

Summary
The intracellular bacterial pathogen Coxiella burnetii is a category B select agent that causes human Q fever. In vivo, C. burnetii targets alveolar macrophages wherein the pathogen replicates in a lysosome-like parasitophorous vacuole (PV). In vitro, C. burnetii infects a variety of cultured cell lines that have collectively been used to model the pathogen’s infectious cycle. However, differences in the cellular response to infection have been observed, and virulent C. burnetii isolate infection of host cells has not been well defined. Because alveolar macrophages are routinely implicated in disease, we established primary human alveolar macrophages (hAMs) as an in vitro model of C. burnetii-host cell interactions. C. burnetii pathotypes, including acute disease and endocarditis isolates, replicated in hAMs, albeit with unique PV properties. Each isolate replicated in large, typical PV and small, non-fused vacuoles, and lipid droplets were present in avirulent C. burnetii PV. Interestingly, a subset of small vacuoles harbored single organisms undergoing degradation. Prototypical PV formation and bacterial growth in hAMs required a functional type IV secretion system, indicating C. burnetii secretes effector proteins that control macrophage functions. Avirulent C. burnetii promoted sustained activation of Akt and Erk1/2 pro-survival kinases and short term phosphorylation of stress-related p38. Avirulent organisms also triggered a robust, early pro-inflammatory response characterized by increased secretion of TNF-α and IL-6, while virulent isolates elicited substantially reduced secretion of these cytokines. A corresponding increase in pro- and mature IL-1β occurred in hAMs infected with avirulent C. burnetii, while little accumulation was observed following infection with virulent isolates. Finally, treatment of hAMs with IFN-γ controlled intracellular replication, supporting a role for this antibacterial insult in the host response to C. burnetii. Collectively, the current results demonstrate the hAM model is a human disease-relevant platform for defining novel innate immune responses to C. burnetii.
Introduction
Alveolar macrophages are the first line defense that confronts pulmonary bacterial pathogens following inhalation of contaminated aerosols. The ability of these cells to rapidly respond to an invading pathogen is central to mounting an effective antibacterial response during initial stages of infection. Upon confronting a pathogen, alveolar macrophages secrete distinct cytokines, including TNF-α, IL-1β, and IL-6, to recruit additional immune cells to the area, promoting pathogen disposal and development of adaptive immunity (Marriott et al., 2007). Following phagocytosis, bacteria-laden phagosomes mature through the phagolysosomal pathway, acquiring features including acid hydrolases, acidic pH, and reactive oxygen species, all of which are detrimental to the harbored organism (Flannagan et al., 2009). Not surprisingly, intracellular bacterial pathogens have evolved sophisticated mechanisms to avoid lysosomal degradation by alveolar macrophages and subvert the host immune response, allowing propagation of the bacterial population. For example, Mycobacterium tuberculosis is a highly prevalent human pathogen that alters macrophage phagolysosomal trafficking to prevent destruction by lysosomal proteases (Deretic et al., 2006).
Coxiella burnetii is a category B select agent that replicates within macrophages and causes human Q fever. C. burnetii has a worldwide distribution and diverse animal reservoir, with livestock serving as the primary source of bacterial spread to humans via contaminated aerosols inhaled while working with infected animals (Maurin et al., 1999). Most symptomatic cases present with acute disease characterized by high fever and pneumonia that can be readily treated with antibiotics (Gikas et al., 2010, Raoult et al., 2005). Persistent infections result from ~ 2% of acute cases and have a higher fatality rate (de Valk, 2012, Raoult et al., 1999), typically presenting as endocarditis in immunocompromised individuals and patients with heart valve conditions. A recent Q fever outbreak in the Netherlands resulted in over 4,000 acute Q fever cases, 250 estimated chronic cases, and 14 deaths to date (Forland et al., 2012, Limonard et al., 2010a, Limonard et al., 2010b, van der Hoek et al., 2012), underscoring our lack of understanding basic C. burnetii biology and virulence mechanisms.
C. burnetii has been isolated from a variety of geographic regions, hosts, and disease conditions since its discovery in the 1930s. C. burnetii isolates are generally placed into one of eight groups with distinct genomic characteristics and disease presentations (Beare et al., 2006). Group I organisms are typified by the Nine Mile I (NMI) reference isolate and typically cause acute disease, while groups IV and V (including the G isolate) are often associated with chronic diseases such as endocarditis. Group VI is composed of organisms collected from rodents in Dugway, UT that are severely attenuated in animal models (Stoenner et al., 1960, Stoenner et al., 1959). Most virulent C. burnetii isolates, including NMI, produce a full length lipopolysaccharide (LPS) and are referred to as being in phase I. However, Nine Mile II (NMII) is a clone derived from NMI C. burnetii that produces a truncated LPS referred to as being in phase II and does not cause disease. Importantly, NMII can infect cells in vitro, providing a model organism for use under biosafety level-2 conditions. Much debate has centered on whether C. burnetii genomic composition or host factors play a larger role in disease presentation (Samuel et al., 1985, Thiele et al., 1994). However, C. burnetii replication inside host cells is a determinant of disease caused by all isolates.
Following inhalation by a mammalian host, C. burnetii is engulfed by macrophages and promotes interactions with autophagosomes, fluid phase endosomes, and lysosomes to establish a host membrane-derived compartment in which to replicate (Voth et al., 2007a). C. burnetii metabolism is activated in the harsh, acidic environment of this parasitophorous vacuole (PV) that decorates with the lysosomal markers LAMP-1, -2, -3 and contains active proteases, including cathepsin D. While replicating in the PV, C. burnetii continuously directs fusion with other host cell compartments to provide membrane for PV expansion and inhibits apoptotic cell death to allow a prolonged infectious cycle (Howe et al., 2003, Vazquez et al., 2010, Voth et al., 2009b, Luhrmann et al., 2007, Voth et al., 2007b). To regulate infection, the pathogen uses a Dot/Icm type IV secretion system (T4SS) to inject effector proteins into the host cytosol (Voth et al., 2009a, Voth et al., 2011, Voth et al., 2009c, Pan et al., 2008, Chen et al., 2010, Luhrmann et al., 2010) and organisms deficient in T4SS-mediated secretion do not replicate in eukaryotic cells or prevent intrinsic apoptosis (Beare et al., 2011, Carey et al., 2011, Beare et al., 2012).
Many eukaryotic cell types, including epithelial cells, monocytes, and macrophages have been used to model the C. burnetii infectious process using NMI or NMII organisms (Voth et al., 2007a). However, nothing is known about the interaction between virulent C. burnetii isolates and their in vivo target, the human alveolar macrophage (hAM). Furthermore, cellular infection has not been addressed for other pathotypes, leaving a substantial void in our understanding of C. burnetii biology. Here, we assessed the intracellular properties of four C. burnetii pathotypes and found that all organisms replicated in primary hAMs with unique PV formation properties. We also determined the cytokine response to infection by each pathotype and assessed the role of IFN-γ in controlling replication. This study is the first to address cellular aspects of endocarditis isolate and naturally attenuated isolate infections and demonstrates the hAM is a human disease-relevant model for studying the unique C. burnetii intracellular lifestyle.
Results
Primary human alveolar macrophages support C. burnetii growth
No studies have addressed the interaction of C. burnetii with its in vivo human target cell, the alveolar macrophage. To address this gap in our understanding of this highly infectious pathogen, primary hAMs were harvested by bronchioalveolar lavage and incubated with one of four C. burnetii isolates (NMII, NMI, G, or Dugway) for 9 days. Every two days, infected cells were harvested, bacterial DNA isolated, and genome equivalents indicative of intracellular organisms determined. As shown in Fig. 1, all C. burnetii isolates productively infected and replicated in hAMs. G and Dugway isolate growth waned slightly through the first 3 days post-infection (dpi), followed by increased growth from 3–9 dpi, while NMII and NMI had similar growth curves indicating avirulent and virulent organisms are not efficiently degraded by hAMs and replicate in these cells similar to studies using primary human dendritic cells (Shannon et al., 2005).
Fig. 1. hAMs support growth of virulent C. burnetii pathotypes.

hAMs were infected with NMII, NMI, G, or Dugway for nine days. Cells were harvested and bacterial genomic DNA isolated every two days in triplicate. Bacterial genomes were quantified using quantitative PCR based on dotA using a DotA-encoding plasmid to generate a standard curve. Samples were analyzed in triplicate and error bars represent the standard deviation from the mean. C. burnetii isolates replicated efficiently in hAMs across a nine-day time course, with G and Dugway growth lagging until after three dpi.
C. burnetii generates a phagolysosome-like PV in hAMs
The hallmark of a C. burnetii infection at the cellular level is biogenesis of a spacious, phagolysosome-like PV required for bacterial growth. Following infection with NMII, NMI, G, or Dugway, hAMs were assessed for the presence of prototypical PV containing replicating organisms. As shown in Fig. 2A, all isolates formed a large vacuole that labeled with the late endosome/lysosome PV marker CD63. Additionally, each isolate replicated in a PV containing cathepsin D, which is indicative of an active lysosome (Fig. 2A and data not shown). These observations agree with recent results demonstrating that NMII and NMI replicate in human monocyte-derived macrophages in a functional lysosomal environment that degrades other bacteria (Howe et al., 2010). Electron microscopy analysis confirmed the presence of large PV in many NMII-infected hAMs similar to those observed in THP-1 human macrophage-like cells (Fig. 2B).
Fig. 2. Biogenesis of a large, spacious PV in hAMs is T4SS-dependent.

hAMs or THP-1 cells were infected for 72 h with the indicated isolate, then processed for fluorescence (A and C) or electron (B) microscopy. The PV markers CD63 or cathepsin D are indicated in the figure and shown in green, C. burnetii in red, and DAPI-stained DNA in blue. Isolates are indicated in panel A and PV and nuclei (N) are denoted in panel B. Each wild type C. burnetii isolate generates a large, phagolysosome-like PV in hAMs while T4SS-deficient organisms (icmD::Tn) are housed in tight-fitting phagolysosomes and fail to replicate (C). Furthermore, electron microscopy analysis (B) shows that large vacuoles in hAMs (left) contain many replicating NMII C. burnetii similar to that seen in THP-1 cells (right).
C. burnetii’s ability to form a replication-permissive PV in cultured cell lines requires a functional Dot/Icm T4SS and accompanying effector proteins (Beare et al., 2011, Carey et al., 2011, Beare et al., 2012). Therefore, we assessed whether the T4SS is needed for PV formation in hAMs. As seen in Fig. 2C, T4SS-deficient C. burnetii (icmD::Tn) was unable to generate a large PV and did not replicate in tight-fitting phagosomes, confirming the importance of the T4SS in primary hAMs.
C. burnetii PV display unique properties in hAMs
In each infected hAM culture, a subset of cells contained small CD63-positive phagosomes harboring individual organisms when observed by immunofluorescence microscopy (Fig. 3A). In cultured cell lines, C. burnetii is only present in a tight-fitting phagosome within the first 12 hpi, at which point the vacuole expands via heterotypic fusion with host compartments (Voth et al., 2007a). In contrast, ~ 50% of NMII-infected hAMs contained at least one large, spacious PV while ~ 50% contained only small phagosomes and no large PV (Fig. 3A). This composition differed substantially from NMII-infected THP-1 cells, where ~ 92% contained large PV and only ~ 8% harbored tight-fitting PV. Additionally, over 90% of hAMs harbored multiple PV of various sizes at 72 hpi (Fig. 3A), while only 20% of THP-1 cells contained multiple PV at this time. Similar phenotypes were observed during infection by NMI, G, and Dugway, with G replicating most efficiently in cells with multiple PV (data not shown). When monitored by electron microscopy, some tight-fitting hAM vacuoles contained a low number of organisms (~ 1–2/phagosome) in various states of degradation as evidenced by disrupted membranes (Fig. 3B).
Fig. 3. C. burnetii generates atypical PV in hAMs.

hAMs were infected for 72 h with NMII C. burnetii, then processed for fluorescence (A) or electron (B) microscopy. The PV marker CD63 is shown in green, C. burnetii in red, and DAPI-stained DNA in blue. Cells containing at least one typical, only atypical, or multiple PV were quantified from at least 250 NMII-infected cells and expressed as a percent of the total population. Samples were analyzed in triplicate and error bars represent the standard deviation from the mean. * indicates a P value < 0.0001 compared to infected THP-1 cells using a Student’s t test. Typical = > 15 μM, atypical = < 15 μM, and multiple = many PV of various sizes in each cell. Approximately 90% of hAMs harbor C. burnetii in multiple phagolysosome-like PV, many that support replication and some of which contain degrading organisms (arrowheads in panel B).
Electron microscopy revealed an unexpected aspect of C. burnetii-host cell interactions, with many PV containing lipid droplets (Fig. 4A). Lipid droplets play a role in cellular infection by intracellular pathogens, including Salmonella spp., M. tuberculosis, and Chlamydia trachomatis (Peyron et al., 2008, Arena et al., 2011, Cocchiaro et al., 2008, Saka et al., 2012, Kumar et al., 2006), but have not previously been reported during C. burnetii infection of other eukaryotic cells. Electron microscopy was confirmed by fluorescence microscopy using the lipid-specific probe BODIPY 493/503, which showed lipid droplets present within the PV at 72 hpi (Fig. 4B), a time when the vacuole is expanding to accommodate replicating organisms. Furthermore, fluorescence intensity analysis confirmed that lipid droplets are harbored within intact PV and some associate with C. burnetii. Thus, lipid droplets represent a novel area of C. burnetii research for future exploration.
Fig. 4. Lipid droplets associate with the PV.

hAMs were infected for 72 h with NMII C. burnetii, then processed for electron (A) or fluorescence microscopy (B). The neutral lipid stain BODIPY 493/503 (green) was used to label lipid droplets, C. burnetii is shown in violet, CD63 is shown in red, and DAPI-stained DNA in blue. L = lipid droplet and arrowheads denote the intact PV membrane. The intensity profile (bottom panel) depicts fluorescence along the white arrow drawn across the PV. In the intensity profile, white arrows indicate the CD63-positive limiting membrane of the PV on each side of a typical vacuole. Lipid droplets are found in and near the PV at 72 hpi and associate with C. burnetii.
C. burnetii modulates hAM pro-survival signaling pathways
Previous studies demonstrated the importance of intracellular signaling cascade activation for C. burnetii PV formation and prevention of apoptosis (Hussain et al., 2010, Voth et al., 2009b, Hussain et al., 2012, MacDonald et al., 2012). The organism inhibits apoptosis by activating kinases such as Akt and Erk1/2, promoting an anti-apoptotic transcriptional program, and preventing cytochrome c release from mitochondria (Voth et al., 2007b, Luhrmann et al., 2007, Luhrmann et al., 2010). To assess the potential in vivo role of host kinase signaling, we probed infected hAMs for phosphorylation (activation) of the cell survival mediators Akt and Erk1/2 and the stress response kinase p38, which have been previously shown in the THP-1 macrophage-like cell line (Voth et al., 2009b). During NMII growth in hAMs, increased phosphorylation of Akt and Erk1/2 occurred from 2–72 hpi (Fig. 5), correlating with phosphorylation profiles in infected THP-1 cells and highlighting the importance of prolonged kinase activation for C. burnetii infection. In contrast, p38 phosphorylation increased substantially at early times post-infection (2–24 hpi) with each isolate, then decreased to near uninfected cell levels by 72–96 hpi, suggesting p38 activation is an early hAM response to C. burnetii.
Fig. 5. C. burnetii modulates intracellular hAM signaling.

hAMs were infected with NMII C. burnetii for the indicated times and whole cell lysates analyzed by immunoblot using antibodies specific for the indicated proteins. C = control, uninfected cells and p = phosphorylated. C. burnetii triggers sustained phosphorylation (indicative of activation) of the pro-survival kinases Akt and Erk1/2 and short term phosphorylation of the stress response protein p38.
hAMs mount a robust inflammatory response to C. burnetii
In a pulmonary innate immune response, hAMs must confront an invading pathogen, traffic the organism to a phagolysosome for degradation, and produce appropriate cytokines to recruit other immune cells, promoting development of an adaptive immune response (Marriott et al., 2007). Although hAMs are unable to efficiently degrade C. burnetii in a phagolysosome, they are predicted to mount a cytokine response to recruit immune cells to the alveoli. Therefore, we monitored secretion of cytokines from hAMs exposed to C. burnetii pathotypes that are likely critical for clearing infection in the host (Fig. 6A). Each isolate elicited increased secretion of TNF-α at 24 hpi; however, the TNF-α response to avirulent C. burnetii was more robust than the response to NMI, G, or Dugway. These results correspond to those reported during C. burnetii infection of primary human dendritic cells (Shannon et al., 2005). Similar to TNF-α, levels of secreted IL-6 increased substantially during infection with each isolate at 24 hpi. IL-10 levels also increased in response to each isolate, representing a potential anti-inflammatory event promoted by the pathogen to establish a persistent infection (Meghari et al., 2008, Honstettre et al., 2003). In contrast to TNF-α, IL-6, and IL-10, IL-1β only increased substantially during NMII infection (Fig. 6B). Using immunoblot analysis, we observed that levels of pro- and mature IL-1β increased substantially above uninfected levels by 24 hpi with NMII organisms and remained elevated through 72 hpi, indicating hAMs respond to replicating intracellular avirulent C. burnetii by activating IL-1β-dependent signaling. Interestingly, pro- and mature IL-1β were undetectable after 24 hpi by NMI, G, or Dugway, suggesting organisms in phase I with full length LPS do not provide the triggers necessary to induce an appropriate inflammatory IL-1β response.
Fig. 6. hAMs mount a robust cytokine response to C. burnetii.

(A) hAMs were infected for 24 h with NMII, NMI, G, or Dugway and supernatants analyzed with a multiplex cytokine assay. Results are from one experiment with infections performed in duplicate and are representative of three independent experiments using hAMs from three different donors. Error bars represent the standard error of the mean and Mock = uninfected cells. hAMs respond to each isolate with increased production of TNF-α, IL-6, and IL-10 at 24 hpi, with levels consistently higher in NMII-infected hAMs. (B) Cells were infected for the indicated times with the isolates shown, then harvested for immunoblot analysis. C = control, uninfected cells and + = uninfected cells treated with E. coli LPS and ATP. Pro and mature IL-1β production was greatest in NMII-infected hAMs and was sustained from 24–72 hpi. Phase I isolates triggered production of low pro- and mature IL1β levels at 24 hpi and levels were undetectable from 48–72 hpi.
IFN-γ-mediated control of C. burnetii replication
In vivo, IFN-γ stimulates macrophage antibacterial responses such as production of reactive oxygen species that promote clearance of intracellular pathogens. IFN-γ is an integral component of the immune response to C. burnetii in animal models (Andoh et al., 2007), implicating its importance in humans. Therefore, we monitored C. burnetii NMII infection of hAMs in the presence of IFN-γ. hAMs were either pre-treated with IFN-γ for 24 h or treated at the time of infection as previously reported (Ghigo et al., 2002, Dellacasagrande et al., 2002) to determine the impact of the cytokine on C. burnetii growth. As seen in Fig. 7A, each treatment condition significantly decreased total infectious C. burnetii numbers as determined by a standard foci forming unit assay. Interestingly, many IFN-γ-treated cells allowed large PV formation, but limited C. burnetii replication in these vacuoles (data not shown). These results underscore the importance of IFN-γ signaling in controlling C. burnetii infection. However, infected hAMs treated with IFN-γ did not undergo high levels of apoptosis to destroy intracellular bacteria (Fig. 7B), as previously reported for C. burnetii-infected THP-1 cells (Dellacasagrande et al., 2002, Dellacasagrande et al., 1999).
Fig. 7. IFN-γ treatment controls C. burnetii growth in hAMs.
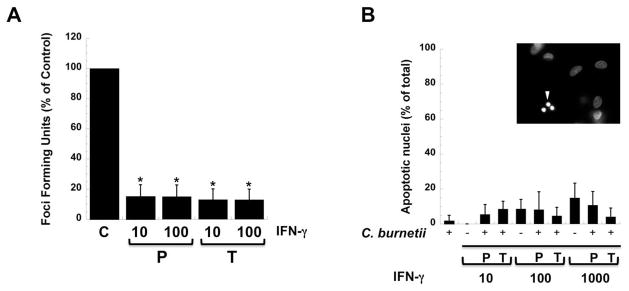
hAMs were either pre-treated for 24 h or treated at the time of infection with NMII C. burnetii with the indicated concentrations of IFN-γ (U/ml) and incubated for 72 h. (A) Infectious bacteria were assessed by plating hAM lysates on Vero cell monolayers and quantifying FFU/ml. Experiments were performed in triplicate and results are expressed as % of control (C) untreated infected cell FFUs. Error bars represent the standard deviation from the mean. P = 24 h pre-treatment with IFN-γ and T = treatment at time of infection. * indicates a P value < 0.0001 compared to untreated cells using a Student’s t test. IFN-γ treatment before or at the time of infection significantly lowers the number of infectious C. burnetii. (B) hAM viability was assessed by quantifying apoptotic nuclei using DAPI staining after treatment with IFN-γ at the indicated concentrations. Inset shows infected cells treated with 1000 U/ml IFN-γ and arrowhead denotes a fragmented nucleus indicative of an apoptotic cell. Samples were analyzed in triplicate and error bars represent the standard deviation from the mean. P values were > 0.05 for all conditions compared to uninfected treated cells using a Student’s t test. IFN-γ controls C. burnetii replication without signifcantly affecting hAM viability.
Discussion
Here, we present the first in vitro study demonstrating that primary hAMs are suitable hosts for replication and study of the highly infectious intracellular bacterial pathogen C. burnetii. The macrophage has been accepted for many years as the pathogen’s in vivo target in humans, but no cellular studies of this unique host-pathogen interaction have been reported. Alternatively, a collection of cultured cell lines and primary cells have been used to provide a wealth of relevant information about the infectious process (Voth et al., 2007a), with monkey alveolar macrophages and human monocyte-derived macrophages serving as the models most similar to the in vivo human condition studied to date (Voth et al., 2007b, Howe et al., 2010). Animal studies consistently report high numbers of C. burnetii in alveolar macrophages (Khavkin et al., 1988, Russell-Lodrigue et al., 2009, Russell-Lodrigue et al., 2006), making the primary hAM a disease-relevant model for probing C. burnetii mechanisms of host cell parasitism. While our hAM results correspond in some respects with those derived using cultured cell lines, we present new evidence of non-fused, atypical PV harboring replicating C. burnetii, lipid droplet recruitment to the PV, and distinct cytokine responses to infection with four different pathotypes. These results suggest hAMs can be used as a valuable model to uncover novel host-pathogen interactions.
hAMs are an unquestionably critical innate defense against pulmonary pathogens. Arguably the most prevalent pulmonary intracellular pathogen, M. tuberculosis, prevents its own destruction in lysosomes, allowing replication in hAMs (Deretic et al., 2006, Clemens et al., 2000, Via et al., 1997, Clemens et al., 1995). To avoid destruction, M. tuberculosis prevents phagolysosome formation by stalling phagosome maturation at the stage of an early phagosome that decorates with Rab5 but fails to recruit LAMPs. However, cell surface interactions with M. tuberculosis elicit an inflammatory cytokine response characterized by secretion of TNF-α and IL-6 (Yang et al., 2007). Legionella pneumophila, the causative agent of Legionnaires’ disease that is closely related to C. burnetii, infects alveolar macrophages and replicates in an endoplasmic reticulum-derived vacuole that does not fuse with lysosomes (Joshi et al., 2001, Clemens et al., 2000). L. pneumophila deploys an arsenal of secreted Dot/Icm T4SS effector proteins to establish its intracellular niche and control host signaling (Hubber et al., 2010). A functional C. burnetii Dot/Icm T4SS is also required for generation of large PV that contain lysosomal cathepsin D in hAMs, as organisms with disrupted icmD are only found in tight-fitting phagosomes and do not replicate. Intracellular bacterial pathogens must also combat the action of IFN-γ and reactive oxygen species (Katti et al., 2008, Yang et al., 2007, Nathan et al., 2000), which comprise a primary hAM defense mechanism. C. burnetii antagonizes production of reactive oxygen species in vitro (Hill et al., 2011, Siemsen et al., 2009) and this response is likely necessary for establishing a productive hAM infection. Additionally, IFN-γ efficiently decreased bacterial replication in hAMs, highlighting the importance of this cytokine in controlling C. burnetii infection.
An early mouse infection study demonstrated the presence of C. burnetii within alveolar macrophages following intranasal challenge (Khavkin et al., 1988). Following this report, other researchers compared differing routes of animal infection and found intranasal or aerosol delivery to be most representative of natural human exposure (Russell-Lodrigue et al., 2009, Russell-Lodrigue et al., 2006, La Scola et al., 1997). Together, these studies support a major role for alveolar macrophages in the initial stages of Q fever. Khavkin and Tabibzadeh also reported that murine alveolar macrophages had the capacity to degrade C. burnetii (Khavkin et al., 1988). However, virulent C. burnetii replicates efficiently in hAMs in lysosome-like PV similar to cultured cell lines such as THP-1 human macrophage-like cells and primary human monocyte-derived macrophages (Howe et al., 2010), suggesting a large percentage of bacteria survive macrophage antibacterial responses. C. burnetii anti-apoptotic activity was originally identified in monkey alveolar macrophages that also support replication, providing evidence of the in vivo importance of preventing macrophage death (Voth et al., 2007b). Likewise, our preliminary studies demonstrate that apoptosis prevention is involved in C. burnetii infection of hAMs, with virulent organisms preventing staurosporine-induced death (MacDonald et al., unpublished results).
Two novel aspects of infection uncovered in this study are the presence of infected cells with multiple atypical PV and trafficking of lipid droplets into the PV. In most cells, C. burnetii forms and maintains a large, spacious PV that fills with hundreds of bacteria (Voth et al., 2007a). It is uncommon to observe cells with multiple PV containing replicating organisms, particularly with the low inocula used in this study. The C. burnetii Priscilla isolate was originally thought to replicate in numerous individual PV in murine cells, but further studies confirmed the presence of one, multi-lobed PV (Hechemy et al., 1993). Compared to our established THP-1 infection model, ~ 70% more hAMs contain numerous PV and many of these provide a suitable replication environment. Each isolate replicates in cells with multiple PV, suggesting this phenotype is representative of an in vivo scenario. It is currently unknown if all tight-fitting PV in hAMs harbor viable bacteria, as some small vacuoles contain only one or two bacteria. These small phagosomes may possess more efficient lysosomal capacity that aids degradation of C. burnetii. Indeed, C. burnetii encased in some small vacuoles contain membranes in various states of degradation. Therefore, it appears that some hAM phagosomes proficiently degrade C. burnetii, and the composition of these small vacuoles is under investigation. In support of these findings, a population of L. pneumophila is killed by hAMs, with surviving bacteria replicating to high numbers (Jacobs et al., 1984).
An intriguing finding in C. burnetii-infected hAMs is the presence of lipid droplets within and adjacent to the PV. Lipid droplets contain sterols and triacylglycerols (Tauchi-Sato et al., 2002) and are involved in eukaryotic cell processes such as vesicular fusion and signaling (Saka et al., 2012), two aspects of cell biology central to intracellular pathogen infections. Lipid droplets associated with C. trachomatis replication vacuoles are predicted to influence chlamydial development (Cocchiaro et al., 2008, Kumar et al., 2006). Although the mechanistic role of these components in chlamydial infection is unknown, they potentially regulate cholesterol transport, membrane traffic, and signaling, events that are also involved in C. burnetii manipulation of host cells. C. burnetii exploits host cholesterol metabolism during intracellular growth, triggering increased cholesterol biosynthesis, and the PV decorates with the sterol probe filipin (Howe et al., 2006). Additionally, the pathogen encodes a sterol reductase predicted to process cholesterol precursors (Gilk et al., 2010). C. trachomatis produces and secretes at least three proteins that associate with, and possibly recruit, lipid droplets (Kumar et al., 2006). Thus, C. burnetii may devote a subset of Dot/Icm substrates to recruitment of lipid bodies during infection.
There is currently a void in C. burnetii research regarding host cell interactions with non-NM isolates, including chronic disease pathotypes and severely attenuated Dugway isolates obtained during studies in the 1950s in Utah (Sidwell et al., 1964, Stoenner et al., 1960). Although virulence in humans is unknown, Dugway is highly attenuated in hamsters, mice, and guinea pigs (Stoenner et al., 1960). Russell-Lodrigue et al. recently showed that Dugway organisms do not infect wild type mice as efficiently as NMI bacteria and cause no detectable fever in guinea pigs, but are able to infect and replicate in immunodeficient SCID mice, causing severe splenomegaly (Russell-Lodrigue et al., 2009). However, no studies have been performed at the cellular level to determine if Dugway attenuation results from an inability to grow in macrophages. Interestingly, growth of Dugway organisms lags slightly in hAMs between 1 and 3 dpi. However, after 3 dpi, Dugway numbers increase through 9 dpi and organisms replicate in hAMs in a lysosome-like PV that decorates with CD63 and cathepsin D. These results indicate that Dugway can replicate in macrophages and attenuation in vivo likely requires an intact immune system.
Alveolar macrophages mount an inflammatory cytokine response upon confrontation with a pathogen to recruit other immune cells. The cytokine response to NMI C. burnetii is typified by production of inflammatory TNF-α and IL-6 in host serum and at the cellular level (Shannon et al., 2009, Russell-Lodrigue et al., 2009), while production of IL-10 may regulate persistent infection (Meghari et al., 2008, Honstettre et al., 2003). Indeed, alveolar macrophages can produce high levels of these cytokines in response to bacteria (Marriott et al., 2007). hAMs demonstrate a robust inflammatory response to NMII organisms at 24 hpi as evidenced by dramatic increases in secreted TNF-α and IL-6. hAMs respond similarly to NMI, G, and Dugway, but overall cytokine levels are substantially reduced compared to NMII, suggesting organisms with phase I LPS activate a less intense inflammatory immune response. This hypothesis is supported by the observation that NMI LPS shields immune mediators on the bacterial cell surface and prevents activation of toll-like receptor 4 signaling (Shannon et al., 2005). Surprisingly, hAMs do not produce high levels of IL-10 in response to virulent C. burnetii as expected based on previous reports supporting an in vivo role for the cytokine (Meghari et al., 2008, Meghari et al., 2006, Honstettre et al., 2003). However, the current study only assessed cytokine secretion at 24 hpi, representing an early stage in the C. burnetii infectious cycle. Additionally, this study only monitored hAM supernatants and not whole host serum levels. Thus, non-macrophage cells may serve as the major source of IL-10 during active disease.
This is the first study to report production and secretion of mature IL-1βduring C. burnetii infection of macrophages. IL-1β is an inflammatory mediator produced by alveolar macrophages in response to pathogenic situations (Descamps et al., 2012). Following stimulation by a pathogen-associated molecular pattern such as LPS, pro-IL-1β is produced and subsequently processed to mature IL-1β by caspase-1 after receiving a second stimulus that activates the inflammasome. Mature IL-1β is then secreted from the cell. hAMs respond to NMII with prolonged production of pro- and mature IL-1β from 24–72 hpi, supporting a role for the cytokine in mediating the innate immune response to avirulent organisms. Interestingly, much lower levels of pro- and mature IL-1β are observed at 24 hpi with phase I C. burnetii isolates and are undetectable from 48–72 hpi, suggesting virulent organisms either combat or fail to fully trigger this inflammatory response. The inflammasome is activated in response to a variety of pathogen products in the cytoplasm and is inhibited by several secreted bacterial proteins (Barker et al., 2011). For example, L. pneumophila flagellin triggers activation of the inflammasome, leading to clearance of the organism from wild type mice (Case et al., 2009), while M. tuberculosis Zmp1 prevents caspase-1 activation and subsequent IL-1β secretion (Master et al., 2008). Although C. burnetii does not produce flagellin, NMII LPS may trigger inflammasome activity during phagocytic uptake by hAMs. Once inside the cell, secreted bacterial proteins are candidates for cytosolic detection, and hAMs may detect C. burnetii T4SS effectors, resulting in inflammasome activation and mature IL-1β production. However, if phase I isolate LPS provides a weak trigger of pro-IL-1β production, only low levels of mature IL-1β would be produced in response to T4SS effectors, allowing virulent organisms to persist in the absence of this critical inflammatory response. This intriguing scenario is supported by our hAM results and is currently under further investigation.
Collectively, the current results indicate the hAM is a disease-relevant model of C. burnetii-host interactions that can be used to test hypotheses about in vivo infection events at the cellular level, particularly those investigating the innate immune response to infection. The hAM model is particularly relevant in regard to virulent C. burnetii isolates associated with differing forms of Q fever. Furthermore, hAMs support replication of the Dugway isolate that is severely attenuated in animal models. Numerous studies support a major role for LPS in the immune response to C. burnetii, with NMII truncated LPS stimulating an immune response that clears the organism from infected animals. However, Dugway produces phase I LPS, suggesting a more complex host immune response to attenuated organisms. Finally, hAMs can be used to probe shared and distinct parasitic mechanisms employed by C. burnetii and related pulmonary pathogens. For example, little is known about growth of the macrophage pathogens L. pneumophila and Chlamydia pneumoniae in their in vivo target cell. Interestingly, an early report showed that > 97% of L. pneumophila is destroyed by hAMs within 30 minutes of uptake, with remaining bacteria surviving, replicating, and eventually killing hAMs (Jacobs et al., 1984). Similarly, C. pneumoniae growth is restricted in primary murine alveolar macrophages compared to cell lines (Redecke et al., 1998, Haranaga et al., 2003). Together with our C. burnetii results, these studies indicate alveolar macrophages elicit distinct responses to related intracellular pathogens and should not be neglected for validating cellular studies of the innate immune response.
Experimental Procedures
C. burnetii and mammalian cell culture
C. burnetii Nine Mile II (RSA439), Nine Mile I (RSA493), G (Q212), and Dugway (7E65-68) were cultured in acidified citrate cysteine medium (ACCM) for 7 days at 37°C in 5% CO2 and 2.5% O2 (Omsland et al., 2009). Isolates were collected by centrifugation and washed with sucrose phosphate buffer prior to use. Work with virulent C. burnetii isolates was performed in the UAMS biosafety level-3 laboratory that is approved by the Centers for Disease Control and Prevention.
Vero (Green monkey kidney) cells and human monocytic THP-1 cells (American Type Culture Collection) were cultured in RPMI1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) at 37°C in 5% CO2. THP-1 cells were incubated with 200 nM phorbol 12-myristate 13-acetate (PMA; EMD Biosciences) overnight to promote differentiation into macrophage-like cells (Voth et al., 2007b). Prior to infection, medium containing PMA was replaced with PMA-free medium. Primary human alveolar macrophages were isolated by bronchioalveolar lavage with phosphate buffered saline (PBS) from lung tissue obtained postmortem from the National Disease Research Interchange. No donors were excluded unless obvious contamination, indicative of infection, was observed after culturing macrophages. All experiments were performed with at least two different donor macrophage populations. Cells were collected from lavage fluid by centrifugation at 1,000 rpm for 5 min. Red blood cells were lysed by resuspending cells in 0.0084% ammonium chloride for 10 min. Ammonium chloride was then neutralized by addition of medium and removed from cells by centrifugation. Cells were then resuspended in Dulbecco’s modified Eagle medium/F-12 medium containing 10% FBS, penicillin (50 U/mL), streptomycin sulfate (50 μg/mL), gentamicin sulfate (50 μg/mL), and amphotericin B (0.25 μg/mL) (Invitrogen) and plated at 4 × 105 cells/well in 24-well plates, or 1.5 × 106 cells/well in 6-well plates. Macrophages were allowed to adhere to plates for 2 h at 37°C in 5% CO2, then non-adherent cells were removed and remaining cells washed with fresh medium. hAMs were routinely assessed for homogeneity by immunofluorescence and immunoblot analysis (Fig. S1) with antibodies directed against the macrophage-specific markers CD11/18b, CD14, and CD68 (Abcam). Lavage cultures typically contained > 90% macrophages. hAMs and THP-1 cells were infected with C. burnetii isolates at a multiplicity of infection (MOI) of ~ 10 in the absence of gentamicin, penicillin, streptomycin, and amphotericin B.
Growth curve analysis
hAMs were infected and samples collected for DNA analysis at 1, 3, 5, 7, and 9 dpi. Cells were harvested by scraping in 1 mL of medium, and DNA was isolated using a bacterial DNA extraction kit according to the manufacturer’s instructions (MO BIO Laboratories, Inc.). Genomes were quantified using quantitative real time PCR and primers (DotA-F 5′-GCG CAA TAC GCT CAA TCA CA-3′ and DotA-R 5′-CCA TGG CCC CAA TTC TCT T-3′; Integrated DNA Technologies) that recognize C. burnetii dotA, and calculated as previously described (Samuel et al., 2009). Platinum Taq and SYBR green (Invitrogen) were used to amplify and detect DNA with an Applied Biosystems 7300 Real Time PCR cycler.
Fluorescence microscopy
Following infection of hAMs or THP-1 cells, cells were fixed and permeabilized with 100% ice-cold methanol for 3 min, then blocked for 1 h in PBS containing 0.5% bovine serum albumin (BSA; Cell Signaling) at room temperature. Cells were then incubated with either mouse anti-CD63 (BD Biosciences), mouse anti-cathepsin D (Enzo Life Sciences), rabbit anti-C. burnetii, or guinea pig anti-C. burnetii primary antibodies for 1 h at room temperature. Cells were then washed 3 times with ice cold PBS and incubated in 0.5% BSA-PBS containing AlexaFluor-488, -594, or 647-conjugated secondary antibodies (Invitrogen) for 1 h at room temperature. Indicated samples were incubated with BODIPY 493/503 (Invitrogen) at a concentration of 1:500 for 30 min to detect lipid droplets. Cells were then incubated with DAPI (4′,6′–diamindino-2-phenylindole dilactate; Invitrogen) for 5 min at room temperature to detect host and bacterial DNA. Fluorescence microscopy was performed using a Ti-U microscope with a 60X oil immersion objective (Nikon). Images were obtained with a D5-QilMc digital camera and analyzed using NIS-Elements software (Nikon). PV and apoptotic nuclei were quantified from 5–10 fields containing ~ 25 cells/field.
Electron microscopy
hAMs or THP-1 cells were infected for 72 h, then washed with PBS. Cells were fixed on ice for 15 min in PBS containing 4% paraformaldehyde and 2.5% glutaraldehyde (Invitrogen). Cells were then gently scraped from plates and incubated in fixative for an additional 45 min at room temperature. Cells were then collected by centrifugation at 1000 rpm for 5 min and resuspended in PBS. Samples were subsequently processed and analyzed by Dr. Wandy Beatty at the Washington University School of Medicine Molecular Imaging Facility.
Immunoblot analysis
C. burnetii-infected hAMs were lysed in buffer containing 1% sodium dodecyl sulfate (SDS) by 10 passages through a 26-gauge needle and boiled for 5 minutes. Total protein concentration was determined using a DC assay (Bio-Rad). Ten μg of total protein was separated by SDS-polyacrylamide gel electrophoresis, transferred to a 0.2-μm-pore-size polyvinylidene fluoride membrane (Bio-Rad), and blocked in Tris-buffered saline (150 mM NaCl, 100 mM Tris-HCl [pH 7.6]) containing 0.1% Tween 20 and 5% nonfat milk for 1 h at room temperature. After blocking, membranes were probed for equal protein loading using a mouse anti-β-tubulin primary antibody (Sigma-Aldrich). After confirming equal loading, lysates were probed for total and phosphorylated p38, Erk1/2 (Thr202/Tyr204) or Akt (Ser473) (Cell Signaling), IL-1β (R & D Systems), or CD14, CD11/18b, or CD68 (Abcam). A positive control for mature IL-1β production was established by treating hAMs with 100 ng/mL of E. coli LPS for 6 h and 5 mM ATP (Adenosine 5′-triphosphate disodium salt hydrate; Sigma-Aldrich) for 30 minutes. In all immunoblots, reacting proteins were detected using anti-mouse or anti-rabbit secondary antibodies conjugated to horseradish peroxidase (Cell Signaling).
Multiplex cytokine analysis
TNF-α, IL-6, and IL-10 concentrations were determined using a multiplex Bio-Plex cytokine assay (Bio-Rad). Supernatants were harvested from E. coli LPS-treated or infected hAMs at 24 h and centrifuged at 14,000 rpm for 15 min to clear any precipitate. Standards and buffers were prepared according to the manufacturer’s instructions and equilibrated to room temperature for 20 min before use. A 96-well filter bottom plate was incubated with assay buffer prior to use and removed using a vacuum manifold. Magnetic coupled beads were prepared and added to all wells followed by addition of standards or samples to individual wells. Plates were shaken at 1,100 rpm for 30 sec, then at 300 rpm for 30 min at room temperature. Wells were then washed three times with wash buffer and detection antibodies added for 30 min. Plates were then washed and Streptavidin PE was added for 10 min. After washing, samples were analyzed using a Bio-Plex 200 system and cytokine concentrations calculated using Bio-Plex Manager 6.0 software.
IFN-γ treatment and foci forming unit assays
hAMs were left untreated, pretreated for 24 h, or treated at the time of infection with 10, 100, or 1000 U/mL IFN-γ (Invitrogen). Cells were then either mock-infected or infected with C. burnetii isolates for 72 h in the presence of IFN-γ. At 24 hpi, hAMs were washed to remove excess organisms and fresh IFN-γ added. At 72 hpi, cells were harvested and sonicated to release intracellular organisms and samples applied to Vero cell monolayers. Infected Vero cells were incubated for 96 h, then processed for fluorescence microscopy using an anti-C. burnetii primary antibody and an AlexaFluor-488-conjugated anti-rabbit secondary antibody (Invitrogen) to detect infectious foci. Foci-forming units (FFUs) were visualized using a 40X objective lens and quantified as FFU/mL as previously described (Cockrell et al., 2008).
Supplementary Material
Following alveolar lavage of lungs post-mortem, cells were allowed to adhere to tissue culture plates for 2h, then washed vigorously to remove non-adherent, non-macrophage cells. Adherent cells were processed for immunofluorescence microscopy and immunoblot using the macrophage-specific markers CD11/18b, CD14, and CD68. Arrowheads indicate non-macrophage cells. Greater than 90% of adherent cells were positive for all three markers by microscopy, indicating a high percentage of hAMs were present in lavage samples. hAMs also produced similar levels of CD14 and CD68 to THP-1 cells (immunoblot) and this macrophage marker was not detectable in HeLa cells (epithelial).
Acknowledgments
This research was supported by funding to D. E. V. from the NIH/NIAID (R01AI087669), the American Heart Association (BGIA3080001), and the Arkansas Biosciences Institute, and to R. C. K. from the UAMS Translational Research Institute (1UL1RR029884). J. G. G. was supported by the UAMS Initiative for Maximizing Student Development.
Footnotes
The authors have no conflicts of interest.
References
- Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun. 2007;75:3245–3255. doi: 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena ET, Auweter SD, Antunes LC, Vogl AW, Han J, Guttman JA, et al. The deubiquitinase activity of the Salmonella pathogenicity island 2 effector, SseL, prevents accumulation of cellular lipid droplets. Infect Immun. 2011;79:4392–4400. doi: 10.1128/IAI.05478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker BR, Taxman DJ, Ting JP. Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol. 2011;23:591–597. doi: 10.1016/j.coi.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, et al. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio. 2011;2:e00175–11. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, Larson CL, Gilk SD, Heinzen RA. Two systems for targeted gene deletion in Coxiella burnetii. Appl Environ Microbiol. 2012;78:4580–4589. doi: 10.1128/AEM.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, Samuel JE, Howe D, Virtaneva K, Porcella SF, Heinzen RA. Genetic diversity of the Q fever agent, Coxiella burnetii, assessed by microarray-based whole-genome comparisons. J Bacteriol. 2006;188:2309–2324. doi: 10.1128/JB.188.7.2309-2324.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey KL, Newton HJ, Luhrmann A, Roy CR. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 2011;7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case CL, Shin S, Roy CR. Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun. 2009;77:1981–1991. doi: 10.1128/IAI.01382-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, et al. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc Natl Acad Sci U S A. 2010;107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med. 1995;181:257–270. doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens DL, Lee BY, Horwitz MA. Mycobacterium tuberculosis and Legionella pneumophila phagosomes exhibit arrested maturation despite acquisition of Rab7. Infect Immun. 2000;68:5154–5166. doi: 10.1128/iai.68.9.5154-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci U S A. 2008;105:9379–9384. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockrell DC, Beare PA, Fischer ER, Howe D, Heinzen RA. A method for purifying obligate intracellular Coxiella burnetii that employs digitonin lysis of host cells. J Microbiol Methods. 2008;72:321–325. doi: 10.1016/j.mimet.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Valk H. Q fever: new insights, still many queries. Euro Surveill. 2012;17:20062. [PubMed] [Google Scholar]
- Dellacasagrande J, Capo C, Raoult D, Mege JL. IFN-gamma-mediated control of Coxiella burnetii survival in monocytes: the role of cell apoptosis and TNF. J Immunol. 1999;162:2259–2265. [PubMed] [Google Scholar]
- Dellacasagrande J, Ghigo E, Raoult D, Capo C, Mege JL. IFN-gamma-induced apoptosis and microbicidal activity in monocytes harboring the intracellular bacterium Coxiella burnetii require membrane TNF and homotypic cell adherence. J Immunol. 2002;169:6309–6315. doi: 10.4049/jimmunol.169.11.6309. [DOI] [PubMed] [Google Scholar]
- Deretic V, Singh S, Master S, Harris J, Roberts E, Kyei G, et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 2006;8:719–727. doi: 10.1111/j.1462-5822.2006.00705.x. [DOI] [PubMed] [Google Scholar]
- Descamps D, Le Gars M, Balloy V, Barbier D, Maschalidi S, Tohme M, et al. Toll-like receptor 5 (TLR5), IL-1beta secretion, and asparagine endopeptidase are critical factors for alveolar macrophage phagocytosis and bacterial killing. Proc Natl Acad Sci U S A. 2012;109:1619–1624. doi: 10.1073/pnas.1108464109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- Forland F, De Carvalho Gomes H, Nokleby H, Escriva A, Coulombier D, Giesecke J, Jansen A. Applicability of evidence-based practice in public health: risk assessment on Q fever under an ongoing outbreak. Euro Surveill. 2012;17:20060. [PubMed] [Google Scholar]
- Ghigo E, Capo C, Tung CH, Raoult D, Gorvel JP, Mege JL. Coxiella burnetii survival in THP-1 monocytes involves the impairment of phagosome maturation: IFN-gamma mediates its restoration and bacterial killing. J Immunol. 2002;169:4488–4495. doi: 10.4049/jimmunol.169.8.4488. [DOI] [PubMed] [Google Scholar]
- Gikas A, Kokkini S, Tsioutis C. Q fever: clinical manifestations and treatment. Expert Rev Anti Infect Ther. 2010;8:529–539. doi: 10.1586/eri.10.29. [DOI] [PubMed] [Google Scholar]
- Gilk SD, Beare PA, Heinzen RA. Coxiella burnetii expresses a functional Delta24 sterol reductase. J Bacteriol. 2010;192:6154–6159. doi: 10.1128/JB.00818-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haranaga S, Yamaguchi H, Ikejima H, Friedman H, Yamamoto Y. Chlamydia pneumoniae infection of alveolar macrophages: a model. J Infect Dis. 2003;187:1107–1115. doi: 10.1086/368168. [DOI] [PubMed] [Google Scholar]
- Hechemy KE, McKee M, Marko M, Samsonoff WA, Roman M, Baca O. Three-dimensional reconstruction of Coxiella burnetii-infected L929 cells by high-voltage electron microscopy. Infect Immun. 1993;61:4485–4488. doi: 10.1128/iai.61.10.4485-4488.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J, Samuel JE. Coxiella burnetii acid phosphatase inhibits the release of reactive oxygen intermediates in polymorphonuclear leukocytes. Infect Immun. 2011;79:414–420. doi: 10.1128/IAI.01011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honstettre A, Imbert G, Ghigo E, Gouriet F, Capo C, Raoult D, Mege JL. Dysregulation of cytokines in acute Q fever: role of interleukin-10 and tumor necrosis factor in chronic evolution of Q fever. J Infect Dis. 2003;187:956–962. doi: 10.1086/368129. [DOI] [PubMed] [Google Scholar]
- Howe D, Heinzen RA. Coxiella burnetii inhabits a cholesterol-rich vacuole and influences cellular cholesterol metabolism. Cell Microbiol. 2006;8:496–507. doi: 10.1111/j.1462-5822.2005.00641.x. [DOI] [PubMed] [Google Scholar]
- Howe D, Melnicakova J, Barak I, Heinzen RA. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell Microbiol. 2003;5:469–480. doi: 10.1046/j.1462-5822.2003.00293.x. [DOI] [PubMed] [Google Scholar]
- Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun. 2010;78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubber A, Roy CR. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol. 2010;26:261–283. doi: 10.1146/annurev-cellbio-100109-104034. [DOI] [PubMed] [Google Scholar]
- Hussain SK, Broederdorf LJ, Sharma UM, Voth DE. Host kinase activity is required for Coxiella burnetii parasitophorous vacuole formation. Front Microbio. 2010;1:137. doi: 10.3389/fmicb.2010.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SK, Voth DE. Coxiella subversion of intracellular host signaling. Adv Exp Med Biol. 2012;984:131–140. doi: 10.1007/978-94-007-4315-1_7. [DOI] [PubMed] [Google Scholar]
- Jacobs RF, Locksley RM, Wilson CB, Haas JE, Klebanoff SJ. Interaction of primate alveolar macrophages and Legionella pneumophila. J Clin Invest. 1984;73:1515–1523. doi: 10.1172/JCI111357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AD, Sturgill-Koszycki S, Swanson MS. Evidence that Dot-dependent and -independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell Microbiol. 2001;3:99–114. doi: 10.1046/j.1462-5822.2001.00093.x. [DOI] [PubMed] [Google Scholar]
- Katti MK, Dai G, Armitige LY, Rivera Marrero C, Daniel S, Singh CR, et al. The Delta fbpA mutant derived from Mycobacterium tuberculosis H37Rv has an enhanced susceptibility to intracellular antimicrobial oxidative mechanisms, undergoes limited phagosome maturation and activates macrophages and dendritic cells. Cell Microbiol. 2008;10:1286–1303. doi: 10.1111/j.1462-5822.2008.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavkin T, Tabibzadeh SS. Histologic, immunofluorescence, and electron microscopic study of infectious process in mouse lung after intranasal challenge with Coxiella burnetii. Infect Immun. 1988;56:1792–1799. doi: 10.1128/iai.56.7.1792-1799.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16:1646–1651. doi: 10.1016/j.cub.2006.06.060. [DOI] [PubMed] [Google Scholar]
- La Scola B, Lepidi H, Raoult D. Pathologic changes during acute Q fever: influence of the route of infection and inoculum size in infected guinea pigs. Infect Immun. 1997;65:2443–2447. doi: 10.1128/iai.65.6.2443-2447.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limonard GJ, Nabuurs-Franssen MH, Weers-Pothoff G, Wijkmans C, Besselink R, Horrevorts AM, et al. One-year follow-up of patients of the ongoing Dutch Q fever outbreak: clinical, serological and echocardiographic findings. Infection. 2010a;38:471–477. doi: 10.1007/s15010-010-0052-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limonard GJ, Peters JB, Nabuurs-Franssen MH, Weers-Pothoff G, Besselink R, Groot CA, et al. Detailed analysis of health status of Q fever patients 1 year after the first Dutch outbreak: a case-control study. Qjm. 2010b;103:953–958. doi: 10.1093/qjmed/hcq144. [DOI] [PubMed] [Google Scholar]
- Luhrmann A, Nogueira CV, Carey KL, Roy CR. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A. 2010;107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrmann A, Roy CR. Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect Immun. 2007;75:5282–5289. doi: 10.1128/IAI.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald LJ, Kurten RC, Voth DE. Coxiella burnetii alters cyclic AMP-dependent protein kinase signaling during growth in macrophages. Infect Immun. 2012;80:1980–1986. doi: 10.1128/IAI.00101-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott HM, Dockrell DH. The role of the macrophage in lung disease mediated by bacteria. Exp Lung Res. 2007;33:493–505. doi: 10.1080/01902140701756562. [DOI] [PubMed] [Google Scholar]
- Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–232. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin M, Raoult D. Q fever. Clin Microbiol Rev. 1999;12:518–553. doi: 10.1128/cmr.12.4.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghari S, Bechah Y, Capo C, Lepidi H, Raoult D, Murray PJ, Mege JL. Persistent Coxiella burnetii infection in mice overexpressing IL-10: an efficient model for chronic Q fever pathogenesis. PLoS Pathog. 2008;4:e23. doi: 10.1371/journal.ppat.0040023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghari S, Capo C, Raoult D, Mege JL. Deficient transendothelial migration of leukocytes in Q fever: the role played by interleukin-10. J Infect Dis. 2006;194:365–369. doi: 10.1086/505227. [DOI] [PubMed] [Google Scholar]
- Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Luhrmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science. 2008;320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008;4:e1000204. doi: 10.1371/journal.ppat.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med. 1999;159:167–173. doi: 10.1001/archinte.159.2.167. [DOI] [PubMed] [Google Scholar]
- Raoult D, Marrie T, Mege J. Natural history and pathophysiology of Q fever. Lancet Infect Dis. 2005;5:219–226. doi: 10.1016/S1473-3099(05)70052-9. [DOI] [PubMed] [Google Scholar]
- Redecke V, Dalhoff K, Bohnet S, Braun J, Maass M. Interaction of Chlamydia pneumoniae and human alveolar macrophages: infection and inflammatory response. Am J Respir Cell Mol Biol. 1998;19:721–727. doi: 10.1165/ajrcmb.19.5.3072. [DOI] [PubMed] [Google Scholar]
- Russell-Lodrigue KE, Andoh M, Poels MW, Shive HR, Weeks BR, Zhang GQ, et al. Coxiella burnetii isolates cause genogroup-specific virulence in mouse and guinea pig models of acute Q fever. Infect Immun. 2009;77:5640–5650. doi: 10.1128/IAI.00851-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell-Lodrigue KE, Zhang GQ, McMurray DN, Samuel JE. Clinical and pathologic changes in a guinea pig aerosol challenge model of acute Q fever. Infect Immun. 2006;74:6085–6091. doi: 10.1128/IAI.00763-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka HA, Valdivia RH. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol. 2012;28:411–437. doi: 10.1146/annurev-cellbio-092910-153958. [DOI] [PubMed] [Google Scholar]
- Samuel JE, Frazier ME, Mallavia LP. Correlation of plasmid type and disease caused by Coxiella burnetii. Infect Immun. 1985;49:775–779. doi: 10.1128/iai.49.3.775-779.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel JE, Hendrix LR. Laboratory maintenance of Coxiella burnetii. Curr Protoc Microbiol. 2009;Chapter 6(Unit6C):1. doi: 10.1002/9780471729259.mc06c01s15. [DOI] [PubMed] [Google Scholar]
- Shannon JG, Cockrell DC, Takahashi K, Stahl GL, Heinzen RA. Antibody-mediated immunity to the obligate intracellular bacterial pathogen Coxiella burnetii is Fc receptor- and complement-independent. BMC Immunol. 2009;10:26. doi: 10.1186/1471-2172-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon JG, Howe D, Heinzen RA. Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc Natl Acad Sci U S A. 2005;102:8722–8727. doi: 10.1073/pnas.0501863102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidwell RW, Lundgren DL, Bushman JB, Thorpe BD. The occurrence of a possible epizootic of Q Fever in fauna of the great salt lake desert of Utah. Am J Trop Med Hyg. 1964;13:754–762. doi: 10.4269/ajtmh.1964.13.754. [DOI] [PubMed] [Google Scholar]
- Siemsen DW, Kirpotina LN, Jutila MA, Quinn MT. Inhibition of the human neutrophil NADPH oxidase by Coxiella burnetii. Microbes Infect. 2009;11:671–679. doi: 10.1016/j.micinf.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoenner HG, Holdenried R, Lackman D, Orsborn JS., Jr The occurrence of Coxiella burnetii, Brucella, and other pathogens among fauna of the great salt lake desert in Utah. Am J Trop Med Hyg. 1959;8:590–596. doi: 10.4269/ajtmh.1959.8.590. [DOI] [PubMed] [Google Scholar]
- Stoenner HG, Lackman DB. The biologic properties of Coxiella burnetii isolated from rodents collected in Utah. Am J Hyg. 1960;71:45–51. doi: 10.1093/oxfordjournals.aje.a120088. [DOI] [PubMed] [Google Scholar]
- Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem. 2002;277:44507–44512. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- Thiele D, Willems H. Is plasmid based differentiation of Coxiella burnetii in ‘acute’ and ‘chronic’ isolates still valid? Eur J Epidemiol. 1994;10:427–434. doi: 10.1007/BF01719667. [DOI] [PubMed] [Google Scholar]
- van der Hoek W, Schneeberger PM, Oomen T, Wegdam-Blans MC, Dijkstra F, Notermans DW, et al. Shifting priorities in the aftermath of a Q fever epidemic in 2007 to 2009 in The Netherlands: from acute to chronic infection. Euro Surveill. 2012;17:20059. [PubMed] [Google Scholar]
- Vazquez CL, Colombo MI. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 2010;17:421–438. doi: 10.1038/cdd.2009.129. [DOI] [PubMed] [Google Scholar]
- Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem. 1997;272:13326–13331. doi: 10.1074/jbc.272.20.13326. [DOI] [PubMed] [Google Scholar]
- Voth DE, Beare PA, Howe D, Sharma UM, Samoilis G, Cockrell DC, et al. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J Bacteriol. 2011;193:1493–1503. doi: 10.1128/JB.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Heinzen RA. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol. 2007a;9:829–840. doi: 10.1111/j.1462-5822.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- Voth DE, Heinzen RA. Coxiella type IV secretion and cellular microbiology. Curr Opin Microbiol. 2009a;12:74–80. doi: 10.1016/j.mib.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Heinzen RA. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect Immun. 2009b;77:205–213. doi: 10.1128/IAI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Howe D, Beare PA, Vogel JP, Unsworth N, Samuel JE, Heinzen RA. The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm-mediated secretion. J Bacteriol. 2009c;191:4232–4242. doi: 10.1128/JB.01656-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Howe D, Heinzen RA. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun. 2007b;75:4263–4271. doi: 10.1128/IAI.00594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Lee HM, Lee JY, Kim JA, Lee SJ, Shin DM, et al. Reactive oxygen species and p47phox activation are essential for the Mycobacterium tuberculosis-induced pro-inflammatory response in murine microglia. J Neuroinflammation. 2007;4:27. doi: 10.1186/1742-2094-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Following alveolar lavage of lungs post-mortem, cells were allowed to adhere to tissue culture plates for 2h, then washed vigorously to remove non-adherent, non-macrophage cells. Adherent cells were processed for immunofluorescence microscopy and immunoblot using the macrophage-specific markers CD11/18b, CD14, and CD68. Arrowheads indicate non-macrophage cells. Greater than 90% of adherent cells were positive for all three markers by microscopy, indicating a high percentage of hAMs were present in lavage samples. hAMs also produced similar levels of CD14 and CD68 to THP-1 cells (immunoblot) and this macrophage marker was not detectable in HeLa cells (epithelial).