway further opens the door for heterologous production of a diversity of related, but novel polyketides in S. cerevisiae, for example by co-expressing combinations of tailoring enzymes such as p450s, glycosyl-, acyl- and prenyl- transferases etc. selected from other secondary metabolite pathways and gene clusters. The genes of such enzymes could efficiently be laid “on top” of the rubrofusarin pathway for example by random assembly and expression on yeast artificial chromosomes [22]. The readily observable phenotype of the developed rubrofusarin-producing strain may also serve as a tool for optimizing yeast for general polyketide production, e.g. via enhanced precursor supply or screening for optimized enzymes such as PPTases.
Conclusions
S. cerevisiae was engineered to produce the orange-brown polyketide pigment rubrofusarin, a common intermediate in a range of biosynthetic pathways found in different filamentous fungal species, by introduction of the three F. graminearum genes PKS12, aurZ and aurJ together with the PKS-activating PPTase gene npgA. A titer of 1.1 mg/L rubrofusarin was detected. The biosynthetic reconstruction further provides the evidence that PKS12 is responsible for formation of the pigment YWA1, whereas AurZ catalyzes the formation of the pigment nor-rubrofusarin from YWA1. The results confirm the previously proposed biosynthetic pathway for the formation of rubrofusarin using a bottom-up approach and show that no other enzymes are necessary. The study also demonstrates that it is possible to produce complex fungal polyketide products in S. cerevisiae, which opens up for the industrial production of a diversity of active and commercially interesting polyketides in the future.
Materials and methods
Strains
Molecular cloning was performed using Escherichia coli XL1 Blue, E. coli XL10 Gold (Stratagene) and E. coli DH5α (Invitrogen) competent cells. Final plasmids carried by strains in the chemical analysis are listed in Table 1. All S. cerevisiae strains constructed had the original genotype mat α, trp1Δ, lys2Δ0, leu2Δ, his3Δ1, ura3Δ0, arg4::loxP and were derived from S. cerevisiae BY4742 (Euroscarf). The constructed strains are listed in Table 1.
Enzymes and oligonucleotides
All restriction enzymes were purchased from New England Biolabs. Oligonucleotides for subcloning and mutagenesis of PKS12 were synthesized by Microsynth AG, while oligonucleotides for the USER friendly cloning were synthesized by Integrated DNA Technologies.
Design and synthesis of genes
A wild type F. graminearum PH1 PKS12 ORF was constructed using USER friendly cloning [30] by the assembly of the six exons predicted by MIPS [31] [FGDB: FGSG_02324] into the recipient vector BGHA-P1 [32] with in vivo ligation in E. coli. All exon fragments were PCR amplified from F. graminearum PH1 genomic DNA using primers designed with the PHUSER web tool (see oligonucleotides in Additional file 1) [33]. An internal HindIII restriction site in the ORF was silently mutated by mixing approx. 200 ng plasmid harboring the gene with 10 nmol of the primer Mut1Fwd and 10 nmol Mut1Rev in an otherwise standard 50 μL PCR mixture using iProof DNA polymerase and HF Buffer (BioRad) as the manufacturer specifies. The reaction was PCR thermocycled for 18 cycles followed by addition of 1 μL DpnI and 3 hours of incubation at 37°C to degrade the dam methylated original (non-mutated) plasmid strands. 2 μL reaction was transformed into 25 μL E. coli XL10 Gold competent cells and plasmids isolated with QIAprep Miniprep (Qiagen). A correctly mutated plasmid was subsequently validated by the absence of HindIII digestion.
The AurZ [FGDB: FGSG_02326] and AurJ [FGDB: FGSG_02325] protein sequences from F. graminearum PH1 were used for synthesizing the two gene ORFs with verification by sequencing and proprietary “GeneGPS” codon optimization for S. cerevisiae by DNA2.0. The A. fumigatus npgA gene [GenBank: XM_750100.1] was synthesized by DNA2.0 with codon-optimization for S. cerevisiae and verification by sequencing.
Cloning of ORFs in pRS vectors
A set of pRS vectors [31] were used in which a unique AscI site had been inserted between the two PvuII sites, thus replacing the original multi-cloning site. All ORFs were subcloned with HindIII and SacII into vectors containing the respective, constitutive promoters and terminators (specified in Table 1) to form vectors with gene cassettes. The PKS12 was provided with flanking HindIII and SacII restriction sites by PCR with iProof DNA polymerase using primers PKS12Fwd and PKS12Rev (see Additional file 1). All cassettes were subsequently cloned into the specified pRS plasmids (Table 1) with AscI. All ligations were performed using T4 DNA Ligase (New England Biolabs) and confirmed by sequencing (Microsynth). The pRS plasmids each harbour a unique auxotrophic marker gene (Table 1) and the ARS/CEN sequences for stable maintenance in S. cerevisiae at approx. one copy per cell, thus allowing strain-to-strain comparability. The PKS12 ORF was controlled by the inducible CUP1 promoter, which is induced from its low basal expression level by addition of Cu2+ to the medium [34].
Transformation of S. cerevisiae strains
The S. cerevisiae strains (Table 1) were constructed by transformation with the specified plasmids (Table 1) according to the protocol described by Gietz & Schiestl, 2007 [35]. Transformants were plated on selective synthetic complete (SC) agar plates lacking the appropriate supplements uracil, histidine, tryptophan and leucine.
Cultivation of the S. cerevisiae strains
The S. cerevisiae strains were grown for characterization on 2% agar plates with selective SC medium plus 500 μM CuSO4 for induction at 30°C. For chemical analysis, the strains were cultured by inoculating 5 mL selective SC medium from single colonies. These pre-cultures were incubated overnight at 30°C, with 160 rpm horizontal shaking. Main cultures of 10 mL selective SC medium, plus 300 μM CuSO4 for induction, were inoculated from the pre-cultures to an OD600 = 0.10 in three replicates of each pre-cultured strain. The main cultures were incubated for 96 hours at 30°C with 160 rpm horizontal shaking.
Cultivation of reference F. graminearum strains
The reference F. graminearum strains ΔaurZ, ΔaurJ and ΔaurF were grown on Potato Dextrose Agar (Difco) plates at 20°C for 7 days.
Chemical extraction and LC-MS analysis
Cells and growth medium of the main cultures were separated by centrifugation. The cells were resuspended in 1 mL dH2O and extracted by vigorous shaking with 5 mL extraction solvent (methanol:dichloromethane:ethyl acetate (1:2:3) + 1% formic acid). The mixtures were sonicated for 1 hour in an ultrasonic bath, centrifuged, filtered through filter paper and dried in a SpeedVac. Eight mL of the growth medium were extracted by vigorous shaking with 10 mL extraction solvent after which the organic phases were dried in a SpeedVac. Three 10 mm plugs were cut out of the mycelium of each reference F. graminearum strain. The plugs of each replicate were mashed in 10 mL ethyl acetate and shaken vigorously to facilitate extraction. The organic phase was recovered following centrifugation and filtering, at which point it was dried in a SpeedVac. The extractions from the F. graminearum reference strains were performed in triplicates.
All samples were reconstituted in 150 μL dimethyl sulfoxide. Five μL was injected and analyzed by LC-UV/ELSD on a Waters Acquity UPLC (Waters Corporation, Milfords, MA, USA), running a gradient of mobile phases A (water + 0.1% formic acid) and B (acetonitrile +0.1% formic acid), going from 1 to 100% B in 15 min, flow rate of 0.4 mL/min. The column was a Waters Acquity BEH C18 (2.1 mm × 100 mm × 1.7 μm (Waters Corporation, Milfords, MA, USA) temperature-controlled at 35°C. Liquid chromatography was coupled to a Bruker Daltronics microQTOF II (Bruker Daltonics, Bremen, Germany) using electrospray ionization source in positive mode: End plate Offset: -500 V, Capillary voltage: 4500 V, Nebulizer: 1.6 Bar, Dry Gas: 8.0 L/min, Dry temperature: 180°C. Extracted ion chromatograms for YWA1 (m/z 277.0707 ± 0.01), nor-rubrofusarin (m/z 259.0601 ± 0.01) and rubrofusarin (m/z 273.0753 ± 0.01) were created using the Bruker DataAnalysis 4.0 software. The integration of the peaks was performed with the DataAnalysis Integrate functions (with default settings).
Quantification of rubrofusarin production
Cells and growth medium from 8 mL main culture were separated by centrifugation. The cell pellet was resuspended in 1 mL dH2O and 5 mL ethyl acetate. The cell pellet solution was sonicated for 30 min. in an ultrasonic bath followed by vigorous shaking. The growth medium supernatant was shaken vigorously with 8 mL ethyl acetate. The ethyl acetate phases of growth medium and cell pellet were collected individually and allowed to evaporate in a fume hood. The extracted compounds of growth medium and cell pellet were reconstituted in 1.5 mL and 1.0 mL methanol, respectively. A rubrofusarin standard, purified from F. graminearum, was purchased from BioViotica Naturstoffe GmbH (Liestal, Switzerland) and reconstituted to 1 mg/mL in dimethyl sulfoxide. A standard curve was prepared by dissolving the rubrofusarin standard in methanol in a range from 4 μg/mL to 10 μg/mL. Twenty μL of each standard concentration and the extracted samples were analyzed by HPLC using an Ascentis C18 column (particle size: 5 μm, length × I.D.: 15 cm × 4.6 mm) (Sigma-Aldrich) running a gradient of mobile phases C (10 mM ammonium formate pH 3.0) and D (acetonitrile) at a flow rate of 1 mL/min, starting at 25% D (0-3 min), going to 80% D (3-15 min), remaining at 80% D (15-16 min), going to 25% D (16-18 min) and remaining at 25% D (18-20 min).
Competing interests
PR, UM and RF declare no competing interests. MN is employed by Evolva SA.
Authors’ contributions
PR and MN planned the experimental procedures of the study. PR carried out the experiments, except for cloning the exons of PKS12, which RF did. PR analyzed and interpreted the data and wrote the manuscript together with RF, MN and UM. All authors read and approved the final manuscript.
Supplementary Material
Oligonucleotides, UV spectra and HPLC chromatograms.
Contributor Information
Peter Rugbjerg, Email: petru@biosustain.dtu.dk.
Michael Naesby, Email: michaeln@evolva.com.
Uffe H Mortensen, Email: um@bio.dtu.dk.
Rasmus JN Frandsen, Email: rasf@bio.dtu.dk.
Acknowledgements
The authors wish to thank Evolva SA for supplying the laboratory facilities and materials including chemical gene synthesis. For helpful technical assistance and guidance we especially wish to thank Anders Hansson, Christophe Folly and Thiamo Boussemghoune, all employed at Evolva. Further, we wish to thank Sailesh Malla for helpful advice regarding compound quantification. RF’s involvement was funded by the Danish Research Council for Independent Research — Technology and Production grant no. 09-069707 and Young Elite Researcher grant no. 09-076147.
References
- Weissman KJ, Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- Tao K, Du L, Sun X, Cai M, Zhu T, Zhou X, Gu Q, Zhang Y. Biosynthesis of aspergiolide A, a novel antitumor compound by a marine-derived fungus Aspergillus glaucus via the polyketide pathway. Tetrahedron Lett. 2009;50:1082–1085. doi: 10.1016/j.tetlet.2008.12.094. [DOI] [Google Scholar]
- Zhang YL, Ge HM, Zhao W, Dong H, Xu Q, Li SH, Li J, Zhang J, Song YC, Tan RX. Unprecedented Immunosuppressive Polyketides from Daldinia eschscholzii, a Mantis-Associated Fungus. Angew Chem. 2008;120:5907–5910. doi: 10.1002/ange.200801284. [DOI] [PubMed] [Google Scholar]
- Mapari SA, Meyer AS, Thrane U, Frisvad JC. Identification of potentially safe promising fungal cell factories for the production of polyketide natural food colorants using chemotaxonomic rationale. Microb Cell Fact. 2009;8:24. doi: 10.1186/1475-2859-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Lin T. Monascus Rice Products. Adv Food Nutr Res. 2007;53:123–159. doi: 10.1016/S1043-4526(07)53004-4. [DOI] [PubMed] [Google Scholar]
- Kealey J, Liu L, Santi D, Betlach M, Barr P. Production of a polyketide natural product in nonpolyketide-producing prokaryotic and eukaryotic hosts. Proc Natl Acad Sci USA. 1998;95:505–509. doi: 10.1073/pnas.95.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattanachaisaereekul S, Lantz AE, Nielsen ML, Andre S. Optimization of Heterologous Production of the Polyketide 6-MSA in Saccharomyces cerevisiae. Biotechnology. 2007;97:893–900. doi: 10.1002/bit.21286. [DOI] [PubMed] [Google Scholar]
- Wattanachaisaereekul S, Lantz AE, Nielsen ML, Nielsen J. Production of the polyketide 6-MSA in yeast engineered for increased malonyl-CoA supply. Metab Eng. 2008;10:246–254. doi: 10.1016/j.ymben.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Zhou H, Qiao K, Gao Z, Vederas JC, Tang Y. Insights into radicicol biosynthesis via heterologous synthesis of intermediates and analogs. J Biol Chem. 2010;285:41412–41421. doi: 10.1074/jbc.M110.183574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Gao Z, Qiao K, Wang J, Vederas JC, Tang Y. A fungal ketoreductase domain that displays substrate-dependent stereospecificity. Nat Chem Biol. 2012;8:331–333. doi: 10.1038/nchembio.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiuchi K, Nakazawa T, Ookuma T, Sugimoto S, Sato M, Tsunematsu Y, Ishikawa N, Noguchi H, Hotta K, Moriya H, Watanabe K. Establishing a New Methodology for Genome Mining and Biosynthesis of Polyketides and Peptides through Yeast Molecular Genetics. Chem Bio Chem. 2012;13:846–854. doi: 10.1002/cbic.201100798. [DOI] [PubMed] [Google Scholar]
- Frandsen RJN, Nielsen NJ, Maolanon N, Sørensen JC, Olsson S, Nielsen J, Giese H. The biosynthetic pathway for aurofusarin in Fusarium graminearum reveals a close link between the naphthoquinones and naphthopyrones. Mol Microbiol. 2006;61:1069–1080. doi: 10.1111/j.1365-2958.2006.05295.x. [DOI] [PubMed] [Google Scholar]
- Ghosal S, Biswas K, Chakrabati D. Toxic naphtho-gamma-pyrones from Aspergillus niger. J Agric Food Chem. 1979;27:1347–1351. doi: 10.1021/jf60226a018. [DOI] [PubMed] [Google Scholar]
- Shibata S, Ohta A, Ogihara Y. The reactions of ustilaginoidin A. Bull: Chem. Pharm; 1963. [DOI] [PubMed] [Google Scholar]
- Branco A, Pinto AC, Braz-Filho R, Silva EF, Grynberg NF, Echevarria A. Rubrofusarin, a natural polyketide as new human topoisomerase II-α inhibitor. Revista Brasileira de Farmacognosia. 2008;18:703–708. [Google Scholar]
- Graham JG, Zhang H, Pendland SL, Santarsiero BD, Mesecar AD, Cabieses F, Farnsworth NR. Antimycobacterial Naphthopyrones from Senna obliqua. J Nat Prod. 2004;67:225–227. doi: 10.1021/np030348i. [DOI] [PubMed] [Google Scholar]
- Brown D, Hauser F, Tommasi R. Structural Elucidation of a Putative Conidial Pigment Intermediate in Aspergillus parasiticus. Tetrahedron Lett. 1993;34:5–8. [Google Scholar]
- Frandsen RJN, Schütt C, Lund BW, Staerk D, Nielsen J, Olsson S, Giese H. Two novel classes of enzymes are required for the biosynthesis of aurofusarin in Fusarium graminearum. J Biol Chem. 2011;286:10419–10428. doi: 10.1074/jbc.M110.179853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Sankawa U, Mayorga E, Timberlake WE, Ebizuka Y. Re-identification of Aspergillus nidulans wA Gene to Code for a Polyketide Synthase of Naphthopyrone. Tetrahedron Lett. 1999;40:91–94. doi: 10.1016/S0040-4039(98)80027-0. [DOI] [Google Scholar]
- Kim J, Han K, Jin J, Kim H, Kim J, Yun S, Lee Y. Putative Polyketide Synthase and Laccase Genes for Biosynthesis of Aurofusarin in Gibberella zeae. Appl Environ Microbiol. 2005;71:1701–1708. doi: 10.1128/AEM.71.4.1701-1708.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukibe K, Hashida K, Yoshida N, Takagi H. Metabolic engineering of Saccharomyces cerevisiae for astaxanthin production and oxidative stress tolerance. Appl Environ Microbiol. 2009;75:7205–7211. doi: 10.1128/AEM.01249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naesby M, Nielsen SV, Nielsen CA, Green T, Tange TO, Simón E, Knechtle P, Hansson A, Schwab MS, Titiz O, Folly C, Archila RE, Maver M, Van Sint Fiet S, Boussemghoune T, Janes M, SS Ka, Sonkar SP, Mitra PP, Benjamin VAK, Korrapati N, Suman I, Hansen EH, Thybo T, Goldsmith N, Sorensen AS. Yeast artificial chromosomes employed for random assembly of biosynthetic pathways and production of diverse compounds in Saccharomyces cerevisiae. Microb Cell Fact. 2009;8:45. doi: 10.1186/1475-2859-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii I, Watanabe A, Sankawa U, Ebizuka Y. Identification of Claisen cyclase domain in fungal polyketide synthase WA, a naphthopyrone synthase of Aspergillus nidulans. Chem Biol. 2001;8:189–197. doi: 10.1016/S1074-5521(00)90068-1. [DOI] [PubMed] [Google Scholar]
- Fujii I, Yasuoka Y, Tsai H-F, Chang YC, Kwon-Chung KJ, Ebizuka Y. Hydrolytic polyketide shortening by ayg1p, a novel enzyme involved in fungal melanin biosynthesis. J Biol Chem. 2004;279:44613–44620. doi: 10.1074/jbc.M406758200. [DOI] [PubMed] [Google Scholar]
- Chiang Y-M, Meyer KM, Praseuth M, Baker SE, Bruno KS, Wang CCC. Characterization of a polyketide synthase in Aspergillus niger whose product is a precursor for both dihydroxynaphthalene (DHN) melanin and naphtho-γ-pyrone. Fungal genetics and biology: FG & B. 2011;48:430–437. doi: 10.1016/j.fgb.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui MS, Thodey K, Trenchard I, Smolke CD. Advancing Secondary Metabolite Biosynthesis in Yeast with Synthetic Biology Tools. FEMS Yeast Res. 2011;12:144–170. doi: 10.1111/j.1567-1364.2011.00774.x. [DOI] [PubMed] [Google Scholar]
- Chiang Y-M, Chang S-L, Oakley BR, Wang CCC. Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr Opin Chem Biol. 2011;15:137–143. doi: 10.1016/j.cbpa.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok JW, Hoffmeister D, Maggio-Hall La, Murillo R, Glasner JD, Keller NP. Genomic mining for Aspergillus natural products. Chem Biol. 2006;13:31–37. doi: 10.1016/j.chembiol.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Geu-Flores F, Nour-Eldin HH, Nielsen MT, Halkier BA. USER fusion: a rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res. 2007;35:e55. doi: 10.1093/nar/gkm106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Walter M, Lee W, Mannhaupt G, Münsterkötter M, Mewes H-W, Adam G, Güldener U. FGDB: revisiting the genome annotation of the plant pathogen Fusarium graminearum. Nucleic Acids Res. 2011;39:D637–D639. doi: 10.1093/nar/gkq1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen BG, Salomonsen B, Nielsen MT, Nielsen JB, Hansen NB, Nielsen KF, Regueira TB, Nielsen J, Patil KR, Mortensen UH. Versatile enzyme expression and characterization system for Aspergillus nidulans, with the Penicillium brevicompactum polyketide synthase gene from the mycophenolic acid gene cluster as a test case. Appl Environ Microbiol. 2011;77:3044–3051. doi: 10.1128/AEM.01768-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen LR, Hansen NB, Bonde MT, Genee HJ, Holm DK, Carlsen S, Hansen BG, Patil KR, Mortensen UH, Wernersson R. PHUSER (Primer Help for USER): a novel tool for USER fusion primer design. Nucleic Acids Res. 2011;39(Suppl 2):W61–W67. doi: 10.1093/nar/gkr394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé S, Thiele D. Copper ion inducible and repressible promoter systems in yeast. Methods Enzymol. 1999;306:145–153. doi: 10.1016/s0076-6879(99)06010-3. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotides, UV spectra and HPLC chromatograms.