

Abstract
Rats are naturally resistant to Toxoplasma gondii infection, particularly the RH strain, while mice are not. Previous studies have demonstrated that inducible nitric oxide synthase (iNOS) and arginase-1 of rodent peritoneal macrophages are linked to the mechanism of resistance. As an increasing number of studies on human and animal infections are showing that pulmonary toxoplasmosis is one of the most severe clinical signs from T. gondii infection, we are interested to know whether T. gondii infection in alveolar macrophages of rats is also linked to the levels of iNOS and arginase-1 activity. Our results demonstrate that T. gondii could grow and proliferate in rat alveolar macrophages, both in vitro and in vivo, at levels higher than resistant rat peritoneal macrophages and at comparable levels to sensitive mouse peritoneal macrophages. Lower activity and expression levels of iNOS and higher activity and expression levels of arginase-1 in rat alveolar macrophages were found to be linked to the susceptibility of T. gondii infection in these cells. These novel findings could aid a better understanding of the pathogenesis of clinical pulmonary toxoplasmosis in humans and domestic animals.
Introduction
Toxoplasma gondii is an obligate intracellular parasitic protozoan causing toxoplasmosis in infected humans and animals. In most cases, T. gondii causes asymptomatic infection in healthy individuals, but severe clinical presentations can be found in congenital toxoplasmosis, ocular toxoplasmosis and in immunocompromised people including AIDS patients [1]. Pulmonary toxoplasmosis has been reported from immunocompromised or immunodeficient patients [2]–[10], pregnant women [11] and immunocompetent individuals [12]–[17]. Additionally, animals with toxoplasmic pneumonia have also been reported in a large number of studies [18]–[21]. However, little attention has been focused on this disease, due to the difficulties of diagnosis, leading to the reporting of relatively few cases [6], [22]. It has been recognized that the lungs are one of the most susceptible organs (following the CNS) to T. gondii infection [23] and there are considerable concerns especially when considering lung transplantation [24].
Alveolar macrophages are one of the most important components of the first line of pulmonary defense against inhaled pathogens and other microorganisms [25]–[28]. Yet, controversy still surrounds the characteristics of alveolar macrophages infected with T. gondii [5], [29]. Chinchilla and colleagues considered that macrophages are important in the mechanism of resistance to T. gondii infection although they did not know what the mechanism was [29]. The mechanism of innate resistance to Toxoplasma infection in macrophages was suggested to be a non-oxidative mechanism [30], [31] and was considered to be related to IFN-γ, TNF-α, IL-12, IL-10, TGF-β and other cytokines [32]. The function of nitric oxide (NO) and arginine in mouse macrophages against pathogen infection has been well documented [33]–[42]. In fact, it is well known that the L-arginine metabolic pathway plays an important role in host defense and the control of inflammatory reactions [43]–[45]. The relationship between NO and L-arginine is integrally linked. The enzyme which produces NO, inducible nitric oxide synthase (iNOS), utilizes L-arginine as a substrate. However, it also competes with the enzyme, arginase-1, for L-arginine as a substrate. Arginase-1 hydrolyzes L-arginine to L-ornithine and urea. L-ornithine promotes parasite growth as it is a precursor for a variety of polyamines via the ornithine decarboxylase (ODC) pathway [46]–[48]. Thus the balance between iNOS expression (pathogen destruction) and arginase-1 expression (pathogen promotion) is postulated to be linked to susceptibility or resistance to Toxoplasma infection.
In our previous work, we demonstrated that the differences in expression levels and activity of iNOS and arginase-1 between the rat peritoneal macrophages (RPMs) and mouse peritoneal macrophages (MPMs) are strongly linked to the resistance and host specificity for Toxoplasma infection in these cells and hosts [49]. As severe clinical signs are commonly observed in the lungs of the host infected with T. gondii, we were interested to know if the infectivity of the rat alveolar macrophages (RAMs) for T. gondii is similar to those from the peritoneal cavity. The answer to this question could significantly benefit our understanding of the mechanism of pulmonary toxoplasmosis in humans and in domestic animals.
Results
Proliferation of T. gondii in Rat Alveolar Macrophages
Using infection studies on peritoneal and alveolar macrophages, T. gondii was found to be able to multiply in MPMs (sensitive cells) but this proliferation was not seen in RPMs (resistant cells) (Figure 1) as has been reported previously in other studies [28], [49], [50]. However, in rat alveolar macrophages from the same inbred line, a significant proliferation of T. gondii was observed and this was similar to the situation seen in the sensitive MPMs (Figure 1). Following infection by T. gondii, a significant increase of T. gondii in RAMs was found from 1 hr (36.07±1.09 per 100 cells) to 24 hrs after infection (296.44±22.00 per 100 cells) (Figure 2A) when the same population of cells were infected. However, a highly significant decrease in the number of T. gondii in RPMs was observed from 1 hr (33.91±1.89 per 100 cells) to 24 hrs (12.79±1.26 per 100 cells) after infection using the same experimental approach. Although the number of T. gondii in RAMs was less than that in MPMs at 24 hrs after infection, the massive proliferation of this parasite in RAMs indicated that this cell type is highly susceptible to T. gondii infection. Moreover, a significant increase in the ratio of infected to non-infected cells in the RAM samples was also found between 1 hr (27.49±1.27 per 100 cells) and 24 hrs (45.22±1.99 per 100 cells) post infection (Figure 2B). This was similar to the situation observed in MPMs at 1 hr (31.24±2.39 per 100 cells) and 24 hrs (54.92±2.96 per 100 cells) after infection. In addition to reporting the high degree of sensitivity of RAMs, these results also confirmed the results reported in our previous publication that the proportion of infected cells was highly significantly decreased in RPMs from 1 hr (32.28±1.44 per 100 cells) to 24 hrs (11.18±1.15 per 100 cells) after infection [49].
Figure 1. Analysis of T. gondii proliferation in rat alveolar macrophages in vitro.

Rat alveolar macrophages (RAMs) were incubated with T. gondii at the ratios of 1∶1 (parasites/macrophages = 1∶1), then the extracellular T. gondii (the non-invaded individuals) were washed from the medium and the time was defined as 1 hr. After 24 hrs, cells from the same cultures were compared. Mouse peritoneal macrophages (MPMs) (susceptible) and rat peritoneal macrophages (RPMs) (resistant) were designated as controls in the infection of T. gondii. (A and B) Different methods of Fluorescence Microscopy or DAPI staining were used in these experiments to observe the infection of T. gondii in macrophages. All the results were observed by fluorescence microscopy using a 350 nm exciting wavelength and 495 nm emitting wavelength. The results are representative of three similar experiments.
Figure 2. Proliferation of T. gondii in RAMs.
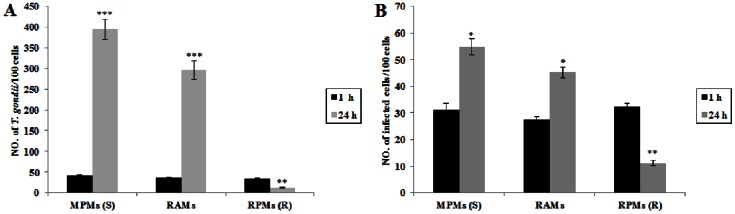
(A) Number of T. gondii/100 cells. (B) Number of infected cells/100 cells. Data in A and B represent a mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities *p<0.05, **p<0.01, and ***p<0.001, (ANOVA), indicate a significant difference from infection at 1 hr in MPMs (S), RAMs and RPMs (R) respectively (S: susceptible; R: resistant).
iNOS/arginase Expression and Activity in Rat Alveolar Macrophages
Since iNOS and arginase are competitors for the same substrate (arginine) in the L-arginine metabolic pathway, we analyzed the level of iNOS/arginase activity and gene expression in non-activated alveolar macrophages freshly isolated from rats. High iNOS/low arginase activity and expression were found in RPMs, while lower iNOS/higher arginase activity and expression were found in RAMs. As a matter of fact, these levels were similar to those found in the sensitive MPMs. Although RAMs could secrete a small amount of NO (in comparison with the non-detectable NO production in MPMs), the concentration of NO production (10.89±0.73 µM) was significantly lower than RPMs (34.86±1.96 µM) especially at 24 hrs after cultivation in vitro (Figure 3A). When comparing the arginase activity between rat peritoneal and alveolar macrophages, our results showed that arginase activity of RAMs (10.47±1.72 µM urea/mg protein) was much higher than that found in RPMs (2.26±0.25 µM urea/mg protein), but lower than that found in MPMs (18.21±1.87 µM urea/mg protein) as the sensitive control (Figure 3B). To investigate whether these differences in activity levels are due to differences in expression of the iNOS and arginase-1 genes, mRNA and protein levels were measured. Our results showed that the level of iNOS mRNA expression in RAMs was much lower than that in RPMs. In contrast, the mRNA expression of arginase-1 was highly expressed in RAMs (Figure 4A). Results from Western-blot analysis also indicated that iNOS protein expression in RAMs was much less than that found in RPMs, while higher expression of arginase-1 protein expression was found in RAMs (Figure 4B).
Figure 3. Comparison of nitric oxide production and arginase activity among RAMs, MPMs and RPMs.
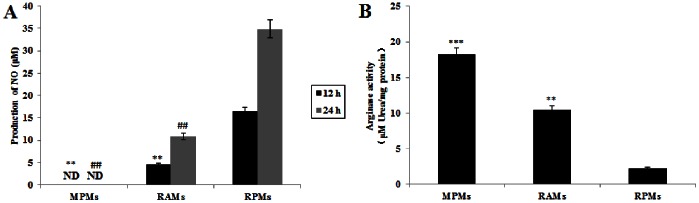
(A) A comparison of NO production between different macrophage types as measured by the Griess reaction, relative to RPMs. Data represent mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities **p<0.01, (ANOVA), show significant differences from NO production of RPMs cultured for 12 hrs; probabilities ## p<0.01, (ANOVA), show significant differences from NO production of RPMs cultured for 24 hrs. (B) A comparison of arginase activity, as measured by a colorimetric assay (enzyme activity is the output of urea secreted from lysed macrophages), between different macrophage types relative to RPMs. Data represent a mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities **p<0.01 and ***p<0.001, (ANOVA), show significant differences from arginase activity of freshly purified RPMs.
Figure 4. iNOS/arginase expression in MPMs, RAMs and RPMs.
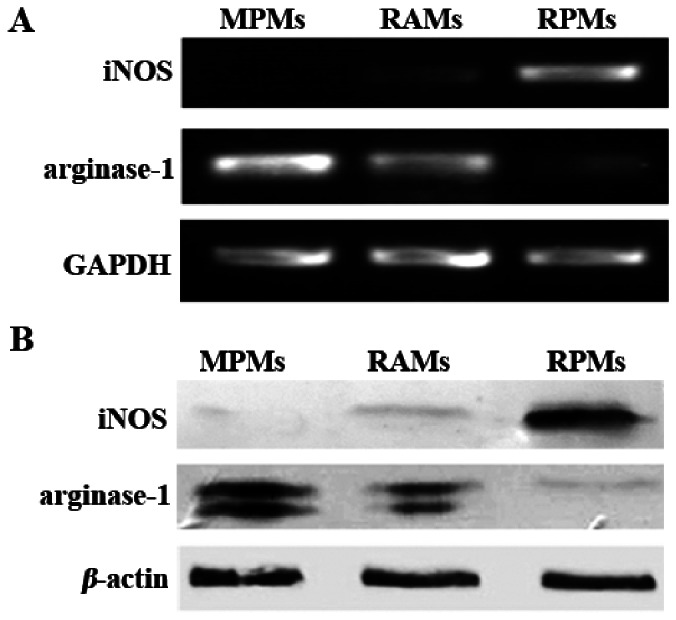
(A) RT-PCR analysis for the expression of iNOS and arginase-1 mRNA in MPMs, RAMs, and RPMs, using a housekeeping gene, GAPDH, as a control. (B) Western-blotting analysis of the expression of iNOS and arginase-1 protein in MPMs, RAMs, and RPMs, using a housekeeping protein, β-actin, as a control. The results are representative of three similar experiments.
Changes in NO Production in RAMs and Response to Infection with T. gondii
Since MPMs were susceptible to T. gondii RH strain infection and the major reason was due to the lack of NO production [49], we wanted to investigate the growth of T. gondii in RAMs stimulated by LPS+IFN-γ (stimulators of NO production) and inhibited by L-NAME. Figure 5A shows that NO production was significantly increased in the cells treated with LPS+IFN-γ but decreased in the macrophages treated with L-NAME. We further demonstrate that the number of T. gondii/100 cells was significantly decreased in the macrophages treated with LPS+IFN-γ but significantly increased in those treated with L-NAME (p<0.05) at 24 hrs after infection (Figure 5B). The profile of the ratio of infected cells to uninfected cells showed a similar significant pattern (Figure 5C).
Figure 5. Effect of NO regulation on the growth of T. gondii in rat alveolar macrophages.
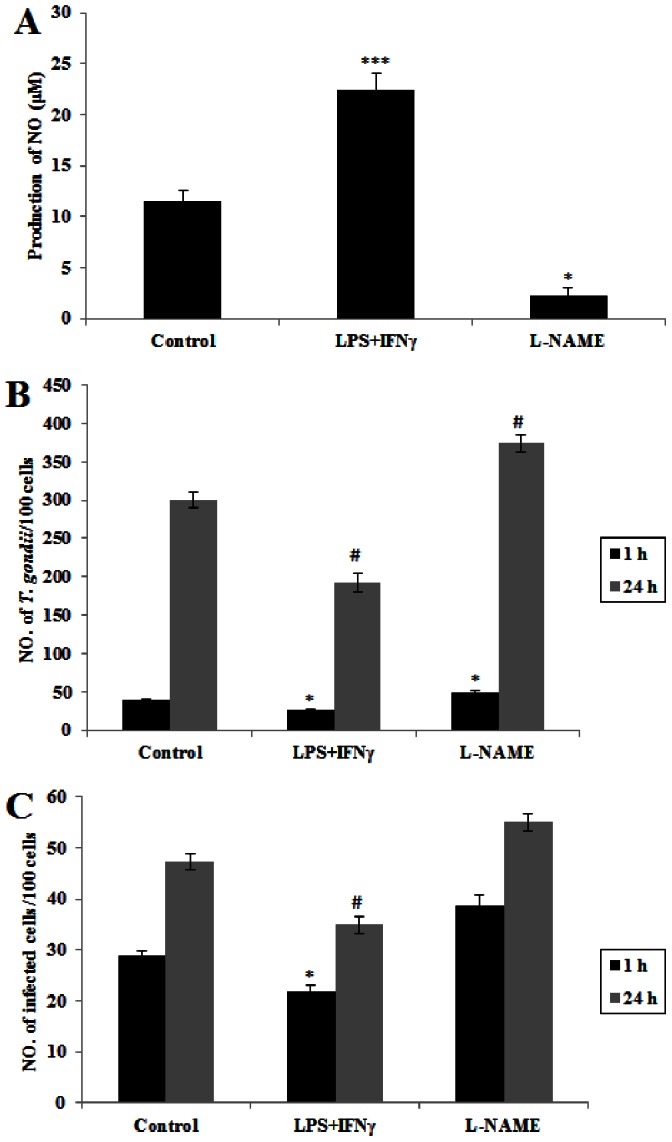
(A) RAMs were treated with LPS+IFN-γ or L-NAME for 12 hrs. NO production of each group was measured by the Griess reaction, using normal cultured RAMs as control. Data represent a mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities *p<0.05 and ***p<0.01, (ANOVA), show significant differences from controls. (B) Comparison of number of T. gondii per 100 cells. (C) Comparison of number of infected cells per 100 cells. Data in B and C represent a mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities *p<0.05 and # p<0.05, (ANOVA), show significant differences from controls of infection at 1 hr and 24 hrs respectively. ND: not detectable.
The Growth of T. gondii in Alveolar Macrophages from Different Strains of Rats in vitro
Previous studies [49] showed that there was variation in iNOS/arginase-1 activity in different inbred lines of rats suggesting genetic control of host enzyme activity and therefore degree of susceptibility to T. gondii infection. Figure 6A and 6B show that the levels of T. gondii infection in alveolar macrophages isolated from different inbred lines of rats (BN, F344, Lewis, SD and Wistar) differ between strains. These results also demonstrate that sensitivity to T. gondii infection by rat alveolar macrophages is not confined to individual inbred lines and therefore suggests a common response in rats. Figure 6C shows that the NO production in non-activated alveolar macrophages from different strains of rats was significantly lower than the controls (RPMs). However, the arginase activity of alveolar macrophages from different strains of rats was significantly higher than the controls (RPMs) (Figure 6D). The susceptibility of these populations of RAMs from different inbred lines of rats, as measured by infection levels, was completely correlated with arginase-1 activity and inversely correlated with NO production.
Figure 6. Growth status, production of NO and arginase activity of T. gondii in alveolar macrophages isolated from different strains of rats (BN, F344, Lewis, SD and Wistar).

Rat alveolar macrophages (RAMs) were incubated with T. gondii at the ratios of 1∶1 (parasite/macrophages = 1∶1), then the extracellular T. gondii (the non-invaded individuals) were washed from the medium and the time was defined as 1 hr. The infection results were observed by fluorescence microscopy. (A) Comparison of number of T. gondii per 100 cells. (B) Comparison of number of infected cells per 100 cells. Data in A and B represents a mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities *p<0.05 and ***p<0.001, (ANOVA), show significant differences from the infection at 1 hr of each type of macrophage respectively. (C) NO production of different rat alveolar macrophages was measured by the Griess reaction. Data represent a mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities **p<0.01 and ***p<0.001, (ANOVA), show significant differences from NO production of control (RPMs cultured for 12 h); probabilities ## p<0.01 and ### p<0.001, (ANOVA), show significant differences from NO production of control (RPMs cultured for 24 h). Control: SD rat peritoneral macrophage. (D) Arginase activity of different rat alveolar macrophages were measured by a colorimetric assay (enzyme activity is the output of urea secreted from lysed macrophages). Data represent mean ± SEM calculated from 3 independent experiments performed in triplicate. Probabilities **p<0.01 and ***p<0.001, (ANOVA), show significant difference from control (SD rat peritoneal macrophages).
Infection of Rat Alveolar Macrophages in vivo by T. gondii
Although we have demonstrated that RAMs are susceptible to T. gondii infection in vitro, we did not know if similar results could be confirmed in vivo. Groups of 6 rats were infected by either the pulmonary route, peritoneal route or were not infected (injected with PBS). Figure 7A shows that we can detect the infection of T. gondii in alveolar macrophages of SD and Wistar rats infected directly from the pulmonary cavity. Different numbers of tachyzoites were observed within cells which could have been generated either by differential infection with T. gondii cells or by post-infection intracellular proliferation. Immunohistochemical tests also confirmed the presence of parasites in the alveolar macrophages (Figure 7B). Tachyzoites were not detected in other organs of these rats which had been infected directly via the pulmonary cavity (Figure 7C) demonstrating that the rat lungs were more susceptible to the parasites. To clarify this, parasites were infected peritoneally, using both SD and Wistar rats, but no parasites were found in any organs (brain, lung, liver, spleen and kidney) in these infected rats (Figure 7D). This confirms that infection in vivo is particularly occurring in the RAMs and not, as has been previously observed, in RPMs. These results show that the in vitro results, on susceptibility of RAMs and resistance of RPMs, are also observed in vivo. These data also show that intraperitoneal infection in rats does not appear to spread, even to lung tissue at least in the case of the T. gondii RH strain. While pulmonary injection results in infection of RAMs but does not appear to, under the conditions used here, spread systemically. Thus, different outcomes of infection can be observed when using the two routes of inoculation. It would be an interesting question to investigate the mechanisms of cell/macrophage invasion during the natural (fecal-oral) infection route. However, it is not suitable for this study because of the complicated life cycle of T. gondii and variability in the efficiency of infection of the host when infected in this way. In addition, technical difficulties would need to be overcome to achieve reproducibility in the rat due to resistance to T. gondii and difficulties in ensuring accurate and comparative delivery doses of oocysts or cysts.
Figure 7. Analysis of T. gondii infection in rat alveolar macrophages in vivo.

Rats were infected by pulmonary infection (n = 6); peritoneal infection (n = 6) and uninfected controls (n = 6; injected with PBS) to determine whether T. gondii could be taken up in vivo. (A) At 24 hrs after the time of infection, the lungs of infected rats were sectioned, directly smeared and then stained with Diff. The results are representative of the replicated experiments. (B) Similarly, the lungs of these infected rats were steeped in 10% polyphosphate formalin for 24–48 hrs, then treated by immunohistochemistry with an specific anti-toxoplasma antibody. The arrows indicate the proliferation of T. gondii in RAMs in vivo. The results were observed by microscopy (100×). The results are representative of the replicated experiments. (C) Diff stained section of brain, liver, spleen and kidney tissue following pulmonary infection showing that parasites could only be found in alveolar macrophages and not in other tissues. The results are representative of the replicated experiments (6 rats per experiment). (D) Diff stained section of brain, lung, liver, spleen and kidney tissue following peritoneal infection showing that parasites could not be found in any tissues. The results are representative of the replicated experiments (n = 6).
Discussion
For a long time, pneumonia caused by T. gondii has been overlooked as a severe problem in immunocompetent hosts and thus few studies have been concerned with the disease. However, more and more reports have indicated that toxoplasmic pneumonia can be frequently found in individuals with normal immune function including both humans [12]–[17] and animals [18]–[21]. It was considered that rats, like humans, have a higher level of resistance to T. gondii infection in comparison to other mammals such as mice, guinea pigs and hamsters which all show a higher degree of susceptibility to this parasite [50]–[53]. Therefore, rats are recognized as a good animal model for the understanding of human toxoplasmosis [52], [54]–[59]. Previous studies from our laboratory demonstrated that the higher expression and activity of iNOS and lower expression and activity of arginase in RPMs are strongly linked with the resistance to the T. gondii RH strain infection in resistant (rats) and susceptible (mice) species [49]. This highlights the mechanism of species specificity to pathogen infection at least in T. gondii. Furthermore, these studies demonstrated that individual variation in susceptibility, based on differences in inbred lines of rats, could also be linked to the balance of expression of iNOS and arginase-1 [49]. Although RPMs were considered to be resistant to T. gondii, particularly the RH strain, interestingly, we have found that RAMs are much more susceptible to this parasite. This phenomenon was also reported by Chinchilla et al. [29] and Badger et al. [27] but the mechanism was not known. They did, however, report that interferon-γ was found to be involved in activating rat alveolar macrophages which in turn appeared to induce antimicrobial activity against T. gondii in vitro [60]. As far as we can ascertain, no other studies have investigated this phenomenon more recently. A key question that has not been addressed is the reason why RPMs show different susceptibility levels to RAMs during T. gondii RH strain infection. Previous studies [49] showed that iNOS and arginase-1 expression are strongly linked to resistance and susceptibility by peritoneal macrophages. This raises the question as to whether the expression and activity levels of iNOS and arginase-1 differ between RAMs and RPMs and could therefore account for differences in susceptibility. Our results clearly demonstrate that, in comparison to RPMs, there is a much lower level of expression of iNOS (and, consequently, a lower production of NO) but higher expression levels of arginase-1 (and, consequently, more recorded urea) in RAMs (Figure 3 and Figure 4). This could explain why the T. gondii RH strain grows well in RAMs but not in RPMs. In comparison with the peritoneal macrophages of mice, which are also highly sensitive to infection with T. gondii, RAMs also produce less NO. The presence of high levels of NO is considered to confer great benefit in preventing the growth of T. gondii and the consequent destruction of host cells [49]. This study shows a strong link between the balance of iNOS and arginase-1 activity in RAMs and their higher susceptibility to T. gondii than RPMs in vitro and in vivo. As far as we can ascertain, this has not been reported before and few studies have considered the interactions between T. gondii and RAMs. The presence of the phylogenetically unrelated parasite, Trypanosoma lewisi, has been shown to have an immunosuppressant effect on T. gondii infection in RAMs [61]. The mechanism for this is not known but may be triggering key generic pathways that suppress the proliferation of T. gondii. A recent study, using indoleamine 2,3-dioxygenase (IDO) gene knockout mice, has shown that increased IDO attenuates acute toxoplasmic infection in lung tissue [62]. This enzyme, which depletes local stocks of L-tryptophan is stimulated by effectors such as interferon-γ. Previous studies have reported associations between NO synthesis, interferon-γ and reduced microbial activity [63], suggesting that the roles of these molecules may also be linked to the regulation of T. gondii proliferation. More research is required to explore such associations.
An important question is why different expression levels of iNOS and arginase-1 exist in these cells from the same body?
It is well known that the lungs are more sensitive and prone to be injured by invading microorganisms, dust particles, harmful chemicals and autoimmune damage. Alveolar macrophages, therefore, are required to remain in a quiescent state to avoid damaging other pneumocytes by expressing minimal quantities of inflammatory cytokines and possessing lower levels of phagocytic activity [28], [64]. In fact, alveolar macrophages play a compensating role in the regulation of the immune defense in mammals. The adaptive immunity is suppressed by alveolar macrophages to prevent nonselective destruction of normal pneumocytes by secreting Th2 cytokines, such as transforming growth factor-β (TGF-β), interleukin-4 (IL-4), interleukin -10 (IL-10) and other small molecules [64]–[66]. Studies have shown that iNOS and arginase-1 are tightly regulated by Th1 cytokines (IFN-γ and TNF-α) and Th2 cytokines (IL-4 and IL-13) respectively [37], [67], [68]. In addition, the L-arginine metabolic pathway is very important for host defense and pathogen infection [69], [70]. Therefore, we presume that the lower expression and activity of iNOS and higher expression and activity of arginase-1 are highly correlated with Th1/Th2 cytokines regulated by alveolar macrophages in the quiescent state. However, other activators of arginase-1 are recognized such as STAT3 signaling cytokines IL-6, GCSF, and IL-10 [71]. Furthermore, it has been reported that an interaction between the T. gondii ROP16 protein kinase and the STAT3 and STAT6 pathways results in an increase in arginase-1 expression [72]. It is possible, therefore, that the T. gondii ROP protein kinase systems are also involved. This quiescent state and immunological down regulation in peritoneal macrophages were not found despite the fact that it is generally thought that both macrophage types originate from the blood mononuclear phagocyte system or bone marrow [73]. Recent studies have shown that some macrophages may develop from sources other than these traditional origins [74] – perhaps RPMs and RAMs are derived from a different source. Moreover, other differences have been reported in phenotype and function of these two macrophage types (peritoneal cavity and lung tissue) such as O2 metabolism, cell surface antigens and modulation of immune cell function [28], [64], [75]–[78]. Therefore, we suggest that the differences between phenotype and function of RAMs and RPMs may also be related to the differences in the L-arginine metabolic pathway.
In the L-arginine metabolic pathway, the competition for L-arginine by iNOS and arginase-1 results in an opposing status in relation to NO and urea (i.e. either higher NO and lower urea or lower NO and higher urea) in macrophages or possibly other types of cells [44], [45], [49]. There is a great deal of evidence to demonstrate that non-activated RPMs display a higher capacity to catalyze L-arginine to NO by means of higher expression of iNOS resulting in a lesser production of urea (or polyamines) [49], [79], [80]. It is well documented that the competition for arginine between iNOS and arginase affects the outcome of T. gondii infection [44], [45], [49] and our study clearly replicates this association when comparing RPMs and RAMs.
The marked differences in susceptibility to T. gondii infection between mice and rats [49] are clearly genetically determined by comparison of inbred mouse strains with inbred rat strains. Additionally, it was clearly demonstrated, by genetic crosses, that differences in infection susceptibility in RPMs from different rat inbred strains are genetically controlled [49]. The differences in susceptibility between RPMs and RAMs are an interesting genetic question. The fact that the RPMs and RAMs are derived from the same inbred rat strains, combined with the fact that different inbred rat strains also exhibit the same phenomenon, leads to the conclusion that differences in susceptibility (and arginine metabolism) are determined epigenetically. This raises interesting questions as to which host genes determine susceptibility to T. gondii infection and how host epigenetic mechanisms determine differences in susceptibility in different tissues. Our data suggest that the iNOS and arginase-1 genes are central to these questions.
In addition to host mechanisms which determine susceptibility or resistance to T. gondii infection, there are clearly parasite derived mechanisms [81]. In this study, strain RH was used as the model parasite due to the very clear differences in responses in rats and mice. RH is, however, a type I strain and is highly virulent, particularly to mice and this strain type is not the predominant type found in humans which tend to be Type II strains. Type II strains are much less virulent in mice and further work would be required to investigate the effect that these strains have on the arginase/iNOS balance in both mice and rats. Recent studies are beginning to shed light on the mechanisms of virulence derived from the parasite in mice and these are thought to involve the ROP2 family of protein kinases [82]. However, little is currently known about these processes in humans. An important future area of research should be aimed at linking the mechanisms determining virulence in the parasite with corresponding host mechanisms to resistance and susceptibility.
In summary, the characteristics of RAMs infected with T. gondii are clearly defined and these cells are susceptible to T. gondii RH strain infection both in vitro and in vivo. It is clear that the native biological characteristics in macrophages from the lungs and peritoneal cavity of rats are strongly linked with their resistance or susceptibility to T. gondii infection. Our results have demonstrated that the lower expression and activity of iNOS but higher activity of arginase (and higher polyamine levels) in RAMs are linked to the susceptibility to T. gondii infection. The question of how these results from rats relate to humans requires further research. The mechanisms of action of human macrophages to microbial challenge differs from that in rats and mice. It is widely reported that iNOS and arginase are not expressed under similar stimulation conditions in human macrophages in vitro [83], however, in vivo, they have been shown to express iNOS and arginase-1 and produce NO. Future studies need to address these differences. Nevertheless, an understanding of the interactions between T. gondii and host macrophages promises to provide great benefit to our understanding of the pathogenesis and mechanisms of pulmonary toxoplasmosis in humans and domestic animals.
Materials and Methods
Ethics Statement
All animal experiments were conducted using the guidelines provided by the Laboratory Animal Use and Care from the Chinese CDC and the Rules for Medical Laboratory Animals (1998) from the Ministry of Health, China. All protocols for animal use in this work were approved by the Laboratory Animal Use and Care Committee of Sun Yat-Sen University under the license number 2012CB53000.
Animals and Parasites
Sprague Dawley (SD), Wistar rats and BALB/c mice were purchased from the Laboratory Animal Center of Sun Yat-Sen University; Brown Norway (BN), Fischer 344 (F344) and Lewis rats were purchased from Vital River Laboratories (Beijing, China). All rats (6 to 8 weeks old, weight 150∼200 g) and mice (6 to 8 weeks old, weight 18∼25 g) were certified to be free of specific pathogens when entering the laboratory and maintained with sterile distilled water and commercial food.
The T. gondii strain RH-GFP, kindly provided by Dr. Xue-Nan Xuan of the Obihiro University of Agriculture and Veterinary Medicine, Japan, was generated as previously described [84]. For the purification of tachyzoites, host cell debris and T. gondii were harvested from the peritoneal cavities of infected BALB/c mice by injection of cold D-Hanks solution three days after infection. The solution containing T. gondii was centrifuged at 50×g for 5 min at 4°C to discard host cells and fragments. The resulting supernatant was then centrifuged at 1350×g for 10 min at 4°C, and then was suspended in RPMI-1640 medium (GIBCO, USA) with 10% FBS and the parasites counted.
Isolation and Cultivation of Peritoneal and Alveolar Macrophages
Rats or mice, anaesthetised by carbon dioxide (CO2), were injected intraperitoneally with 15 mL (rat) or 5 mL (mouse) of cold calcium and magnesium-free Dulbecco’s phosphate buffered saline (D-Hanks) containing 100 U of penicillin and 100 µg of streptomycin per ml. To obtain the peritoneal macrophages, 11–14 ml (rat) and 3–4 ml (mouse) of peritoneal cavity fluid was collected from each animal. For each experiment, the cells from 1–2 rats, or 3–4 mice, were pooled and then cultivated as described below.
Resident alveolar macrophages were obtained from the lungs in situ by the methods described by Myrvik et al. [85] and Catterall et al. [5]. Briefly, rats were injected peritoneally with 2.5% hydral at 50 mg/kg and a sterile #26 pipe conveying fluid was used. The lungs were lavaged with cold (4°C) D-Hanks containing 100 U of penicillin and 100 µg of streptomycin per 1 ml with a 10 ml plastic syringe. This process was repeated until a total of 50 mL lavage fluid was collected. For each experiment, cells from 5–6 rats were pooled and then cultivated as described below.
The lavage fluid or harvested peritoneal macrophages were centrifuged at 250×g, for 10 min at 4°C. The pellet was resuspended in 5 mL of RPMI-1640 medium (GIBCO Laboratories, USA) with 10% fetal bovine serum (FBS; GIBCO Laboratories, USA) and penicillin (100 U/mL) and streptomycin (100 µg/mL). In all experiments, the viability of cells was higher than 95 percent as measured by the trypan blue exclusion test. More than 92% of rat or mouse peritoneal lavage cells and >95% of rat alveolar lavage cells were assessed to be phagocytes as determined by the uptake of neutral red. Cells were counted and seeded in a 96 well plate (2×105 cells/well) or 6 well plate (2×106 cells/well) for 2 h at 37°C, 90% humidity and 5% CO2. After 2 h, wells were washed with sterile DPBS (pH 7.2) to remove non-adherent cells and fresh culture medium was added. Macrophages were incubated with or without lipopolysaccharide (LPS; 10 µg/ml, Sigma, St. Louis, USA) plus IFN-γ (100 U/ml; Sigma, USA) or with the NOS specific inhibitor Nω –nitro-L -arginine methyl ester (L-NAME; 10 mM; Sigma, St. Louis, USA). Cells were then cultured overnight and then used for further experiments.
Parasite Infection and Proliferation
Rat alveolar and peritoneal macrophages isolated and cultivated, as described above, were challenged with T. gondii tachyzoites at the ratio of 1∶1 (T. gondii/macrophage) only once. Extracellular T. gondii were then washed out 1 hr after incubation with the cells. This time point was defined as 1 hr for the start of the experiment. Thereafter, the results were observed under an inverted fluorescence microscope or stained with DAPI at each desired time point. When time point analysis was carried out (e.g. 1 hr and 24 hrs), the same population of cells was sampled. Infection results from 96 well plates were observed and analyzed by merged photographs of normal and fluorescence pictures. Six well plates and cell slides were used for the DAPI experiments. All the results were observed by fluorescence microscopy using a 350 nm exciting wavelength and 495 nm emitting wavelength. The number of T. gondii within the cells and the infected cells were counted in 100 macrophages which were randomly selected from different fields of view at least 3 times and an average was determined. Samples from 1 hr to 24 hrs in infection experiments were observed in independent 96 or 6 well plates respectively. These experiments were performed no less than twice.
Measurement of Production of Nitric Oxide and Arginase Activity
Nitrite content as a reflection of NO production was determined by the Griess reaction as previously described [86]. Briefly, 100 µl supernatant or standard solution (NaNO2) were incubated with 100 µl of Griess reagent (0.5% sulfanilamide, 0.05% naphthyldiamine dihydrochloride in 5% H3PO4) for 10 min. The plates were read at 550 nm in an ELISA reader (Multiskan MK3, Thermo Labsystems, Finland). All experiments were carried out in triplicate.
Arginase activity in purified macrophages was measured by a colorimetric method as described [87]. Briefly, 10 mM MnCl2 and 0.5 M L-arginine were successively added to macrophage lysates for 1 hr at 37°C. The reaction was stopped by the addition of an acid solution (H2SO4:H3PO4:H2O = 1∶3∶7) and the urea generated by arginase was analyzed by addition of α-isonitrosopropiophenone at 100°C for 45 min. The colored product was quantified by absorption at 550 nm in an ELISA reader. Arginase activity was determined as the amount of urea produced from total protein in the peritoneal and alveolar macrophages.
RT-PCR Analysis
Total RNA from treated and nontreated macrophages was extracted using Trizol Reagent (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. Total RNA was reverse transcribed to cDNA using a set of oligo (dT) primer and SuperScript™ III First-Strand Synthesis System according to the instructions by the manufacturer (Invitrogen, Carlsbad, USA). cDNA (1 µg) was used as a template for amplifying the iNOS, arginase-1 and GAPDH (used as internal standard) genes by PCR using the following primers: rat-iNOS, 5′-CTA CCT ACC TGG GGA ACA CCT GGG-3′ and 5′-GGA GGA GCT GAT GGA GTA GTA GCG G-3′ 442 bp; mouse-iNOS, 5′-GCC TCG CTC TGG AAA GA-3′ and 5′-TCC ATG CAG ACA ACC TT-3′, 499 bp; arginase-1, 5′-AAG AAA AGG CCG ATT CAC CT-3′ and 5′-CAC CTC CTC TGC TGT CTT CC-3′, 201 bp; GAPDH, 5′-AAT GCK TCC TGY ACC ACC AAC TGC-3′ and 5′-TTA GCC AWA TTC RTT GTC RTA CCA GG-3′, 513 bp. For semi-quantitative PCR, the cycling conditions were: 94°C for 1 min, 60°C for 1.5 min and 72°C for 1.8 min. For rat-iNOS, mouse-iNOS and arginase-1, 27 cycles were used, but for GAPDH, only 20 cycles were used. Amplified DNA products were separated on 1% agarose gel and were photographed using an electronic documentation system (Biostep, Germany) after staining with ethidium bromide.
Western Blotting
Cells were lysed in SDS loading buffer, fractionated in SDS-PAGE and transferred onto immunoblot polyvinylidene difluoride membrane (Pall, USA). The membrane was probed using the rabbit polyclonal iNOS antibody (Thermo, USA ) and rabbit polyclonal arginase-1 antibody (Santa Cruz, USA). β-actin was stained with antibody (NOVUS, USA) as a sampling control. Horseradish peroxidase-labeled secondary antibodies (Cell Signaling, USA) and BeyoECL Plus Detection Kit (Beyotime, China) were used for antibody detection.
Infection of Toxoplasma in Rat Alveolar Macrophages in vivo
SD or Wistar rats were randomly divided into 3 groups (6 rats in each group) and were infected by syringe into the lung tissue and peritoneal cavity with 2×107 tachyzoites of RH strain per rat. The rats in the control group were injected with PBS only. Rats from each group were anaesthetised and dissected 24 hrs after infection. Brains, hearts, lungs, livers and spleens were removed from the rats. A part of each organ was smeared with a tip and then stained with Diff after being dried and fixed in methyl alcohol. The rest of the tissues of the rats were fixed in 10% polyphosphate formalin for not more than 48 hrs and were then prepared for examination by immunohistochemistry with anti-T. gondii antibody (Abcam, USA) to analyze infection in rat alveolar macrophages.
Statistical Analysis
Results are expressed as a mean ± SEM. In the case of the in vitro macrophage studies, experiments were conducted in triplicate using the same sample at different time points. In vivo studies were conducted on 6 rats per study group. Statistical differences were designated by * (p<0.05), ** (p<0.01) and *** (p<0.001) or in some cases where multiple comparisons are used # (p<0.05), ## (p<0.01) and ### (p<0.001). Multiple data comparisons were derived by one-way ANOVA by SPSS 17.0 software (SPSS Inc., Chicago, USA).
Acknowledgments
The authors are thankful to Dr. Xue-Nan Xuan of National Research Center for Protozoan Diseases, Obihiro University, Hokkaido, Japan for providing the Toxoplasma gondii RH-GFP strain and the people who were working in the authors’ laboratories and provided significant help during the experiments but were not listed in this paper. We also thank the anonymous reviewers for critical comments and suggestions on our manuscript.
Funding Statement
The work was supported by National Basic Research Program of China (973 Program) (No. 2010CB530000) and the National Special Research Programs for Non-Profit Trades (Agriculture) (No. 200803017), http://www.973.gov.cn/English/Index.aspx. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Montoya JG, Liesenfeld O (2004) Toxoplasmosis. Lancet 363: 1965–1976. [DOI] [PubMed] [Google Scholar]
- 2. Ruskin J, Remington JS (1976) Toxoplasmosis in the compromised host. Ann Intern Med 84: 193–199. [DOI] [PubMed] [Google Scholar]
- 3. Mendelson MH, Finkel LJ, Meyers BR, Lieberman JP, Hirschman SZ (1987) Pulmonary toxoplasmosis in AIDS. Scand J Infect Dis 19: 703–706. [DOI] [PubMed] [Google Scholar]
- 4. Tourani JM, Israël-Biet D, Venet A, Andrieu JM (1985) Unusual pulmonary infection in a puzzling presentation of AIDS. Lancet 1: 989. [DOI] [PubMed] [Google Scholar]
- 5. Catterall JR, Hofflin JM, Remington JS (1986) Pulmonary toxoplasmosis. Am Rev Respir Dis 133: 704–705. [DOI] [PubMed] [Google Scholar]
- 6. Oksenhendler E, Cadranel J, Sarfati C, Katlama C, Datry A, et al. (1990) Toxoplasma gondii pneumonia in patients with the acquired immunodeficiency syndrome. Am J Med 88: 18N–21N. [PubMed] [Google Scholar]
- 7. Pomeroy C, Filice GA (1992) Pulmonary toxoplasmosis: a review. Clin Infect Dis 14: 863–870. [DOI] [PubMed] [Google Scholar]
- 8. Rottenberg GT, Miszkiel K, Shaw P, Miller RF (1997) Case report: fulminant Toxoplasma gondii pneumonia in a patient with AIDS. Clin Radiol 52: 472–474. [DOI] [PubMed] [Google Scholar]
- 9. Delhaes L, Mraz JC, Fréalle E, Durand-Joly I, Magro L, et al. (2010) Severe pulmonary toxoplasmosis after allo-SCT in two patients: from Toxoplasma genotyping to clinical management. Bone Marrow Transplant 45: 580–583. [DOI] [PubMed] [Google Scholar]
- 10. Monaco SE, Monaghan SA, Stamm JA, Khalbuss WE, Nichols L, et al. (2012) Toxoplasmosis in a post-transplant bronchoalveolar lavage: a case report. Diagn Cytopathol 40: 629–634. [DOI] [PubMed] [Google Scholar]
- 11. Candolfi E, de Blay F, Rey D, Christmann D, Treisser A, et al. (1993) A parasitologically proven case of Toxoplasma pneumonia in an immunocompetent pregnant woman. J Infect 26: 79–81. [DOI] [PubMed] [Google Scholar]
- 12. Bossi P, Caumes E, Paris L, Dardé ML, Bricaire F (1998) Toxoplasma gondii-associated Guillain-Barré syndrome in an immunocompetent patient. J Clin Microbiol 36: 3724–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carme B, Aznar C, Motard A, Demar M, de Thoisy B (2002) Serologic survey of Toxoplasma gondii in noncarnivorous free-ranging neotropical mammals in French Guiana. Vector Borne Zoonotic Dis 2: 11–17. [DOI] [PubMed] [Google Scholar]
- 14. Bossi P, Bricaire F (2004) Severe acute disseminated toxoplasmosis. Lancet 364: 579. [DOI] [PubMed] [Google Scholar]
- 15. Leal FE, Cavazzana CL, de Andrade HF Jr, Galisteo AJ Jr, de Mendonça JS, et al. (2007) Toxoplasma gondii pneumonia in immunocompetent subjects: case report and review. Clinical infectious diseases 44: e62–66. [DOI] [PubMed] [Google Scholar]
- 16. Carme B, Demar M, Ajzenberg D, Dardé ML (2009) Severe acquired toxoplasmosis caused by wild cycle of Toxoplasma gondii, French Guiana. Emerg Infect Dis 15: 656–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simanaityte D, Le Gouellec N, Ajana F, Baclet N, Poissy J, et al. (2010) Primary pulmonary toxoplasmosis in an immunocompetent patient. Med Mal Infect. 40: 713–715. [DOI] [PubMed] [Google Scholar]
- 18. Ferguson HW, Ellis WA (1979) Toxoplasmosis in a calf. Vet Rec 104: 392–393. [DOI] [PubMed] [Google Scholar]
- 19. Foster SF, Martin P, Allan GS, Barrs VR, Malik R (2004) Lower respiratory tract infections in cats: 21 cases (1995–2000). J Feline Med Surg 6: 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thiptara A, Kongkaew W, Bilmad U, Bhumibhamon T, Anan S (2006) Toxoplasmosis in piglets. Ann N Y Acad Sci 1081: 336–338. [DOI] [PubMed] [Google Scholar]
- 21.Juránková J, Opsteegh M, Neumayerová H, Kovařčík K, Frencová A, et al. (2012) Quantification of Toxoplasma gondii in tissue samples of experimentally infected goats by magnetic capture and real-time PCR. Vet Parasitol doi:pii: S0304-4017(12)00600-0.10.1016/j.vetpar.2012.11.016 [DOI] [PubMed]
- 22. Kovari H, Ebnöther C, Schweiger A, Berther N, Kuster H (2010) Pulmonary toxoplasmosis, a rare but severe manifestation of a common opportunistic infection in late HIV presenters: report of two cases. Infection 38: 141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knani L, Bouslama K, Varette C, Gonzalez Canali G, Cabane J, et al. (1990) Pulmonary toxoplasmosis in AIDS. Report of 3 cases. Ann Med Interne (Paris) 141: 469–471. [PubMed] [Google Scholar]
- 24. Luong ML, Morrissey O, Husain S (2010) Assessment of infection risks prior to lung transplantation. Curr Opin Infect Dis 23: 578–583. [DOI] [PubMed] [Google Scholar]
- 25. Herscowitz HB (1985) In defense of the lung: paradoxical role of the pulmonary alveolar macrophage. Ann Allergy 55: 634–650. [PubMed] [Google Scholar]
- 26. Fels AO, Cohn ZA (1986) The alveolar macrophage. J Appl Physiol 60: 353–369. [DOI] [PubMed] [Google Scholar]
- 27. Badger AM, Hutchman JS, Sung CP, Bugelski PJ (1987) Activation of rat alveolar macrophages by gamma interferon to inhibit Toxoplasma gondii in vitro. J Leukoc Biol 42: 447–454. [DOI] [PubMed] [Google Scholar]
- 28. Lambrecht BN (2006) Alveolar macrophage in the driver’s seat. Immunity 24: 366–368. [DOI] [PubMed] [Google Scholar]
- 29. Chinchilla M, Guerrero OM, Solano E (1982) Lack of multiplication of toxoplasma in macrophages of rats in vitro. J Parasitol 68: 952–955. [PubMed] [Google Scholar]
- 30. Catterall JR, Sharma SD, Remington JS (1986) Oxygen-independent killing by alveolar macrophages. J Exp Med 163: 1113–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Catterall JR, Black CM, Leventhal JP, Rizk NW, Wachtel JS, et al. (1987) Nonoxidative microbicidal activity in normal human alveolar and peritoneal macrophages. Infect Immun 55: 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexander J, Scharton-Kersten TM, Yap G, Roberts CW, Liew FY, et al. (1997) Mechanism of innate resistance to Toxoplasma gondii infection. Philos Trans R Soc Lon B, Biol Sci 352: 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adams LB, Hibbs JB Jr, Taintor RR, Krahenbuhl JL (1990) Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from L-arginine. J Immunol 144: 2725–2729. [PubMed] [Google Scholar]
- 34. Langermans JA, Van der Hulst ME, Nibbering PH, Hiemstra PS, Fransen L, et al. (1992) IFN-gamma-induced L-arginine-dependent toxoplasmastatic activity in murine peritoneal macrophages is mediated by endogenous tumor necrosis factor-alpha. J Immunol 148: 568–574. [PubMed] [Google Scholar]
- 35. Wang WW, Jenkinson CP, Griscavage JM, Kern RM, et al. (1995) Co-induction of arginase and nitric oxide synthase in murine macrophages activated by lipopolysaccharide. Biochem Biophys Res Commun 210: 1009–1016. [DOI] [PubMed] [Google Scholar]
- 36. Fang FC (1997) Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Invest 99: 2818–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Munder M, Eichmann K, Modolell M (1998) Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol 160: 5347–5354. [PubMed] [Google Scholar]
- 38. Yap GS, Sher A (1999) Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-gamma- and tumor necrosis factor (TNF)-alpha-dependent host resistance to the intracellular pathogen, Toxoplasma gondii . J Exp Med 189: 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gobert AP, Mersey BD, Cheng Y, Blumberg DR, Newton JC, et al. (2002) Cutting edge: urease release by Helicobacter pylori stimulates macrophage inducible nitric oxide synthase. J Immunol 168: 6002–6006. [DOI] [PubMed] [Google Scholar]
- 40. Seabra SH, de Souza W, DaMatta RA (2002) Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp Parasitol 100: 62–70. [DOI] [PubMed] [Google Scholar]
- 41. Seabra SH, DaMatta RA, de Mello FG, de Souza W (2004) Endogenous polyamine levels in macrophages is sufficient to support growth of Toxoplasma gondii . J Parasitol 90: 455–460. [DOI] [PubMed] [Google Scholar]
- 42. Lüder CG, Algner M, Lang C, Bleicher N, Gross U (2003) Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Int J Parasitol 33: 833–844. [DOI] [PubMed] [Google Scholar]
- 43. Koschorreck S, Wenzel F, Fuhrmann M, Racké K (2003) Effects of phosphodiesterase inhibitors on L-arginine pathways in rat alveolar macrophages. Eur J Pharmacol 471: 229–236. [DOI] [PubMed] [Google Scholar]
- 44. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, et al. (2008) Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9: 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Das P, Lahiri A, Lahiri A, Chakravortty D (2010) Modulation of the arginase pathway in the context of microbial pathogenesis: a metabolic enzyme moonlighting as an immune modulator. PLoS Pathog. 6: e1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abdallahi OM, Bensalem H, Augier R, Diagana M, De Reggi M, et al. (2001) Arginase expression in peritoneal macrophages and increase in circulating polyamine levels in mice infected with Schistosoma mansoni. Cell Mol Life Sci 58: 1350–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Igarashi K, Kashiwagi K (2000) Polyamines: mysterious modulators of cellular functions. Biochem Biophys Res Commun 271: 559–564. [DOI] [PubMed] [Google Scholar]
- 48. Roberts SC, Tancer MJ, Polinsky MR, Gibson KM, Heby O, et al. (2004) Arginase plays a pivotal role in polyamine precursor metabolism in Leishmania. Characterization of gene deletion mutants. J Biol Chem 279: 23668–23678. [DOI] [PubMed] [Google Scholar]
- 49. Li Z, Zhao ZJ, Zhu XQ, Ren QS, Nie FF, et al. (2012) Differences in iNOS and arginase expression and activity in the macrophages of rats are responsible for the resistance against T. gondii infection. PLoS ONE 7: e35834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krahenbuhl JL, Blazkovec AA (1975) Toxoplasma gondii: immunopathology of cutaneous hypersensitivityreactions in guinea pigs injected with living parasites. Exp Parasitol 37: 83–91. [DOI] [PubMed] [Google Scholar]
- 51. Chinchilla M, Frenkel JK (1978) Mediation of immunity to intracellular infection (Toxoplasma and Besnoitia) within somatic cells. Infect Immun. 19: 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dubey JP, Frenkel JK (1998) Toxoplasmosis of rats: a review, with considerations of their value as an animal model and their possible role in epidemiology. Vet Parasitol 77: 31–32. [DOI] [PubMed] [Google Scholar]
- 53. Dubey JP, Shen SK, Kwok OC, Frenkel JK (1999) Infection and immunity with the RH strain of Toxoplasma gondii in rats and mice. J Parasitol 85: 657–662. [PubMed] [Google Scholar]
- 54. Santoro F, Auriault C, Leite P, Darcy F, Capron A (1987) Infection of the athymic rat by Toxoplasma gondii . C R Acad Sci III 304: 297–300. [PubMed] [Google Scholar]
- 55. Darcy F, Zenner L (1993) Experimental models of toxoplasmosis. Res Immunol 144: 16–23. [DOI] [PubMed] [Google Scholar]
- 56. Zenner L, Darcy F, Capron A, Cesbron-Delauw MF (1998) Toxoplasma gondii: kinetics of the dissemination in the host tissues during the acute phase of infection of mice and rats. Exp Parasitol 90: 86–94. [DOI] [PubMed] [Google Scholar]
- 57. Zenner L, Foulet A, Caudrelier Y, Darcy F, Gosselin B, et al. (1999) Infection with Toxoplasma gondii RH and Prugniaud strains in mice, rats and nude rats: kinetics of infection in blood and tissues related to pathology in acute and chronic infection. Pathol Res Pract 195: 475–485. [DOI] [PubMed] [Google Scholar]
- 58. Freyre A, Falcón J, Méndez J, González M (2008) Toxoplasma gondii: an improved rat model of congenital infection. Exp Parasitol 120: 142–146. [DOI] [PubMed] [Google Scholar]
- 59. Freyre A, Falcón J, Mendez J, Gonzalez M (2008) Toxoplasma gondii: protection against colonization of the brain and muscles in a rat model. Exp Parasitol 119: 252–255. [DOI] [PubMed] [Google Scholar]
- 60. Badger AM, Meunier PC, Weiss RA, Bugelski PJ (1988) Modulation of rat bronchoalveolar lavage cell function by the intratracheal delivery of interferon-gamma. J Interferon Res 8: 251–260. [DOI] [PubMed] [Google Scholar]
- 61. Carrera NJ, Carmona MC, Guerrero OM, Castillo AC (2009) The immunosuppressant effect of T lewisi (Kinetoplastidae) infection on the multiplication of Toxoplasma gondii (Sarcocystidae) on alveolar and peritoneal macrophages of the white rat. Rev Biol Trop 57: 13–22. [PubMed] [Google Scholar]
- 62. Murakami Y, Hoshi M, Hara A, Takemura M, Arioka Y, et al. (2012) Inhibition of increased indoleamine 2,3-dioxygenase activity attenuates Toxoplasma gondii replication in the lung during acute infection. Cytokine 59: 245–251. [DOI] [PubMed] [Google Scholar]
- 63. Lovchik JA, Lyons CR, Lipscomb MF (1995) A role for gamma interferon-induced nitric oxide in pulmonary clearance of Cryptococcus neoformans . Am J Respir Cell Mol Biol 13: 116–124. [DOI] [PubMed] [Google Scholar]
- 64. Holt PG, Oliver J, Bilyk N, McMenamin C, McMenamin PG, et al. (1993) Down regulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med 177: 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lacraz S, Nicod L, Galve-de Rochemonteix B, Baumberger C, Dayer JM, et al. (1992) Suppression of metalloproteinase biosynthesis in human alveolar macrophages by interleukin-4. J Clin Invest 90: 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bunn HJ, Hewitt CR, Grigg J (2002) Suppression of autologous peripheral blood mononuclear cell proliferation by alveolar macrophages from young infants. Clin Exp Immunol 128: 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Munder M, Eichmann K, Morán JM, Centeno F, Soler G, Modolell M (1999) Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol 163: 3771–3777. [PubMed] [Google Scholar]
- 68. Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, et al. (2004) Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol 172: 7565–7573. [DOI] [PubMed] [Google Scholar]
- 69. Brunet LR (2001) Nitric oxide in parasitic infections. Int Immunopharmacol 1: 1457–1467. [DOI] [PubMed] [Google Scholar]
- 70. Nathan C, Shiloh MU (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA 97: 8841–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Qualls JE, DeFreitas A, Smith AM, Watowich SS, Murray PJ (2009) Direct and indirect type-1 arginase (Arg1) induction following Mycobacterium bovis (BCG) infection. J Immunol 182: 43.1. [Google Scholar]
- 72. Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, et al. (2011) Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokineinhibition and arginase-1-dependent growth control. PLoS Pathog 7: e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Naito M (2008) Macrophage differentiation and function in health and disease. Pathol Int 58: 143–155. [DOI] [PubMed] [Google Scholar]
- 74. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, et al. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336: 86–90. [DOI] [PubMed] [Google Scholar]
- 75. Simon LM, Robin ED, Phillips JR, Acevedo J, Axline SG, et al. (1977) Enzymatic basis for bioenergetic differences of alveolar versus peritoneal macrophages and enzyme regulation by molecular O2 . J Clin Invest 59: 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boltz-Nitulescu G, Foerster O (1979) Antigenic differences between alveolar and peritoneal macrophages of the rat. Lack of population-specific determinants. Immunology 38: 621–630. [PMC free article] [PubMed] [Google Scholar]
- 77. Dörger M, Münzing S, Allmeling AM, Messmer K, Krombach F (2001) Phenotypic and functional differences between rat alveolar, pleural, and peritoneal macrophages. Exp Lung Res 27: 65–76. [DOI] [PubMed] [Google Scholar]
- 78. Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, et al. (2009) Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 296: L936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Müllner N, Lázár A, Hrabák A (2002) Enhanced utilization and altered metabolism of arginine in inflammatory macrophages caused by raised nitric oxide synthesis. Int J Biochem Cell Biol 34: 1080–1090. [DOI] [PubMed] [Google Scholar]
- 80. Hrabák A, Bajor T, Csuka I (2006) The effect of various inflammatory agents on the alternative metabolic pathways of arginine in mouse and rat macrophages. Inflamm Res 55: 23–31. [DOI] [PubMed] [Google Scholar]
- 81. Wang L, Chen H, Liu D, Huo X, Gao J, et al. (2013) Genotypes and Mouse Virulence of Toxoplasma gondii Isolates from Animals and Humans in China. PLoS One 8: e53483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dubremetz JF, Lebrun M (2012) Virulence factors of Toxoplasma gondii . Microbes Infect 14: 1403–1410. [DOI] [PubMed] [Google Scholar]
- 83. Schneemann M, Schoedon G, Hofer S, Blau N, Guerrero L, et al. (1993) Nitric oxide synthase is not a constituent of the antimicrobial armature of human mononuclear phagocytes. J Infect Dis 167: 1358–1363. [DOI] [PubMed] [Google Scholar]
- 84. Kempf MC, Cesbron-Delauw MF, Deslee D, Gross U, Herrmann T, et al. (1999) Different manifestations of Toxoplasma gondii infection in F344 and LEW rats. Med Microbiol Immunol 187: 137–142. [DOI] [PubMed] [Google Scholar]
- 85. Myrvik Q, Leake ES, Fariss B (1961) Studies on pulmonary alveolar macrophages from the normal rabbit: a technique to procure them in a high state of purity. J Immunol 86: 128–132. [PubMed] [Google Scholar]
- 86. Ding AH, Nathan CF, Stuehr DJ (1988) Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol 141: 2407–2412. [PubMed] [Google Scholar]
- 87. Corraliza IM, Campo ML, Soler G, Modolell M (1994) Determination of arginase activity in macrophages: a micromethod. J Immunol Methods 174: 231–235. [DOI] [PubMed] [Google Scholar]