

Abstract
Treatment of [FeCp(CO)2Cl] with 1 equiv of the amidophosphine ligands Li[R2PNR′] (R = Ph, iPr, R′ = iPr, tBu, Cy) afforded complexes of the type [FeCp(CO)(κ2(C,P)-(C O)–NiPr-PPh2)] (1a), [FeCp(CO)(κ2(C,P)-(C O)-NtBu-PPh2)] (1b), and [FeCp(CO)(κ2(C,P)-(C O)-NCy-PiPr2)] (1c) in 40–50% yields. Complex 1a was also formed when [FeCp(CO)2(PPh2NHiPr)]+ (2) was reacted with 1 equiv of KOtBu. These complexes feature a four-membered carboxamido-phospha-ferracycle as a result of an intramolecular nucleophilic attack of the amidophosphine ligand on coordinated CO. Upon treatment of 1a with the electrophile [Me3O]BF4 the aminocarbene complex [FeCp(CO)(κ2(C,P) C(OMe)-NiPr-PPh2)]+ (3) was obtained bearing an aza-phospha-carbene moiety. Upon treatment of cis,trans,cis-[Fe(CO)2(Ph2PNHiPr)2(Br)2] (4a) and cis,trans,cis-[Fe(CO)2(Ph2PNHtBu)2(Br)2] (4b) with KOtBu the carboxamido-phospha-ferracycles trans-[Fe(CO)2(κ2(C,P)-(C O)-NiPr-PPh2)(Ph2PNHiPr)Br] (5a) and trans-[Fe(CO)2(κ2(C,P)-(C O)-NtBu-PPh2)(Ph2PNHtBu)Br] (5b) were formed in moderate yield. Finally, representative structures were determined by X-ray crystallography.
Keywords: Iron, Cyclopentadienyl, Carbon monoxide, Aminophosphines, Nucleophilic attack, Ferracycles
Graphical abstract
The synthesis of several iron(II) complexes featuring a four-membered carboxamido-phospha-ferracycle moiety as a result of an intramolecular nucleophilic attack of a deprotonated aminophosphine ligand on coordinated CO is described.

Highlights
-
•
Four-membered carboxamido-phospha-ferracycles were prepared.
-
•
Intramolecular nucleophilic attack of an amidophosphine ligand on coordinated CO takes place.
-
•
Carboxamido-phospha-ferracycles react with electrophiles to afford cyclic amino carbenes.
1. Introduction
Aminophosphines of the type PR2NHR’ containing a direct polar P(III)–N bonds have received considerable attention in recent years as versatile ligands for transition metals [1], (a), (b), (c), (d), (e). They are accessible in large quantities through the use of relatively simple condensation processes from inexpensive starting materials, i.e., primary amines and PR2Cl compounds which contain dialkyl or diaryl substituents as well as achiral and chiral P–O and P–N containing phosphine units. Thus, variations of electronic, steric, and stereochemical parameters may be achieved in a very facile fashion. Due to their soft/hard donor atoms as well their acidic NH hydrogen, these polyfunctional ligands exhibit numerous coordination modes as illustrated in Scheme 1. As middle and late transition metals M are concerned PR2NHR’ ligands typically coordinate in κ1(P)-fashion I [2], (a), (b), (c), (d), (e), (f), (g), (h), (i), while κ1(N)-coordination is unknown. Upon deprotonation of the PR2NHR’ ligands, anionic amidophosphines [PR2NR’]− are readily obtained which exhibit a higher affinity toward electropositive metals due to their increased nucleophilicity at the N-site. Thus, in conjunction with early transition metals M’, amidophosphine ligands were shown to display κ2(P,N) coordination II, and, albeit less common, also κ1(N)-coordination III, while in the presence of both early and middle/late transition metals, amidophosphine ligands were shown to act as μ2 bridging ligand thereby forming heterobimetallic complexes of the type IV [3], (a), (b), (c), [4], (a), (b), (c), (d).
Scheme 1.

Most common bonding modes of aminophosphine and amidophosphine ligands.
Besides interesting structural features, PR2NHR’ ligands display various reactivities opening up a range of synthetically useful transformations [5]. For instance, we have recently shown [6] that in vinylidene and allenylidene complexes [RuCp(PPh2NHR)2( C (C)n = CHR′)]+ and [RuTp(PPh2NHR)2( C (C)n = CHR′)]+ (n = 0, 1; R = Ph, n-Pr; R′ = alkyl, aryl; Tp = trispyrazoloylborate) an intramolecular addition of the NHR’ moiety to the α-carbon of the cumulene moiety takes place (Scheme 2) resulting in the formation of novel four-membered aza-phospha-carbenes (Scheme 3, A). Complexes of the types [RuCp(PPh2NHPh)(CH3CN)2]+ and [RuCp*(PR2NHR’)(CH3CN)2]+ (R = Ph, i-Pr, R′ = Ph, C6F5) have been found to react with terminal alkynes and diynes to give amido butadiene complexes involving PR2NHR’ ligand migration (Scheme 3, B) [7,8]. Bunten et al. reported [9] the synthesis of the dinuclear ruthenium complex [Ru2(CO)3(μ2-PPh2)(μ2-Ph2PNMePPh2)(κ2(C,P)–C( O)NMePPh2)] containing a carboxamido-phospha-ruthen-acyclic moiety (Scheme 3, C). Herberhold et al. reported on the preparation of half sandwich complexes of the type [MCp(CO)2(C( O)N(S-NHPtBu2)PtBu2)] (M = Cr, Mo, W) by reacting [MCp(CO)3H] with PtBu2PN = S=NPtBu2
Scheme 2.
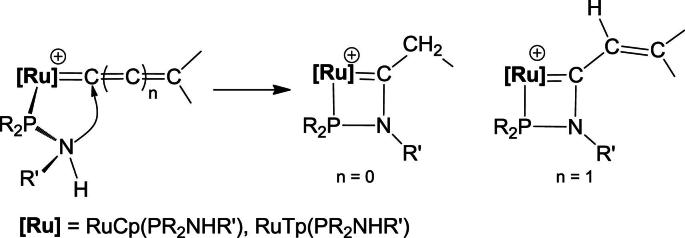
Intramolecular addition of the NHR’ moiety of an aminophosphine to the α-carbon of a cumulene moiety.
Scheme 3.

Aminophosphine ligands displaying various transformations to give unusual transition metal complexes.
(Scheme 3, D) [10]. As iron complexes are concerned, [FeCp(CO)2(PPh2NHNMe2]+ bearing a hydrazinophosphine ligand, which is closely related to PR2NHR’ ligands, was shown to react with nBuLi to give the carboxamido-phospha-ferracycle [FeCp(CO)(κ2(C,P)-(C O)-NNMe2-PPh2)] (Scheme 3, E) [11].
In the present paper we report on the synthesis of iron(II) complexes containing aminophosphine ligands of the type PR2NHR’ with R = Ph, iPr, R′ = iPr, tBu, Cy. We describe the reactivity of these complexes yielding, upon treatment with strong bases, novel cyclic four-membered carboxamido-phospha-ferracycle formed via intramolecular addition of the amine moiety of the bifunctional aminophosphine ligand according to Scheme 4.
Scheme 4.

Intramolecular addition of the NHR’ moiety of a aminophosphine ligand to coordinated CO.
2. Results and discussion
Treatment of [FeCp(CO)2Cl] with 1 equiv of the amidophosphine ligands [R2PNR’]− (R = Ph, iPr, R′ = iPr, tBu, Cy), prepared in situ by the reaction of R2PNHR’ with nBuLi in THF at −20 °C, afforded complexes of the type [FeCp(CO)(κ [2](C,P)-(C O)–NiPr-PPh2)] (1a), [FeCp(CO)(κ2(C,P)-(C O)-NtBu-PPh2)] (1b), and [FeCp(CO)(κ2(C,P)-(C O)-NCy-PiPr2)] (1c) in reasonable isolated yields (40–50%) (Scheme 5). Although no intermediate could be detected spectroscopically, it is most likely that the deprotonated R2PNHR’ ligand first forms a complex with a κ1(P)-bound rather than a κ1(N)-bound amidophosphine. It has to be noted that κ1(N) coordinated [PR2NR’]− ligands to late transition metals are, according to our knowledge, unknown.
Scheme 5.

Synthesis of complexes 1a–c.
Subsequently, intramolecular, chelate assisted, nucleophilic attack of the amido moiety of the PR2NR’ ligand at one of the two CO ligands took place resulting in the formation of novel four-membered carboxamido-phospha-ferracycle. This is also supported by the fact that 1a is readily formed if the cationic dicarbonyl complex [FeCp(CO)2(PPh2NHiPr)]+ with either Br- (2) or BF4− (2’) as counterions was treated with KOtBu as base according to Scheme 6. In the absence of base, even at elevated temperatures, no reaction took place. Complexes 2 and 2’ were readily obtained by the treatment of [FeCp(CO)2Br] with 1 equiv of the PPh2NHiPr ligand in the absence or presence of NaBF4, respectively, in THF at room temperature (Scheme 6).
Scheme 6.
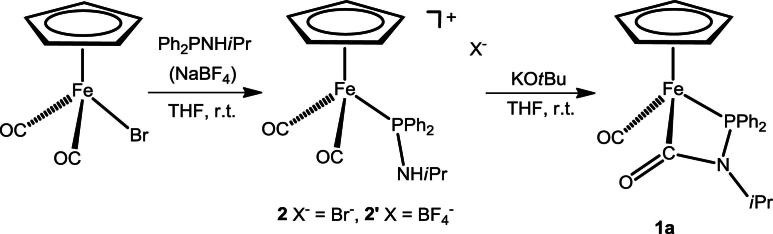
Synthesis of complex 1a via intermediate 2.
All complexes are thermally robust orange solids that are air stable both in the solid state and in solution for several days. Their identity was unequivocally established by 1H, 13C{1H} and 31P{1H} NMR, IR spectroscopy, and elemental analysis. In addition, the molecular structures of complexes 1a and 2 were determined by X-ray crystallography. Structural views are depicted in Figs. 1 and 2 with selected bond distances and angles reported in the captions.
Fig. 1.

Molecular structure of [FeCp(CO)(κ2(C,P)-(C O)-NiPr-PPh2)] (1a) showing 50% displacement ellipsoids. Selected distances and angles (Å, °): <Fe1–CCp> = 2.101(1), Fe1–P1 2.1773(3), Fe1–C6 1.739(1), Fe1–C7 1.981(1), C6–O1 1.158(2), C7–O2 1.219(2), N1–C7 1.404(2), P1–N1 1.6915(10), C7–Fe1–P1 69.43(4), Fe1–C7–O2 133.8(1), Fe1–C7–N7 103.7(1), Fe1–P1–N1 86.95(3), P1–N1–C7 99.7(1).
Fig. 2.

Molecular structure of 2 showing 50% displacement ellipsoids. Selected distances and angles (Å, °): <Fe1–CCp> = 2.104(2), Fe1–P1 2.2223(4), Fe1–C6 1.780(2), Fe1–C7 1.785(2), C6–O1 1.141(2), C7–O2 1.139(2), P1–N1 1.6473(13), N1–C20 1.477(2), P1–Fe1–C6 90.73(5), P1–Fe1–C7 93.83(5), C6–Fe1–C7 96.28(7), C20–N1–P1 125.14(10), N1 Br1 3.3854(13).
Complexes 1a–c display a single resonance in the 31P{1H} NMR at 111.6, 113.0, and 140.0 ppm, respectively. In the 13C{1H} NMR spectra, the Cp ring gives rise to a singlet in the range of about 80–82 ppm. The CO ligands exhibit a low-field doublet at 220.9, 221.0, and 221.0 ppm with coupling constants JPC of 24.4–26.4 Hz, while the resonance of the carboxamido carbon atoms give rise to a doublet centered at 200.4 (JPC = 39.0 Hz), 201.1 (JPC = 45.8 Hz), and 199.8 ppm (JPC = 37.8 Hz), respectively. In the IR spectrum, the characteristic CO stretching frequency of the CO ligand and the carboxamido unit was observed at 1937, 1919, and 1914 cm−1 and 1617, 1605, and 1618 cm−1, respectively. For comparison, the IR stretching frequency of the carboxamido unit in [FeCp(CO)(κ2(C,P)-(C O)-NNMe2-PPh2)] was found at 1642 cm−1 [11].
Complex 1a adopts a typical three legged piano stool conformation with the C and P atoms of the C( O)NiPr-PPh2 moiety and the C atom of the CO ligand as the legs (Fig. 1). The Fe1–C(Cp) distance on average is 2.101(1) Å. The iron carbon bond distances Fe1–C6 and Fe1–C7 are 1.739(1) and 1.981(1) Å, respectively, the latter being typical for an iron carbon single bond. Thus, also in agreement with the NMR and IR spectroscopic data, 1a is best described as a carboxamido complex (A) rather than an aminocarbene complex (B) (Scheme 7). The Fe1-P1 bond distance of 2.1773(3) Å is typical for iron phosphine complexes but slightly shorter than Fe1–P1 = 2.2223(4) Å in the parent complex 2 (Fig. 2). The C7–Fe1–P1 bite angle of the chelating C( O)NiPr-PPh2 ligand is 69.43(4)o. For comparison, in the related complex [FeCp(CO)(κ2(C,P)-(C O)-NNMe2-PPh2)] the bite angle is 70.5(1)o.
Scheme 7.

Resonance structures of complexes 1a–c.
Since acyl and carbamoyl ligands are typically nucleophilic at the carbonyl oxygen, i.e., resonance structure B may play a role, the reaction with carbon-based electrophiles may lead to aminocarbene complexes. It has to be kept in mind however that an electrophilic attack may occur also at the N atom of the carboxamido moiety resulting in P–N bond cleavage with concomitant N-alkylation.
Treatment of 1a with [Me3O]BF4 (1 equiv) at room temperature for 4 h in CH2Cl2 as the solvent results in the formation of the aza-phospha-carbene complex [FeCp(CO)(κ2(C,P) = C(OMe)-NiPr-PPh2)]+ (3) in 58% isolated yield (Scheme 8). This class of complexes belongs to a rare series of transition metal complexes in which the carbene moiety is part of a four-membered chelate ligand coordinated in a κ2 [2](C,P) mode [6], [7], (a), (b), (c), [12]. Complex 3 was characterized by elemental analysis and by 1H, 13C{1H}, and 31P{1H} NMR spectroscopy. Characteristic features comprise, in the 13C{1H} NMR spectrum, a marked low-field doublet resonance at 238.0 ppm (d, JCP = 34.8 Hz) assignable to the carbene carbon atom of the four-membered aza-phospha-carbene moiety. The carbonyl resonance gives rise to a doublet centered at 216.3 ppm (d, JCP = 22.2 Hz). The 31P{1H} NMR spectrum of 3 reveals a singlet at 115.0 ppm (cf. 111.6 ppm in 1a).
Scheme 8.

Reactivity of complex 1a toward the electrophile [Me3O]BF4 and the base NEt3.
On the other hand, protonation of 1a with the Brønsted acid [HNEt3]+ led to clean formation of complex 2a in 95% isolated yield (Scheme 8). Selective N-protonation took place which was associated with C–N bond cleavage and reformation of the CO and Ph2PNHiPr ligands.
In search of related iron systems where carboxamido-phospha-cycles may also be formed via nucleophilic attack of a κ1(P)-coordinated PR2NHR’ ligand, we prepared complexes of the type cis,trans,cis-[Fe(CO)2(Ph2PNHiPr)2(Br)2] (4a) and cis,trans,cis-[Fe(CO)2(Ph2PNHtBu)2(Br)2] (4b). These compounds were readily obtained as red, air stable solids by reacting cis-[Fe(CO)4(Br)2] with 2 equivs of the respective PR2NHR’ ligands in 85 and 88% yield (Scheme 9) in CH2Cl2 at room temperature. In these complexes the CO and bromide ligands are in a mutual cis position, whereas the phosphine ligands are trans to one another.
Scheme 9.

Synthesis of complexes 5a and 5b via intermediates 4a and 4b.
Treatment of 4a with 2 equivs of KOtBu in the presence of CO in THF at room temperature afforded the isomeric complexes trans-[Fe(CO)2(κ2(C,P)-(C O)–NiPr-PPh2)(Ph2PNHiPr)Br] (5a) and cis-[Fe-(CO)2(κ2(C,P)-(C O)–NiPr-PPh2)(Ph2PNHiPr)Br] (5a’) in a roughly 5:1 ratio in 50% overall yield. In agreement with experimental data DFT/B3LYP calculations confirm that the isomer with a trans CO arrangement is slightly more stable by 2.5 kcal/mol (free energy) than the corresponding cis isomer. In the case of 4b only one isomer, viz trans-[Fe(CO)2(κ2(C,P)-(C O)-NtBu-PPh2)(Ph2PNHtBu)Br] (5b), was formed in 40% yield (Scheme 9). These reactions again involve attack of the amido moiety of a deprotonated PR2NHR’ ligand on coordinated CO. It has to be mentioned that the yields of 5a and 5b were independent of whether 1 or 2 equivs of KOtBu were used, i.e., complexes such as [Fe(CO)2(κ2(C,P)-(C O)–NiPr-PPh2)2], were not observed. Complexes 5a and 5b are thermally robust red solids that are air stable in the solid state but slowly decompose in solution. Their identity was unequivocally established by 1H, 13C{1H} and 31P{1H} NMR, IR spectroscopy, and elemental analysis.
In the 13C{1H} NMR spectrum of the major isomer 5a the most noticeable resonances are a low-field signal of the two trans CO ligands observed as triplet centered at 212.4 ppm with a P–C coupling constants of 22.7 Hz. The resonance of the carboxamido moiety give rise to a doublet of doublets centered at 206.0 ppm with P–C couplings constant of 9.6 and 13.4 Hz. The 31P{1H} NMR spectrum of 5a reveals two doublets centered at 95.6 and 85.8 ppm with a large coupling constant of 84.7 Hz which is indicative for the phosphorus atoms being in a mutual trans position. Most 1H and 13C{1H} NMR of resonances of the minor isomer 5a' could not be reliably assigned since they were superimposed by the signals of the major isomer 5a. In the 31P{1H} NMR spectrum, however, 5a’ also exhibits two doublets centered at 94.8 (d, JPP = 115.9 Hz) and 90.0 ppm (d, JPP = 115.9 Hz). The large coupling constant again is consistent with a trans-P,P configuration. Similar NMR spectra were observed for 5b, and are thus not discussed here. In the IR spectrum of 5a and 5b the two CO ligands give rise to two bands at 1966 and 1960 cm-1 (5a) and 1955 and 1950 cm−1 (5b) which can be assigned to the asymmetric CO stretching frequency (cf 2143 cm−1 in free CO). As expected the symmetric CO stretching frequency is IR inactive which is also confirmed by DFT/B3LYP calculations. The appearance of two the resonances, which are only 5–6 cm−1 apart, may be due to intermolecular interactions, e.g. C O∙∙∙HN bonds, in the solid state (b), [13], (a). The CO vibration of the carboxamido phospha cycle is shifted to lower wavenumbers observed at 1619 and 1616 and cm−1, respectively. The scaled calculated frequencies νCO together with the experimentally observed values are given in Table 1 and show a reasonably good agreement.
Table 1.
Comparison of the DFT/B3LYP calculated and experimental νCO absorptions of 5a.
| νCOsym/cm−1 | νCOasym/cm−1 | νNC=O/cm−1 | |
|---|---|---|---|
| Calcd. | 2022 | 1980 | 1615 |
| Exptl. | Not observed | 1966/1960 | 1616 |
The molecular structure of complex 5a was determined by X-ray crystallography. A structural view is depicted in Fig. 3 with selected bond distances and angles reported in the caption. The geometry about the metal center is distorted octahedral with the two phosphorus and carbon atoms in trans position and the bromide and carbon atom of carboxamido moiety atoms in cis position (Fig. 3). The C32–Fe1–C33 bond angle is 170.6(2)o. The chelate system Fe1–P1–N1–(C31 O1)–Fe1 resembles closely in bond lengths and bond angles the corresponding system in complex 1a, except for the bond Fe1-P1, which is 2.1773(3) Å in 1a while it is 2.2480(9) Å in 5a due to the trans-influence of phosphorus P2 (Fe1–P2 = 2.2651(9) Å). The short intramolecular hydrogen bond N2–H2n∙∙∙O1 in 5a (N2∙∙∙O1 = 2.839(4) Å) has no effect on the bond angle Fe1-C31–O1 = 134.2(2)° as evident from the corresponding angle 133.8(1)o in complex 1a which lacks this interaction.
Fig. 3.

Molecular structure of trans-[Fe(CO)2(κ2(C,P)-(C O)-NiPr-PPh2)(Ph2PNHiPr)Br] (5a) (major isomer) showing 40% displacement ellipsoids. Selected distances and angles (Å, °): Fe1–Br1 2.4752(6), Fe1–P1 2.2480(9), Fe1–P2 2.2651(9), Fe1–C33 1.799(3), Fe1–C32 1.826(3), Fe1–C31 1.997(3), P2–N2 1.650(3), P1–N1 1.694(3), C31–O1 1.221(4), C31–N1 1.409(4), C32–Fe1–C33 170.6(2), P1–Fe1–P2 167.1(1), P1–Fe1–C31 68.9(1), Fe1–P1–N1 85.3(1), Fe1–C31–O1 134.2(2), Fe1–C31–N1 103.6(2), O1–C31–N1 122.2(3), P1–N1–C31 101.5(2), P1–N1–C13 132.4(2), N2 O1 2.839(4).
3. Conclusion
In the present study iron(II) complexes featuring one or two aminophosphine ligands of the type PR2NHR’ with R = Ph, iPr and R = iPr, tBu, Cy were synthesized. We demonstrated that upon treatment of [FeCp(CO)2X] (X = Cl, Br) with the anionic amidophosphine ligands [R2PNR’]−, or upon deprotonation of the PR2NHR’ ligand in complexes [FeCp(CO)2(PR2NHR’2)]+ and cis,trans,cis-[Fe(CO)2(Ph2PNHR’)2(Br)2] complexes featuring four-membered carboxamido-phospha-ferracycles were obtained. In the course of these reactions the highly nucleophilic amido nitrogen atom reacted readily in an intramolecular fashion with the electrophilic carbon atom of a CO ligand. We have further demonstrated that the carboxamido-phospha-ferracycles react with the carbon-based electrophile [Me3O]+ to afford an aza-phospha-carbene. This is a relatively rare type of transition metal complexes in which the carbene moiety is part of a four-membered chelate ligand coordinated in a κ2(C,P) mode. In the presence of protons, the carboxamido-phospha-ferracycle underwent clean C–N bond cleavage thereby reforming the starting complex [FeCp(CO)2(PR2NHR’2)]+.
4. Experimental
4.1. General
All manipulations were performed under an inert atmosphere of argon by using Schlenk techniques. The solvents were purified according to standard procedures [14]. The ligands PPh2NHiPr, PPh2NHtBu, and PPh2NHCy (b), [15], (a) and the complexes [FeCp(CO)2Cl], of [FeCp(CO)2Br] [16], and cis-[Fe(CO)4Br2] [17] were prepared according to the literature. The deuterated solvents were purchased from Aldrich and dried over 4 Å molecular sieves [1]. H, 13C{1H}, and 31P{1H} NMR spectra were recorded on Bruker AVANCE-250 and AVANCE-300 DPX spectrometers and were referenced to SiMe4 and H3PO4 (85%), respectively.
4.2. Syntheses
4.2.1. [FeCp(CO)(κ2(C,P)-(C O)–NiPr-PPh2)] (1a)
A solution of PPh2NHiPr (300 mg, 1.23 mmol) in THF (20 mL) was cooled to −20 °C and nBuLi (500 μL, 1.23 mmol, 2.5 M solution in n-hexane) was slowly added. The solution was stirred for 2 h at −20 °C and an additional hour at room temperature. After that, [FeCp(CO)2Cl] (262 mg, 1.23 mmol) was added and the mixture was stirred for 8 h at room temperature. The solvent was then removed under reduced pressure. The residue was redissolved in toluene (10 mL) and the solution was filtered through Celite. After removal of the solvent under reduced pressure, an orange solid was obtained which was washed with n-pentane (10 mL) and dried under vacuum. Yield: 207 mg (40%). Anal. calcd. for C22H22FeNO2P: C, 63.03; H, 5.29; N, 3.34. Found: C, 63.09; H, 5.12; N, 3.28. 1H NMR (δ, CDCl3, 20 °C): 7.87 (m, 4H, Ph), 7.51 (m, 6H, Ph), 4.38 (s, 5H, Cp), 3.49 (m, 1H, CH(CH3)2), 1.29 (d, J = 5.8 Hz, 3H, CH(CH3)2), 0.99 (d, J = 6.7 Hz, 3H, CH(CH3)2). 13C{1H} NMR (δ, CDCl3, 20 °C): 220.9 (d, JPC = 26.0 Hz, CO), 200.4 (d, JPC = 39.0 Hz, NCO), 137.2 (d, JPC = 40.6 Hz, Ph), 133.8 (d, JPC = 13.5 Hz, Ph), 131.8 (d, JPC = 2.7 Hz, Ph), 130.7 (d, JPC = 13.5 Hz, Ph), 130.4 (d, JPC = 2.7 Hz, Ph), 128.6 (d, JPC = 9.5 Hz, Ph), 128.4 (d, JPC = 7.5 Hz, Ph), 82.1 (s, Cp), 50.7 (d, JPC = 8.1 Hz, CH(CH3)2), 23.0 (s, CH(CH3)2), 21.4 (s, CH(CH3)2). 31P{1H} NMR (δ, CDCl3, 20 °C): 111.6. IR (ATR, 25 °C): 1937 (νC=O), 1617 (νC O).
4.2.2. [FeCp(CO)(κ2(C,P)-(C O)-NtBu-PPh2)] (1b)
This complex was prepared analogously to 1a with [FeCp(CO)2Cl] (400 mg, 1.55 mmol) and Ph2PNHtBu (400 mg, 1.55 mmol), and nBuLi (630 μL, 1.48 mmol, 2.5 M solution in n-hexane) as starting materials. Yield: 335 mg (50%). Anal. calcd for C23H24FeNO2P: C, 63.76; H, 5.58; N, 3.23. Found: C, 63.66; H, 5.63; N, 3.19. 1H NMR (δ, CDCl3, 20 °C): 7.90 (m, 4H, Ph), 7.48 (m, 6H, Ph), 4.39 (s, 5H, Cp), 1.20 (s, 9H, C(CH3)3). 13C{1H} NMR (δ, CDCl3, 20 °C): 221.0 (d, JPC = 24.4 Hz, CO), 201.1 (d, JPC = 45.8 Hz, NCO), 136.4 (d, JPC = 39.3 Hz, Ph), 133.8 (d, JPC = 41.3 Hz, Ph), 132.7 (d, JPC = 12.5 Hz, Ph), 131.2 (d, JPC = 3.2 Hz, Ph), 130.7 (d, JPC = 11.7 Hz, Ph), 130.3 (d, JPC = 3.2 Hz, Ph), 128.6 (d, JPC = 7.4 Hz, Ph), 128.4 (d, JPC = 5.8 Hz, Ph), 82.8 (s, Cp), 65.8 (s, CH(CH3)2), 29.5 (s, CH(CH3)2). 31P{1H} NMR (δ, CDCl3, 20 °C): 113.0. IR (ATR, 25 °C): 1919 (νC O), 1605 (νC O).
4.2.3. [FeCp(CO)(κ2(C,P)-(C O)-NCy-PiPR2)] (1c)
This complex was prepared analogously to 1a with [FeCp(CO)2Cl] (370 mg, 1.75 mmol), Ph2PNHCy (376 mg, 1.75 mmol), and nBuLi (700 μL, 1.75 mmol, 2.5 M solution in n-hexane) as starting materials. Yield: 307 mg (45%). Anal. calcd for C19H30FeNO2P: C, 58.48; H, 7.49; N, 3.59. Found: C, 58.53; H, 7.40; N, 3.62. 1H NMR (δ, CDCl3, 20 °C): 4.61 (s, 5H, Cp), 2,93 (m, 1H, NCH), 2.54 (m, 1H, CH(CH3)2), 2.18 (m, 1H, CH(CH3)2), 1,69 (bs, 4H, Cy), 1,53 (bs, 6H, Cy), 1.32 (d, J = 11.0 Hz, 3H, CH(CH3)2), 1.30 (s, 1H, Cy), 1.25 (d, J = 7.3 Hz, 3H, CH(CH3)2). 13C{1H} NMR (δ, CDCl3, 20 °C): 221.0 (d, JPC = 26.4 Hz, CO), 199.8 (d, JPC = 37.8 Hz, NCO), 80.8 (s, Cp), 58.5 (d, JPC = 8.1 Hz, NC), 32.7 (d, JPC = 71.9 Hz, CH(CH3)2), 30.6 (d, JPC = 19.3 Hz, Cy), 27.9 (d, JPC = 15.9 Hz, Cy), 26.8 (s, Cy), 25.3 (s, Cy), 20.4 (d, JPC = 5.7 Hz, CH(CH3)2), 18.7 (s, Cy), 18.0 (d, JPC = 5.74 Hz, CH(CH3)2). 31P{1H} NMR (δ, CDCl3, 20 °C): 140.0. IR (ATR, 25 °C): 1914 (νC O), 1618 (νC O).
4.2.4. [FeCp(CO)2(Ph2PNHiPr)]Br (2)
To a solution of [FeCp(CO)2Br] (1.00 g, 3.89 mmol) in THF (10 mL) Ph2PNHiPr (995 mg, 4.01 mmol) was added and the reaction mixture was stirred overnight at room temperature. Removal of the solvent afforded 2 as a yellow solid. Yield: 1.75 g (90%). Anal. calcd for C22H23BrFeNO2P: C, 52.94; H, 4.44; N, 2.81. Found: C, 53.04; H, 4.39; N, 2.85. 1H NMR (δ, CDCl3, 20 °C): 7.61-7.53 (m, 10H, Ph), 5.96 (d, J = 10.2 Hz, 1H, NH), 5.27 (s, 5H, Cp), 2.99 (bs, 1H, CH(CH3)2), 1.16 (d, J = 5.4 Hz, 6H, CH(CH3)2).). 13C{1H} NMR (δ, CDCl3, 20 °C): 210.3 (d, JPC = 27.1 Hz, CO), 135.0 (d, JPC = 59.1 Hz, Ph), 131.7 (d, JPC = 10.4 Hz, Ph), 131.3 (d, JPC = 10.7 Hz, Ph), 129.0 (d, JPC = 10. 9 Hz, Ph), 88.7 (s, Cp), 48.8 (d, JPC = 9.6 Hz, CH(CH3)2), 24.8 (d, JPC = 3.52 Hz, CH(CH3)2). 31P{1H} NMR (δ, CDCl3, 20 °C): 100.6. IR (ATR, 25 °C): 2040 (νC O), 1993 (νC O).
4.2.5. [FeCp(CO)2(Ph2PNHiPr)]BF4 (2’)
This complex was prepared analogously to 2 with [FeCp(CO)2Br] (350 mg, 1.65 mmol) and Ph2PNHiPr (400 mg, 1.65 mmol) as starting materials but in the presence of NaBF4 (182 mg, 1.65 mmol). Yield: 600 mg (72%). Anal. calcd for C22H23BF4FeNO2P: C, 52.11; H, 4.17; N, 2.76. Found: C, 52.14; H, 4.08; N, 2.80. NMR and IR spectra were identical to those of 2.
4.2.6. Reaction of [FeCp(CO)2(Ph2PNHiPr)]BF4 with KOtBu. Formation of [FeCp(CO)(κ2(C,P)-(C O)–NiPr-PPh2)] (1a)
A solution of [FeCp(CO)2(Ph2PNHiPr)]BF4 (2a’) (220 mg, 0.44 mmol) in THF (10 mL) was treated with KOtBu (55 mg, 0.48 mmol) and was stirred for 8 h. The solvent was removed under vacuum and the crude product was redissolved in toluene and filtered through Celite. After removal of the solvent under reduced pressure, 1a was obtained which was washed with n-pentane (10 mL) and dried under vacuum. Yield: 87 mg (48%).
4.2.7. [FeCp(CO)(κ2(C,P) C(OMe)-NiPr-PPh2)]BF4 (3)
A solution of 1a (500 mg, 1.19 mmol) in CH2Cl2 (10 mL) was treated with [Me3O]BF4 (177 mg, 1.19 mmol). After stirring for 4 h, insoluble materials were removed by filtration through Celite. On removal of the solvent, 5 was obtained as an orange solid which was washed with n-hexane, and dried under vacuum. Yield: 360 mg (58%). Anal. calcd for C35H39BBrF4FeN2O3P: C, 53.27; H, 4.98; N, 3.55. Found: C, 53.19; H, 4.89; N, 3.64. 1H NMR (δ, CDCl3, 20 °C): 7.58 (m, 10H, Ph), 4.80 (s, 5H, Cp), 4.33 (s, 3H, OCH3), 3.93 (m, 1H, CH(CH3)2), 1.40 (d, J = 6.4 Hz, 3H, CH(CH3)2), 0.92 (d, J = 6.4 Hz, 3H, CH(CH3)2). 13C{1H} NMR (δ, CDCl3, 20 °C): 238.0 (d, JPC = 34.8 Hz, Fe C), 216.3 (d, JPC = 22.2 Hz, CO), 134.7 (d, JPC = 22.3 Hz, Ph), 134.4 (d, JPC = 14.2 Hz, Ph), 134.2 (d, JPC = 22.2 Hz, Ph), 132.8 (d, JPC = 2.59 Hz, Ph), 131.9 (d, JPC = 2.5 Hz, Ph), 131.3 (d, JPC = 11.1 Hz, Ph), 129.9 (d, JPC = 11.3 Hz, Ph), 83.1 (s, Cp), 64.9 (s, OCH3), 55.8 (d, JPC = 5.5 Hz, CH(CH3)2), 21.8 (s, CH(CH3)2), 20.8 (s, CH(CH3)2). 31P{1H} NMR (δ, CDCl3, 20 °C): 115.0. IR (ATR, 25 °C): 2004 (νC O).
4.2.8. Protonation of 1a with [HNEt3]Cl. Formation of 2
A solution of 1a (500 mg, 1.19 mmol) in CH2Cl2 (10 mL) was treated with [HNEt3]Cl (165 mg, 1.19 mmol). After stirring for 4h, insoluble materials were removed by filtration through Celite. On removal of the solvent, 2 was obtained as a yellow solid which was collected on a glass frit, washed with n-hexane, and dried under vacuum. Yield: 514 mg (95%).
4.2.9. cis,trans,cis-[Fe(CO)2(Ph2PNHiPr)2(Br)2] (4a)
To a solution of cis-[Fe(CO)4Br2] (1.00 g, 3.05 mmol) in CH2Cl2 (10 mL) Ph2PNHiPr (1.52 g, 6.25 mmol) was added at 0 °C and the mixture was stirred overnight at room temperature. The solution was then filtered through Celite. After removal of the solvent under reduced pressure, an orange solid was obtained which was washed with diethyl ether (10 mL) and dried under vacuum. Yield: 1.96 g (85%). Anal. calcd for C32H36Br2FeN2O2P2: C, 50.69; H, 4.79; N, 3.69. Found: C, 50.72; H, 4.86; N, 3.60. 1H NMR (δ, CDCl3, 20 °C): 7.99 (bs, 8H, Ph), 7.48 (bs, 12H, Ph), 3.42 (bs, 2H, NH), 2.29 (m, 2H, CH(CH3)2), 0.91 (d, JPC = 6.3 Hz, 12H, CH(CH3)2). 13C{1H} NMR (δ, CDCl3, 20 °C): 212.7 (t, JPC = 22.7 Hz, CO), 133.6 (d, JPC = 25. Hz, Ph), 133.0 (dd, JPC = 5.1 Hz, Ph), 131.7 (dd, JPC = 5.4 Hz, Ph), 130.8 (s, Ph, 127.9 (dd, JPC = 4.6 Hz, Ph), 46.4 (dd, JPC = 4.9 Hz, CH(CH3)2), 24.7 (s, CH(CH3)2). 31P{1H} NMR (δ, CDCl3, 20 °C): 80.6. IR (ATR, 25 °C): 2041 (νC O), 1986 (νC O).
4.2.10. cis,trans,cis-[Fe(CO)2(Ph2PNHtBu)2(Br)2] (4b)
This complex was prepared analogously to 4a with cis-[Fe(CO)4Br2] (845 mg, 2.58 mmol) and Ph2PNHtBu (1.33 g, 5.16 mmol) as starting materials. Yield: 1.7 mg (88%). Anal. calcd for C34H40Br2FeN2O2P2: C, 51.94; H, 5.13; N, 3.56. Found: C, 52.04; H, 5.20; N, 3.46. 1H NMR (δ, acetone-d6, 20 °C): 8.36 (bs, 8H, Ph), 7.48 (bs, 12H, Ph), 3.7 (bs, 2H, NH), 0.87 (s, 18H, C(CH3)3). 13C{1H} NMR (δ, CDCl3, 20 °C): 213.3 (t, JPC = 24.0 Hz, CO), 134.2 (dd, JPC = 25.7 Hz, Ph), 133.5 (dd, JPC = 5.3 Hz, Ph), 130.7 (s, Ph), 127.7 (dd, JPC = 4.7 Hz, Ph), 55.5 (s, C(CH3)3), 31.7 (s, C(CH3)3). 31P{1H} NMR (δ, acetone-d6, 20 °C): 72.2. IR (ATR, 25 °C): 2035 (νC O), 1980 (νC O).
4.2.11. trans-[Fe(CO)2(κ2(C,P)-(C O)–NiPr-PPh2)(Ph2PNHiPr)Br] (5a) and cis-[Fe(CO)2(κ2(C,P)-(C O)–NiPr-PPh2)(Ph2PNHiPr)Br] (5a')
A Schlenk tube was charged with 4a (400 mg, 0.53 mmol) and KOtBu (122 mg, 1.06 mmol). Under a CO atmosphere THF (10 mL) was added and the mixture was stirred overnight at room temperature. The red solution was then filtered through Celite and the solvent was removed under reduced pressure. The residue was redissolved in CH2Cl2. Insoluble materials were removed by filtration. After evaporation of the solvent in vacuo, a red solid was obtained which was washed with n-pentane (10 mL) and dried under vacuum. Yield: 185 mg (50%). Anal. calcd for C33H35BrFeN2O3P2: C, 56.19; H, 5.00; N, 3.97. Found: C, 56.00; H, 5.12, N, 4.01. Major isomer: 1H NMR (δ, CDCl3, 20 °C): 7.85 ( d, J = 6.79 Hz, 8H, Ph), 7.50 (d, J = 8.38 Hz, 8H, Ph), 7.43 (s, 4H, Ph), 4.70 (bs, 1H, NH), 3.46 ( m, 1H, CH(CH3)2), 3.12 ( m, 1H, CH(CH3)2), 1.31 (d, J = 6.4 Hz, 6H, CH(CH3)2), 0.94 (d, J = 6.4 Hz, 6H, CH(CH3)2). 13C{1H} NMR (δ, CDCl3, 20 °C): 212.4 (t, JPC = 22.7 Hz, CO), 206.0 (dd, JPC = 9.6 Hz, JPC = 13.4 Hz, NCO), 135.1 (d, JPC = 37.5 Hz, Ph), 133.3 (d, JPC = 12.1 Hz, Ph), 133.1 (d, JPC = 13.5 Hz, Ph), 131.8 (s, Ph), 130.2 (s, Ph), 128.7 (d, JPC = 11.1 Hz, Ph), 128.0 (d, JPC = 11.1 Hz, Ph), 45.7 (t, JPC = 12.7 Hz, Ph), 25.5 (s, CH(CH3)2), 22.0 (s, CH(CH3)2). 31P{1H} NMR (δ, CDCl3, 20 °C): 95.6 (d, JPP = 84.7 Hz), 85.8 (d, JPP = 84.7 Hz). IR (ATR, 25 °C): 1966 (νC O), 1960 (νC O), 1616 (νC O). Minor isomer cis-[Fe(CO)2(κ2(C,P)-(C O)–NiPr-PPh2)(Ph2PNHiPr)Br] (5a’): Most 1H and 13C NMR resonances are superimposed by the signals of the major isomer and could not reliably assigned. 31P{1H} NMR (δ, CDCl3, 20 °C): 94.8 (d, JPP = 115.9 Hz), 90.0 (d, JPP = 115.9 Hz).
4.2.12. trans-[Fe(CO)2(κ2(C,P)-(C O)-NtBu-PPh2)(Ph2PNHtBu)Br] (5b)
This complex was prepared analogously to 5a with 4b (0.70 g, 0.89 mmol) as starting material. Yield: 260 mg (40%). Anal. calcd for C35H39BrFeN2O3P2: C, 57.32; H, 5.36; N, 3.82. Found: C, 57.40; H, 5.29; N, 3.79. 1H NMR (δ, CDCl3, 20 °C): 7.98 (bs, 8H, Ph), 7.42 (bs, 12H, Ph), 4.84 ( bs, 1H, NH), 3.46 ( m, 1H, CH(CH3)2), 1.28 (s, 9H, C(CH3)3), 1.05 (s, 9H, C(CH3)3). 13C{1H} NMR (δ, CDCl3, 20 °C): 213.3 (dd, JPC = 23.4 Hz, JPC = 22.9 Hz CO), 206.0 (dd, JPC = 9.6 Hz, JPC = 13.4 Hz, NCO), 136.2 (d, JPC = 47.7 Hz, Ph), 136.1 (d, JPC = 47.7 Hz, Ph), 134.5 (d, JPC = 10.8 Hz, Ph), 132.8 (d, JPC = 10.8 Hz, Ph), 132.6 (d, JPC = 11.6 Hz, Ph), 131.4 (d, JPC = 2.3 Hz, Ph), 129.9 (d, JPC = 2.3 Hz, Ph), 128.5 (d, JPC = 10.6 Hz, Ph), 127.6 (d, JPC = 10.0 Hz, Ph), 62.3 (d, JPC = 7.6 Hz, C(CH3)3), 55.9 (d, JPC = 13.9 Hz, C(CH3)3), 32.1 (d, JPC = 3.1 Hz, C(CH3)3), 29.5 (d, JPC = 1.5 Hz, C(CH3)3). 31P{1H} NMR (δ, CDCl3, 20 °C): 90.4 (d, JPP = 79.3 Hz), 87.3 (d, JPP = 79.3 Hz. IR (ATR, 25 °C): 1955 (νC=O), 1950 (νC O),1619 (νC O).
4.3. X-ray structure determinations
Single crystals for X-ray diffraction were obtained as follows: [FeCp(CO)(κ2(C,P)-(C O)–NiPr-PPh2)] (1a), by vapor diffusion of n-pentane into a THF solution, [FeCp(CO)2(Ph2PNHiPr)]Br (2) by vapor diffusion of Et2O into a CH2Cl2 solution; trans/cis-[Fe(CO)2(κ2(C,P)-(C O)–NiPr-PPh2)(Ph2PNHiPr)Br] (5a/5a') by vapor diffusion of Et2O into a CH2Cl2 solution. The color of the crystals varied from orange to red brown. X-ray diffraction data were collected at T = 100 K on a Bruker Kappa APEX-2 CCD diffractometer with an Oxford Cryosystems cooler using graphite-monochromatised Mo-Kα radiation (λ = 0.71073 Å) and fine sliced φ- and ω-scans covering complete spheres of the reciprocal space. After data integration with program SAINT corrections for absorption and detector effects were applied with the program SADABS [18] The structures were solved by direct methods (SHELXS97) and refined on F [2] with the program SHELXL97 [19] Non-hydrogen atoms were refined anisotropically. Most H atoms were placed in calculated positions and thereafter refined as riding. In (5a) Br was partly substituted by CO and vice versa (84% trans- and 16% in cis-dicarbonyl configuration), and the subordinately occupied sites were refined with distance and displacement parameter restraints. All crystal structures were checked with the program PLATON [20] Molecular graphics was generated with program MERCURY [21] Crystal data and experimental details are given in Table 2.
Table 2.
Details for the crystal structure determinations of compounds 1a, 2, and 5a.
| 1a | 2 | 5a | |
|---|---|---|---|
| Formula | C22H22FeNO2P | C22H23BrFeNO2P | C33H35BrFeN2O3P2 |
| fw | 419.23 | 500.14 | 705.33 |
| Cryst.size, mm | 0.45 × 0.26 x 0.14 | 0.59 × 0.20 × 0.18 | 0.25 × 0.20 × 0.18 |
| Color, shape | Red plate | Yellow prism | Red prism |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n (no. 14) | P21/c (no. 14) | P21/c (no. 14) |
| a, Å | 10.0648(3) | 13.2697(2) | 10.1667(4) |
| b, Å | 14.5893(5) | 17.5534(2) | 17.7127(7) |
| c, Å | 13.4024(4) | 9.2565(2) | 18.9313(8) |
| α, deg | 90 | 90 | 90 |
| β, deg | 92.871(2) | 92.768(2) | 105.453(2) |
| γ, deg | 90 | 90 | 90 |
| V, Å3 | 1965.52(11) | 2153.59(6) | 3285.9(2) |
| T, K | 100(2) | 100(2) | 100(2) |
| Z | 4 | 4 | 4 |
| ρcalc, g cm-3 | 1.417 | 1.543 | 1.426 |
| μ, mm−1 (MoKα) | 0.865 | 2.647 | 1.807 |
| F(000) | 872 | 1016 | 1448 |
| Absorption corrections | Multi-scan, 0.89–0.76 | Multi-scan, 0.65–0.43 | Multi-scan, 0.74–0.58 |
| θ range, deg | 2.06–30.11 | 1.93-30.00 | 2.08-27.50 |
| No. of rflns measd | 65996 | 39753 | 50035 |
| Rint | 0.034 | 0.023 | 0.076 |
| No. of rflns unique | 5771 | 6253 | 7526 |
| No. of rflns I > 2σ(I) | 4787 | 5578 | 5356 |
| No. of params/restraints | 246/0 | 258/0 | 387/136 |
| R1 (I > 2σ(I)) a | 0.0253 | 0.0269 | 0.0406 |
| R1 (all data) | 0.0366 | 0.0320 | 0.0742 |
| wR2 (I > 2σ(I)) | 0.0593 | 0.0682 | 0.0891 |
| wR2 (all data) | 0.0642 | 0.0713 | 0.1081 |
| Diff.Four.peaks min/max, eÅ−3 | −0.32/0.40 | −0.45/0.82 | −0.60/0.77 |
R1 = Σ||Fo|–|Fc||/Σ|Fo|, wR2 = {Σ[w(Fo2 – Fc2)2]/Σ[w(Fo2)2]}½, GooF = {Σ[w(Fo2 – Fc2)2]/(n–p)}½
4.4. Computational details
Calculations were performed using the Gaussian 09 software package [22], and the B3LYP functional (c), [23], (a), (b) without symmetry constraints. The optimized geometries were obtained with the Stuttgart/Dresden ECP (SDD) basis set c), [24], a), b) to describe the electrons of the iron atom. For all other atoms the 6–31g** basis set was employed [25], (a), (b), (c), (d), (e), (f). Frequency calculations were performed to confirm the nature of the stationary points. A scaling factor of 0.9521 was applied for the CO frequencies [26].
Acknowledgments
Financial support by the Austrian Science Fund (FWF) is gratefully acknowledged (Project No. P24202-N17).
References
- 1.(a) Gopalakrishnan J. Appl. Organomet. Chem. 2009;23:291. [Google Scholar]; (b) Ansell J., Wills M. Chem. Soc. Rev. 2002;31:259. doi: 10.1039/b200392a. [DOI] [PubMed] [Google Scholar]; (c) Fei Z., Dyson P.J. Coord. Chem. Rev. 2005;249:2056. [Google Scholar]; (d) Appleby T., Woollins J.D. Coord. Chem. Rev. 2002;235:121. [Google Scholar]; (e) Balakrishna M.S., Sreenivasa Reddy V., Krishnamurthy S.S., Nixon J.F., Burckett J.C.T.R., Laurent St. Coord. Chem. Rev. 1994;129:1. [Google Scholar]
- 2.(a) Priya S., Balakrishna M.S., Mague J.T. J. Organomet. Chem. 2003;679:116. [Google Scholar]; (b) Priya S., Balakrishna M.S., Mobin S.M., McDonald R. J. Organomet. Chem. 2003;688:227. [Google Scholar]; (c) Gaw K.G., Slawin A.M.Z., Smith M.B. Organometallics. 1999;18:3255. [Google Scholar]; (d) Fenske D., Maczek B., Maczek K. Z. Anorg. Allg. Chem. 1997;623:1113. [Google Scholar]; (e) Kühl O., Junk P.C., Hey-Hawkins E. Z. Anorg. Allg. Chem. 2000;626:1591. [Google Scholar]; (f) Kühl O., Blaurock S., Sieler J., Hey-Hawkins E. Polyhedron. 2001;20:111. [Google Scholar]; (g) Kühl O., Koch T., Somoza F.B., Jr., Junk P.C., Hey-Hawkins E., Plat D., Eisen M.S. J. Organomet. Chem. 2000;604:116. [Google Scholar]; (h) Suss-Fink G., Pellingeelli M.A., Tiripicchhio A. J. Organomet. Chem. 1987;320:101. [Google Scholar]; (i) Ainscough E.W., Brodie A.M., Wong S.T. J. Chem. Soc. Dalton Trans. 1977:915. [Google Scholar]
- 3.For recent examples see:; (a) Kuppuswawamy S., Bezpalko M.W., Powers T.M., Turnbull M.M., Foxman B.M., Thomas C.M. Inorg. Chem. 2012;51:8225. doi: 10.1021/ic300776y. [DOI] [PubMed] [Google Scholar]; (b) Evers D.A., Bluestein A.H., Foxman B.M., Thomas C.M. Dalton Trans. 2012;41:8111. doi: 10.1039/c2dt00034b. [DOI] [PubMed] [Google Scholar]; (c) Cooper B.G., Fafard C.M., Foxman B.M., Thomas C.M. Organometallics. 2010;29:5179. [Google Scholar]
- 4.(a) Nagashima H., Sue T., Oda T., Kanemitsu A., Matsumoto T., Motoyama Y., Sunada Y. Organometallics. 2006;25:1987. [Google Scholar]; (b) Sunada Y., Sue T., Matsumoto T., Nagashima H. J. Organomet. Chem. 2006;691:3176. [Google Scholar]; (c) Sue T., Sunada Y., Nagashima H. Eur. J. Inorg. Chem. 2007:2897. [Google Scholar]; (d) Tsutsumi H., Sunada Y., Shiota Y., Yoshizawa K., Nagashima H. Organometallics. 2009;28:1988. [Google Scholar]
- 5.Kühl O. Coord. Chem. Rev. 2006;250:2867. For a review of the chemistry of N-phosphinoamides see. [Google Scholar]
- 6.Pavlik S., Mereiter K., Puchberger M., Kirchner K. Organometallics. 2005;24:3561. [Google Scholar]
- 7.Pavlik S., Mereiter K., Schmid R., Kirchner K. Organometallics. 2003;22:1771. [Google Scholar]
- 8.Jimenez-Tenorio M., Puerta M.C., Valerga P. Eur. J. Inorg. Chem. 2005:2631. doi: 10.1021/ic201912b. [DOI] [PubMed] [Google Scholar]
- 9.Bunten K.A., Farrar D.H., Poë A.J., Lough A.J. Organometallics. 2000;19:3674. [Google Scholar]
- 10.Herberhold M., Ehrenreich W., Guldner K., Jellen W., Thewalt U., Klein H.P. Z. Naturforsch. 1983;38B:1383. [Google Scholar]
- 11.Harlan C.J., Wright T.C., Atwood J.L., Bott S.G. Inorg. Chem. 1991;30:1955. [Google Scholar]
- 12.For related cyclic aminocarbenes, see; (a) Ruiz J., García L., Perandones B.F., Vivanco M. Angew. Chem. Int. Ed. 2011;50:3010. doi: 10.1002/anie.201007937. [DOI] [PubMed] [Google Scholar]; (b) Zhang S., Xu Q., Sun J., Chen J. Organometallics. 2001;20:2387. [Google Scholar]; (c) Yin J., Chen J., Xu W., Zhang Z., Tang Y. Organometallics. 1988;7:21. [Google Scholar]
- 13.(a) Braunstein P., Taquet J.-P., Siri O., Welter R. Angew. Chem. Int. Ed. 2004;43:5922. doi: 10.1002/anie.200461175. [DOI] [PubMed] [Google Scholar]; (b) Camiolo S., Coles S.J., Gale P.A., Hursthouse M.B., Mayer T.A., Paver M.A. Chem. Commun. 2000:275. [Google Scholar]
- 14.Perrin D.D., Armarego W.L.F. third ed. Pergamon; New York: 1988. Purification of Laboratory Chemicals. [Google Scholar]
- 15.(a) Sisler H.H., Smith N.L. J. Org. Chem. 1961;26:611. [Google Scholar]; (b) Poetschke N., Nieger M., Khan M.A., Niecke E., Ashby M.T. Inorg. Chem. 1997;36:4087. [Google Scholar]
- 16.Piper T.S., Cotton F.A., Wilkinson G.J. Inorg. Nucl. Chem. 1955;1:165. [Google Scholar]
- 17.Hieber W., Wirsching A.Z. Anorg. Allg. Chem. 1940;245:305. [Google Scholar]
- 18.Bruker Computer Programs: APEX2, SAINT, SADABS, and SHELXTL. Bruker AXS Inc; Madison, WI: 2012. [Google Scholar]
- 19.Sheldrick G.M. Acta Crystallogr. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 20.Spek A.L. J. Appl. Crystallogr. 2003;36:7. [Google Scholar]
- 21.Macrae C.F., Edgington P.R., McCabe P., Pidcock E., Shields G.P., Taylor R., Towler M., van de Streek J. J. Appl. Crystallogr. 2006;39:453. [Google Scholar]
- 22.Frisch M.J., et al. Gaussian, Inc.; Wallingford, CT: 2009. Gaussian 09, Revision A.02. [Google Scholar]
- 23.(a) Becke A.D. J. Chem. Phys. 1993;98:5648. [Google Scholar]; (b) Miehlich B., Savin A., Stoll H., Preuss H. Chem. Phys. Lett. 1989;157:200. [Google Scholar]; (c) Lee C., Yang W., Parr G. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 24.a) Haeusermann U., Dolg M., Stoll H., Preuss H. Mol. Phys. 1993;78:1211. [Google Scholar]; b) Kuechle W., Dolg M., Stoll H., Preuss H. J. Chem. Phys. 1994;100:7535. [Google Scholar]; c) Leininger T., Nicklass A., Stoll H., Dolg M., Schwerdtfeger P. J. Chem. Phys. 1996;105:1052. [Google Scholar]
- 25.(a) McLean A.D., Chandler G.S. J. Chem. Phys. 1980;72:5639. [Google Scholar]; (b) Krishnan R., Binkley J.S., Seeger R., Pople J.A. J. Chem. Phys. 1980;72:650. [Google Scholar]; (c) Wachters A.J.H. Chem. Phys. 1970;52:1033. [Google Scholar]; (d) Hay P.J. J. Chem. Phys. 1977;66:4377. [Google Scholar]; (e) Raghavachari K., Trucks G.W. J. Chem. Phys. 1989;91:2457. [Google Scholar]; (f) Curtiss L.A., McGrath M.P., Blaudeau J.-P., Davis N.E., Binning R.C., Jr., Radom L. J. Chem. Phys. 1995;103:6104. [Google Scholar]; (g) McGrath M.P., Radom L. J. Chem. Phys. 1991;94:511. [Google Scholar]
- 26.Yu L., Srinivas G.N., Schwartz M. J. Mol. Struct. (Theochem) 2003;625:215. [Google Scholar]