

Abstract
Altered copper homeostasis and hypercholesterolemia have been identified independently as risk factors for Alzheimer's disease (AD). Abnormal copper and cholesterol metabolism are implicated in the genesis of amyloid plaques and neurofibrillary tangles (NFT), which are two key pathological signatures of AD. Amyloidogenic processing of a sub-population of amyloid precursor protein (APP) that produces Aβ occurs in cholesterol-rich lipid rafts in copper deficient AD brains. Co-localization of Aβ and a paradoxical high concentration of copper in lipid rafts fosters the formation of neurotoxic Aβ:copper complexes. These complexes can catalytically oxidize cholesterol to generate H2O2, oxysterols and other lipid peroxidation products that accumulate in brains of AD cases and transgenic mouse models. Tau, the core protein component of NFTs, is sensitive to interactions with copper and cholesterol, which trigger a cascade of hyperphosphorylation and aggregation preceding the generation of NFTs. Here we present an overview of copper and cholesterol metabolism in the brain, and how their integrated failure contributes to development of AD.
Keywords: copper, cholesterol, Alzheimer's disease, ApoE, amyloid precursor protein, Aβ, tau, lipid rafts
Introduction
Since the first case description of Alzheimer's disease (AD) by Alois Alzheimer, AD has steadily gained increasing recognition as the most common neurodegenerative disorder worldwide, and as such, it has been the focus of intense biomedical research efforts. The main brain regions affected in AD include the enterohinal cortex, hippocampus, basal forebrain and amygdala, which exhibit synaptic loss resulting in extensive brain atrophy (Mattson, 2004). Currently, confirmation of an AD diagnosis requires post-mortem examination of the brain for the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs) (Alzheimer, 1907), which are the key identifying pathological signatures of AD. Clinically, AD patients present with symptoms of memory loss, altered personality and behavior, and impaired executive function. Medical advances that have contributed to increased longevity and an aging world population is propelling the number of AD cases on a trajectory to quadruple to 106.2 million over the next 40 years (Brookmeyer et al., 2007). This is projected to lead to an ~500% increase in medical and health care costs (http://www.alz.org). In addition, AD can cost patients' family and friends their physical and emotional health, as well as significant financial loss. Therefore, there is a huge demand for an effective disease-modifying therapy for AD that will extend the productive and healthy lifetime of individuals, and reduce its negative socio-economic impact on society.
AD cases can be broadly classified as familial or sporadic. Early-onset familial AD (FAD), which comprises 5–10% of AD cases, is caused by genetic mutations in the genes encoding the amyloid precursor protein (APP) and presenilins 1 and 2 (PSEN1 and PSEN2). The remaining AD cases are late-onset and sporadic, with diverse biochemical and clinical presentations. Common to both types of AD is the increased level of Aβ peptide, especially the neurotoxic Aβ42 species, as a consequence of enhanced cleavage of APP by β- and γ-secretases. The complex and multifactorial biochemistry that underlies sporadic AD presents a challenge to the discovery of a specific etiology and hence the development of an effective universally applicable therapy. Aging is acknowledged as the leading risk factor for AD, with numerous genetic and environmental factors also found to associate with AD (Pappolla et al., 2003; Cannon and Greenamyre, 2011; Hollingworth et al., 2011; Naj et al., 2011; Bertram and Tanzi, 2012). However, detailed understanding of the contribution of most of these risk factors to the etiology of sporadic AD is yet to be achieved. Amongst the various risk factors for AD, corrupted copper and cholesterol homeostasis have attracted considerable attention independently over the last two decades as potential therapeutic targets. Both copper and cholesterol are critical to normal brain development and function, which include neurotransmission, myelination, and synaptogenesis. Mounting evidence indicates a connection between copper and cholesterol pathways in the pathogenesis of AD. Here we present an overview of copper and cholesterol metabolism in the brain, and the first description of how their integrated failure may lead to the development of AD.
Brain copper homeostasis
There is an increasing appreciation of a role for copper in normal brain development and function. The importance of copper for normal brain function is underscored by the profound neurodegeneration in Menkes and Wilson diseases, rare genetic disorders of copper deficiency and overload, respectively (Burkhead et al., 2011; Kaler, 2011). More commonly, copper dyshomeostasis is evident in a number of major neurodegenerative disorders, which include AD and Parkinson's disease (PD). Compromised copper regulation in these disorders potentiates inappropriate interactions with aggregation-prone proteins such as Aβ and α-synuclein, respectively, to form neurotoxic copper-protein aggregates (Atwood et al., 1998, 2004; Drew et al., 2008; Gunn et al., 2012; Camponeschi et al., 2013).
The estimated copper content in a healthy human adult brain is 7–10% of total body copper, similar to that found in the liver, the major organ of copper homeostasis (Cartwright and Wintrobe, 1964; Linder, 1991). The redox cycling of copper between Cu2+ and Cu+ oxidation states is exploited by many metabolic processes to catalyze various enzyme reactions. Some of the key cuproenzymes include ceruloplasmin (CP; iron transport and radical scavenging), Cu,Zn-superoxide dismutase 1 (SOD1; antioxidant defense), dopamine-β-hydroxylase (neurotransmission), cytochrome c oxidase (CCO; electron transport and oxidative phosphorylation), and tyrosinase (pigmentation) [reviewed in (Linder and Hazegh-Azam, 1996; Nevitt et al., 2012)]. Paradoxically, the utilization of copper's redox activity in Fenton or Haber–Weiss reactions promotes the production of toxic reactive oxygen species (ROS) (Halliwell and Gutteridge, 1984). Oxidative stress, such as that evident in AD, ensues when the production of ROS overwhelms available antioxidant defenses. The brain is particularly vulnerable to oxidative stress with disproportionately low levels of antioxidants relative to its high rate of oxidative metabolism (Floyd, 1999). Therefore, the dichotomous nature of copper demands a precise regulation to maintain an appropriate level and distribution in the brain, and to prevent inadvertent interactions with other cellular components.
In a human adult brain, copper is particularly enriched in the hippocampus (Dobrowolska et al., 2008) and substantia nigra (Davies et al., 2013). In the hippocampus, following neuronal depolarization, about 15 μM of copper is released into the glutamatergic synaptic cleft from synaptic vesicles (Rajan et al., 1976; Hartter and Barnea, 1988; Kardos et al., 1989; Barnea et al., 1990; Hopt et al., 2003). This release of copper during neurotransmission involves copper-independent trafficking of the copper transporter ATP7A to neuronal processes after glutamate excitation of synaptic N-methyl-D-aspartate (NMDA) receptors (Schlief et al., 2005, 2006). This ATP7A-mediated copper release has been proposed to be a neuroprotective mechanism, whereby synaptic copper catalyzes S-nitrosylation of NMDA receptors to down-regulate their activity, and thus protects neurons from excitotoxicity (Schlief et al., 2006). The ability of copper to mediate suppression of long-term potentiation (LTP) in hippocampal slices further supports a role for copper as a neuromodulator (Doreulee et al., 1997). In contrast to a role for copper in hippocampal glutamatergic synapse neurotransmission, the role of copper in the substantia nigra remains obscure. In the substantia nigra, there is a concomitant elevation of copper, iron and zinc compared to other brain regions (Davies et al., 2013). The substantia nigra is therefore, more susceptible to oxidative stress that can be induced by redox-active copper and iron. Paradoxically, greater concentration of copper and zinc is likely to be associated with a higher demand for copper- and zinc-dependent antioxidant activity of SOD1 in the substantia nigra to counter oxidative stress (Saggu et al., 1989; Davies et al., 2013).
Brain copper import and export
Copper is generally acquired through dietary intake of copper-rich foods such as animal liver, nuts, and shellfish. Dietary copper (Cu2+) is absorbed into the body by intestinal mucosal cells, and delivered to the liver via the portal blood bound to albumin, transcuprein, amino acids, and small peptides (Bearn and Kunkel, 1954; Neumann and Sass-Kortsak, 1967; Weiss and Linder, 1985; Wirth and Linder, 1985). In hepatic cells, copper is incorporated into CP, which carries the majority of copper in the blood for circulation to extrahepatic tissues (Campbell et al., 1981; Lee et al., 1993). Excess dietary copper is secreted into the bile from hepatic cells via biliary canaliculi and eliminated in feces.
Emerging studies of brain copper homeostasis is beginning to shed light on the mechanism of brain copper import, distribution and export. A concentration difference of up to 100-fold between plasma (11–25 μM) (Tietz, 1987) and cerebrospinal fluid (CSF; ~0.25 μM) copper signifies restricted copper uptake into the brain (Kjellin, 1963; Lentner, 1981). A rat brain perfusion study suggested that non-protein bound free copper ion is the predominant copper species that enters the brain via both the blood-brain barrier (BBB) and the blood-cerebrospinal fluid barrier (BCB) (Choi and Zheng, 2009). The greater uptake of copper into the brain parenchyma compared to CSF suggests that the BBB is the main site through which copper enters the brain. An enrichment of copper transporters CTR1, ATP7A, and ATP7B in the brain capillaries and choroid plexus, implicates their involvement in controlling copper influx into the brain parenchyma and CSF (Iwase et al., 1996; Qian et al., 1998; Kuo et al., 2006; Niciu et al., 2006; Choi and Zheng, 2009; Donsante et al., 2010; Davies et al., 2013).
The high affinity copper transporter CTR1 is the primary gatekeeper of copper entry into the brain via brain capillary endothelial cells that form the BBB and choroid plexus epithelial cells that form the BCB. Consistent with a role for CTR1 in cellular copper uptake, it is localized predominantly at the apical membrane in human choroid plexus epithelial cells (Davies et al., 2013). Furthermore, CTR1 heterozygous mice have lower brain copper concentration relative to wild-type controls, and copper deficiency induces an up-regulation of choroid plexus CTR1 expression in mice (Kuo et al., 2001, 2006; Lee et al., 2001; Gybina and Prohaska, 2006). An alternate route of copper uptake into the brain is via the divalent metal transporter, DMT1 (Arredondo et al., 2003). However, the extent to which DMT1 participates in brain copper import is unclear, particularly in the wake of a recent study disputing copper transport by DMT1 (Illing et al., 2012). In conjunction with CTR1, ATP7A, and ATP7B regulate the rate-limiting step of copper influx into the brain. Immunohistochemical stains revealed that in choroid plexus epithelial cells, ATP7A predominantly localizes to the basolateral membrane, whereas ATP7B localizes to the apical membrane (Davies et al., 2013). The distinct membrane localization of these two homologous copper transporters may be a regulatory mechanism related to their respective enzyme kinetics to provide a strict control over copper transport across the BCB and BBB (Tsivkovskii et al., 2002; Barnes et al., 2005; Hung et al., 2007). The comparatively slower rate of copper movement into the CSF relative to copper influx into choroid plexus may be explained by the presence of kinetically slower ATP7B at the CSF-facing apical membrane of choroid plexus epithelial cells (Choi and Zheng, 2009). The kinetically faster ATP7A is more favorably localized to the basolateral membrane to facilitate copper efflux into the blood (Davies et al., 2013). Therefore, BCB serves as the main exit route for excess copper from the brain. Conversely, BBB as the main entry point for copper into the brain has lower ATP7A expression with a brain capillary-facing basolateral localization to enable rapid but limited import of copper into the brain to counterbalance any sudden brain copper depletion (Choi and Zheng, 2009).
Intracellular copper transport in brain cells
There have been limited studies on brain cellular copper transport. It has been largely assumed that the homeostatic mechanisms that regulate cellular copper movement in brain cells parallel that of well-defined pathways in peripheral cells. Indeed, recent studies in astrocytes support this notion (Scheiber et al., 2010, 2012). CTR1, in addition to mediating the initial copper uptake into the brain, is implicated also in general copper uptake into neuronal and glial cells. However, differential expression of CTR1 in different brain cells pertains to cell-specific copper requirements (Davies et al., 2013).
To prevent the generation of ROS by free copper ions inside cells, three well-characterized metallochaperone pathways have evolved to escort intracellular copper: (1) ATOX1; (2) CCS; and (3) COX17. The copper chaperone ATOX1 is widely expressed in the brain, which include the hippocampus, thalamus, cerebellum, and corpus callosum (Klomp et al., 1997; Naeve et al., 1999; Davies et al., 2013). It delivers copper to cuproenzymes in the secretory pathway, and in particular, to the copper transporters ATP7A and ATP7B at the trans-Golgi network (TGN). In addition to its role as a copper chaperone, it regulates the catalytic activity of ATP7B (Walker et al., 2002). ATOX1 is essential to ATP7A copper efflux activity. It stimulates ATP7A translocation from the TGN to the cell surface via direct interactions and copper exchange (Hamza et al., 2003). Consistent with the original identification of ATOX1 in the yeast Saccaromyces cerevisae as an antioxidant protein (Lin and Culotta, 1995), its over-expression in neuronal cell lines confers protection against oxidative insults (Kelner et al., 2000).
The copper chaperone for superoxide dismutase (CCS) loads copper into SOD1 located in the cytoplasm (Culotta et al., 1997). It is expressed in the mammalian brain, but its expression relative to brain regional distribution is unknown (Gybina and Prohaska, 2006). Interestingly, the expression of CCS is sensitive to copper deficiency in the cerebellum, but not in the choroid plexus (Gybina and Prohaska, 2006). In addition to its interactions with SOD1, CCS also transports copper to BACE1 (Angeletti et al., 2005), the β-secretase involved in APP cleavage that results in the production of amyloid plaque-forming Aβ peptide. CCS deficiency promotes Aβ production, which implicates CCS in the regulation of BACE1 activity (Gray et al., 2010). Furthermore, BACE1 competes with SOD1 for binding to CCS, which may contribute to oxidative stress evident in AD (Angeletti et al., 2005; Dingwall, 2007).
COX 17 is the copper chaperone responsible for delivery of copper to COX11, SCO1, and SCO2 in the mitochondria for the metallation of CCO (Glerum et al., 1996a,b; Amaravadi et al., 1997; Beers et al., 1997; Horvath et al., 2000; Horng et al., 2004). It is highly expressed in the cerebral cortex, cerebellum and brain stem, with low expression in the hippocampus and hypothalamus (Kako et al., 2000).
In addition to the metallochaperone-mediated intracellular copper transport, copper can bind also to glutathione (GSH) for transfer to cystein-rich metallothioneins (MT) for storage (Freedman et al., 1989). Notably, astrocyte secretion of MT3 into the synaptic cleft regulates the availability of copper ions released during neurotransmission (Uchida et al., 2002), and confers neuroprotection by removal of copper from redox-active Aβ:Cu2+ complexes abundant in AD brains (Meloni et al., 2008). The importance of GSH in neuronal copper homeostasis is highlighted by the exquisite sensitivity of cultured primary cortical neurons to trace amounts of extracellular copper after GSH depletion. The neurotoxic effect is postulated to involve the generation of Cu+ and ensuing free radical mediated oxidative stress (White et al., 1999a; White and Cappai, 2003).
Intracellular copper concentration is further maintained by the export of excess copper via P-type ATPases ATP7A and ATP7B. In the brain, the expression and distribution of ATP7A and ATP7B are developmentally regulated (Barnes et al., 2005; El Meskini et al., 2005; Niciu et al., 2006). In an adult brain, their expression is particularly abundant in neurons from copper-rich brain regions such as the hippocampus, olfactory bulb, cerebellum and choroid plexus (Iwase et al., 1996; Saito et al., 1999; Barnes et al., 2005; Niciu et al., 2006; Choi and Zheng, 2009; Davies et al., 2013). Notably, the co-enrichment of copper and ATP7A in the hippocampus highlights their critical participation in neurotransmission, and potential involvement in learning and memory. Low levels of ATP7A are present also in a sub-population of astrocytes, microglia, myelinating oligodendrocytes, tanycytes, and endothelial cells (Niciu et al., 2006). In the rodent cerebellum, ATP7A and ATP7B are reported to have a distinct cell and developmental specific expression. ATP7B is expressed constitutively in Purkinje neurons, where it functions to deliver copper to CP; whereas there is a developmental shift in ATP7A expression from the Purkinje neurons to Bergmann glia (Barnes et al., 2005). However, in the post-mortem human adult brain, both ATP7A and ATP7B are strongly expressed in Purkinje neurons, but not in Bergmann glia (Davies et al., 2013). At a cellular level, ATP7A, and ATP7B are localized at the TGN, and undergo copper-responsive redistribution to the basolateral and apical cell surface, respectively, where they mediate copper efflux (Niciu et al., 2006; Davies et al., 2013). Additionally, ATP7A localizes to the axon in maturing olfactory receptor neurons, where it has a putative role in axonal outgrowth and synaptogenesis (El Meskini et al., 2005, 2007).
Brain cholesterol homeostasis
Cholesterol is a fundamental component of the brain. It is an integral structural constituent of cellular membranes of all body cells and a precursor of a number of sterols, hormones, and vitamins. Perturbed brain cholesterol balance can affect membrane dynamics and stability, and contributes to neuronal degeneration, loss of synaptic plasticity and neurotransmission (Koudinov and Koudinova, 2001, 2005). Indeed, neurons may require a threshold level of cholesterol synthesis as statin treatment reduced synapse density and impaired synaptic vesicle release from hippocampal neurons (Mailman et al., 2011). Therefore, it is vital for synaptogenesis, dendritogenesis, axonal guidance, and brain cell signaling events (Mauch et al., 2001; Hering et al., 2003; Goritz et al., 2005; Ko et al., 2005; Guirland and Zheng, 2007; Stottmann et al., 2011). Furthermore, the importance of cholesterol to normal brain development, structure and function is exemplified in neurodegenerative disorders such as Niemann-Pick type C (NP-C) disease, Tangier disease, Smith-Lemi-Opitz Syndrome, Huntington's disease, PD, and AD, that are associated with either genetic or regulatory defects in cholesterol metabolic pathways.
The brain has a disproportionately high cholesterol content to total body mass ratio. The human brain is ~2% of total body mass, yet it contains about a quarter of total body cholesterol. Almost the entire pool of brain cholesterol (>99.5%) is present in the unesterified form, with a distribution rank order of myelin (70%) > glia (20%) > neurons (10%) [reviewed in Björkhem and Meaney (2004) and Dietschy and Turley (2004)]. Brain cholesterol represents a distinct pool from that in the peripheral circulation, which is efficiently segregated by the BBB. Experimental evidence, using 14C- or 2H-labeled cholesterol, demonstrated that there is negligible exchange of cholesterol between the brain and plasma. In animal models with an intact BBB, uptake of dietary 2H-labeled cholesterol into the brain is less than 1% (Meaney et al., 2000). Moreover, in terminally ill human patients with potentially compromised BBB, the detectable influx of 14C-labeled cholesterol from peripheral circulation was negligible (Chobanian and Hollander, 1962). Thus, the primary source of brain cholesterol is de novo synthesis in both neuronal and glial cells, via the mevalonate pathway, mainly in the endoplasmic reticulum (ER) [reviewed in Buhaescu and Izzedine (2007) and Goldstein and Brown (1990)].
Brain cholesterol synthesis
The rate of brain cholesterol synthesis is developmentally regulated. The highest rate of cholesterol synthesis occurs during the early postnatal period and early life, a critical time for brain development (Zhang et al., 1996; Dietschy and Turley, 2004). The primary site of cholesterol synthesis is in cholesterol-rich oligodendrocytes that form the insulating myelin sheath around axons. High levels of cholesterol in myelin is required to increase axonal electrical resistance but decrease the capacitance of the surrounding plasma membrane, effectively acting as a barrier to dissipation of electrical impulses across the membrane, whilst promoting transmission down the axon (Snipes and Suter, 1997; Saher et al., 2005). Thus, cholesterol and myelination are essential to neurotransmission that can influence cognitive and motor activities. Therefore, it is no coincidence that the most productive period of myelination parallels that of cholesterol synthesis (Saher et al., 2005). The impact of cholesterol-deficiency and hypomyelination on brain development is evident in neurodegenerative disorders of cholesterol metabolism such as Smith-Lemi-Opitz Syndrome, cerebrotendinous xanthomatosis, hereditary spastic paresis type SPG5, and NP-C, which present clinical symptoms that include mental retardation, dementia, and ataxia [reviewed in Björkhem et al. (2010) and Ikonen (2006)].
The rate of brain cholesterol synthesis declines with brain maturation and age. To preserve a constant level of cholesterol, it has been postulated that in the adult brain, cholesterol is efficiently recycled. Daily sterol turnover in both mouse and human brains is about 20-fold lower compared with that in whole body (Dietschy and Turley, 2004). The estimated half-life of cholesterol in rat brains is about four to six months (Andersson et al., 1990; Björkhem et al., 1997), and in human brains, is estimated to be 5 years (Björkhem et al., 1998). In comparison, in the periphery, the half-life of rat hepatic cholesterol is only about 4 days (Andersson et al., 1990) and 14C-labeled cholesterol in human plasma has a reported half-life of about 6 days (Bekersky et al., 2001).
In the adult brain, cholesterol synthesis is not limited to oligodendrocytes, the major center of brain cholesterol production. Attenuated cholesterol synthesis occurs in neurons and astrocytes, but the extent of endogenous cholesterol production in these cells is poorly understood. It has been proposed that in the post-developmental period, it is more energetically efficient for neurons to minimize cholesterol production in favor of electrical impulse propagation that is vital to neurotransmission; and to satisfy neuronal cholesterol requirements, cholesterol is imported from glial cells (Pfrieger, 2003). This hypothesis is supported by experiments involving in vivo neuronal conditional inactivation of squalene synthase, the rate-limiting enzyme in the cholesterol synthesis pathways dedicated to sterol formation (Fünfschilling et al., 2007, 2012). Neighboring astrocytes rescued defects in neuronal projection caused by the enforced neuronal cholesterol deficiency. In contrast, the mouse embryonic brain showed a greater demand for cholesterol and it is highly sensitive to cholesterol deficiency. Exogenous cholesterol imported from microglia only partially compensated for the embryonic neuronal cholesterol deficit. Consequently, the mouse pups were born with extensive neuronal degeneration at birth (Fünfschilling et al., 2012). Nevertheless, these observations do not preclude endogenous cholesterol synthesis in neurons. In the adult mouse brain cortical, hippocampal and cholinergic neurons, high expression of key enzymes involved in the first and last step of cholesterol synthesis, 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) and 7-dehydrocholesterol reductase (DHCR7), respectively, suggest cholesterol synthesis occurs in these neurons (Korade et al., 2007). At present, the requirement for endogenous cholesterol production in these neurons is unclear.
Brain cholesterol transport: ApoE
The inter- and intra-cellular transfer of cholesterol in the brain involves a multitude of apolipoproteins, lipoprotein receptors and lipid transporters. Apolipoprotein E (ApoE), a 299-amino acid glycoprotein, is the primary apolipoprotein involved in the regulation of lipid transport in both the plasma and the CSF [reviewed in Hauser et al. (2011), Holtzman et al. (2012) and Leduc et al. (2010)]. In humans, there are three major ApoE isoforms: E2, E3, and E4, encoded by polymorphic APOE alleles ε2, ε3, ε4, respectively. These isoforms are distinguished by their combination of cysteine and arginine residues at amino acid positions 112 and 158. ApoE3 with the highest allelic frequency (50–90%) is generally considered to be the “parental” form with C112 and R158, whereas in ApoE2, both of these amino acid residues are cysteines, and in ApoE4, both are arginines (Utermann et al., 1984; Mahley, 1988). In contrast, only an ApoE3-equivalent form of ApoE is present in all other species (Hatters et al., 2006). The APOE-ε4 genotype is the strongest known genetic risk factor for sporadic late-onset AD (Mahley, 1988; Bertram and Tanzi, 2012). Hence, ApoE is the best characterized of all known apolipoproteins, which include ApoA-I, ApoD, and ApoJ (clusterin) (Borghini et al., 1995). Of these other apolipoproteins, ApoJ has gained increasing research interest in recent years with its emergence as another key genetic risk factor for AD from genome-wide association studies (Bertram and Tanzi, 2009; Harold et al., 2009).
Similar to cholesterol, ApoE in the brain derives primarily from de novo synthesis in glial cells, mostly in astrocytes (Boyles et al., 1985; Pitas et al., 1987a), and there is effectively no exchange with ApoE from the plasma due to lack of permeability through the BBB under normal conditions (Linton et al., 1991). Paradoxically, the stability of the BBB requires ApoE, which exerts an isoform-dependent influence on the integrity of cerebrovasculature (Methia et al., 2001; Nishitsuji et al., 2011; Bell et al., 2012). Minor amounts of ApoE are produced in neurons and microglia under normal conditions, but selective up-regulation of neuronal ApoE can occur under pathological conditions such as stroke and AD (Xu et al., 1999; Aoki et al., 2003). The functional importance of neuronal ApoE under such conditions is not well understood, but indicates a potential role for neuron-derived ApoE to enhance local cholesterol recycling to facilitate neuronal repair.
Astrocyte-derived ApoE has a dual role in cholesterol transport. It is instrumental to the delivery of cholesterol to distal axons for synaptogenesis, dendritogenesis, axonal guidance, signaling, and release of synaptic vesicles in neurotransmission (de Chaves et al., 1997; Posse De Chaves et al., 2000; Mauch et al., 2001; Hering et al., 2003; Goritz et al., 2005). The concomitant neuronal uptake of ApoE and cholesterol present in spherical and discoidal high-density lipoprotein (HDL)-like particles is via a family of low-density lipoprotein receptors, which include low-density lipoprotein receptor (LDLR), LDLR-related protein 1 (LRP1), and ApoE receptor 2 (ApoER2). The structural conformation of ApoE, as dictated by its isoform and lipidation status, determines its binding affinity to LDL receptors (Hatters et al., 2006). Experimental evidence revealed that a positively charged amino acid at position 158 in ApoE is essential for effective binding to LDL receptors (Innerarity et al., 1984; Dong et al., 1996). It is noteworthy that ApoE2 is defective in binding to LDLR, which is due to reduced positive electrostatic potential with a cysteine at this position (Weisgraber et al., 1982; Lund-Katz et al., 2001), yet it is the most protective ApoE isoform against AD. ApoE3 and ApoE4 bind to LDLR with similar affinity. Furthermore, lipidation of ApoE is essential for binding to LDL receptors, and an in vitro lipid binding experiment demonstrated that ApoE4 has the highest lipid binding affinity, although all isoforms have a similar lipid binding capacity (Saito et al., 2003).
In addition to its role in neuronal cholesterol delivery, ApoE is essential also to cholesterol efflux from neurons and astrocytes. In this pathway, ApoE functions as an acceptor of cellular cholesterol and other lipids from cell surface ATP-binding cassette transporters such as ABCA1 and ABCG1 (Kim et al., 2007, 2008; Minagawa et al., 2009). This process of ApoE lipidation then increases ApoE affinity for LDL receptors for delivery of cholesterol to neurons. The effectiveness of ApoE as a cholesterol acceptor is isoform-specific. In vitro experimental evidence showed that highest cholesterol efflux activity is achieved in both neuronal and astrocyte cultures in the presence of exogenous ApoE2 (Michikawa et al., 2000). Therefore, the neuroprotective capacity of ApoE2 may be partially explained by its proficiency in facilitating cellular cholesterol efflux, despite being ineffective in cholesterol delivery due to inefficient binding to LDLR.
Intraneuronal cholesterol transport
The intracellular fate of ApoE-bound cholesterol in neurons is believed to follow that of the classical clathrin-dependent receptor-mediated endocytic pathway [reviewed in Goldstein and Brown (2009)]. HDL-like particles containing cholesterol-bound ApoE/LDLR complex associate with the clathrin-coated vesicles, which initially fuse with the sorting endosomes, where the complex dissociates and LDLR is then either recycled back to the cell surface or delivered to lysosomes for degradation. HDL-like particles are further transported to the late-endosomes/lysosomes, where the acidic environment of these compartments promotes the dissociation of the HDL-like particles, liberating ApoE and cholesterol. The subsequent mobilization of cholesterol from late-endosomes/lysosomes involves NPC1 and NPC2 proteins (Vanier, 2010). Fibroblast cells extracted from NP-C patients with a deficiency in either NPC1 and/or NPC2 proteins show an entrapment of unesterified cholesterol in late-endosomes/lysosomes (Pentchev et al., 1985; Kruth et al., 1986). This abnormal cholesterol accumulating phenotype is present also in both NP-C human and mouse model brains (Karten et al., 2002; Distl et al., 2003; Treiber-Held et al., 2003). More recently, in vitro experiments that demonstrate direct binding and interactions between NPC1, NPC2, and cholesterol provided further support for a critical role for NPC1 and NPC2 in intracellular cholesterol transport (Infante et al., 2008a,b,c; Kwon et al., 2009; Wang et al., 2010). It has been postulated that NPC1 and NPC2 mediated cholesterol egress from late-endosomes/lysosomes can be diverted to either the ER for esterification by acetyl-coenzyme A:cholesterol acetyltransferase (ACAT) for storage of the excess in cytosolic lipid droplets, or to the plasma membrane for export by cholesterol transporters (e.g., ABCA1 and ABCG1). Genetic defects in ABCA1 causes Tangier disease (Kolovou et al., 2006), which exhibits a cholesterol accumulating phenotype similar to that found in NP-C and is characterized by HDL-deficiency. There are limited reports on the effects of ABCA1 mutations in the central nervous system (Pietrini et al., 1990; Negi et al., 2013), with premature atherosclerosis and neuropathy being the best documented symptoms of Tangier disease.
Brain cholesterol export
Cholesterol cannot be degraded. At present, there are two known pathways for brain cholesterol elimination. One route of brain cholesterol removal is via ApoE secretion into the CSF and export from the brain at a rate of 1–2 mg/day by an unknown mechanism (Pitas et al., 1987b; Xie et al., 2003). Cholesterol removed by this pathway is believed to originate primarily from glial cells. Alternatively, the main route of brain cholesterol elimination, primarily of neuronal origin, is via conversion of cholesterol to BBB-permeable oxidized cholesterol species (oxysterols). The primary brain oxysterol species is 24S-hydroxycholesterol (24OHC), the product of 24S-hydroxylase (CYP46) catalyzed addition of a hydroxyl group to the 24-position of cholesterol (Lutjohann et al., 1996; Björkhem et al., 1997, 1998; Lund et al., 1999, 2003; Lutjohann and von Bergmann, 2003). The rate of 24OHC movement from the brain into the circulation is estimated at 6–7 mg/day (Lutjohann et al., 1996; Björkhem et al., 1998). A corresponding rate of uptake in the liver has led to the hypothesis that 24OHC is an exclusive product of the brain, and its concentration in the circulation is reflective of brain cholesterol metabolism (Bretillon et al., 2000; Lutjohann and von Bergmann, 2003). In the liver, 24OHC is further converted to bile acids for excretion from the body (Björkhem et al., 2001). By analogy with 24OHC penetration of the BBB, 27-hydroxycholesterol (27OHC) produced by peripheral cells can enter the brain from circulation at a rate of 5 mg/day (Heverin et al., 2005). There are no known oxysterol transporters, and the transfer of oxysterols across cell membranes is hypothesized to be driven by the concentration gradient (Björkhem et al., 2009).
Oxysterols play an important part in regulation of whole body cholesterol homeostasis. They can activate liver X receptors (LXRs), LXRα, and LXRβ [reviewed in Björkhem (2013)], which regulate a number of genes in the cholesterol metabolic pathway including ABCA1, ABCG1, SREBP-1, and ApoE. Interestingly, cultured glial cells respond to a greater extent to LXR agonist stimulation of LXR target gene expression and cholesterol efflux compared with cultured neuronal cells (Whitney et al., 2002). Thus, LXR and 24OHC are connected in a feedback mechanism to maintain a net cholesterol export, where an increased export of neuronal cholesterol in the form of 24OHC activates cholesterol export from glial cells for delivery to neurons (Pfrieger, 2003). In addition to LXRs, other members of the same nuclear receptor family that also regulate genes in the cholesterol metabolic pathway include peroxisome proliferator-activated receptor γ (PPARγ) and retinoid X receptors (RXRs).
Crosstalk between copper and cholesterol metabolism
The interaction between copper and cholesterol metabolic pathways in the brain is an area of limited research. The majority of current investigations center mainly on peripheral changes. Dietary manipulations in animal studies revealed an inverse relationship between copper and cholesterol in their respective peripheral concentrations. Rats and rabbits fed a high cholesterol diet have decreased hepatic copper concentration compared with animals on a control diet (Klevay, 1988; de Wolf et al., 2003a,b). Increased bilirubin secretion has been proposed as the potential mechanism underlying cholesterol induced hepatic copper reduction (de Wolf et al., 2003b). However, in the Watanabe heritable hyperlipidemic rabbits, an animal model for familial hypercholesterolemia, there is no inverse correlation with copper, which is significantly elevated (Allain et al., 1989). These observations indicate that dietary and genetic changes in cholesterol have differential effects on copper metabolism. It is conceivable that dietary cholesterol alters cholesterol homeostasis in a manner that affects cellular membrane fluidity and dynamic, which impacts on the structure and function of membrane-localized copper transporters. For example, pharmacological depletion of cellular cholesterol levels using the cholesterol sequestering agent methyl-β-cyclodextrin (MβCD) resulted in a shift of the copper transporter CTR1 in HeLa cells from a perinuclear localization to a more diffused vesicular and plasma membrane distribution (Klomp et al., 2002), and thus cause a change in cellular copper balance. However, the exact mechanism underlying the impact of altered cholesterol metabolism on copper homeostasis remains to be elucidated.
In animals fed a copper deficient diet, peripheral cholesterol synthesis is stimulated via up-regulation of HMG-CoA reductase and SREBP-1, and they develop hypercholesterolemia (Carr and Lei, 1989; Yount et al., 1990; Lei, 1991; al-Othman et al., 1994; Tang et al., 2000). In contrast, brain cholesterol synthesis and myelination is sensitive to dietary and genetic copper deficiencies (Figure 1). Mice given dietary supplement of cuprizone, a copper chelator, showed a significant down-regulation of HMG-CoA reductase mRNA expression and cholesterol synthesis, and consequently, severe demyelination, affecting particularly the corpus callosum (Jurevics et al., 2001, 2002). These effects are reversible by removal of cuprizone from the diet. Similarly, profound brain copper deficiency in Menkes disease, due to absent or defective activity of the ATP7A copper transporter, is associated also with significantly reduced brain cholesterol and extensive myelin defects (Hara and Taketomi, 1986). Conversely, copper overload in the Atp7b−/− mouse model of Wilson disease is associated also with reduced brain and hepatic concentrations of cholesterol, lathosterol, desmosterol, 8-hydrocholesterol, and 7-dehydrocholesterol (Sauer et al., 2011). These changes are consistent with down-regulation of cholesterogenic gene expression and reduction in nuclear receptor transcription factors farnesoid X receptor (FXR) and glucocorticoid receptor (GR) in the liver of the same animal model (Huster et al., 2007; Wilmarth et al., 2012). However, the decrease in serum cholesterol observed in animal models of Wilson disease is not recapitulated in a study of human Wilson disease patients (Seessle et al., 2011). Also inconsistent with in vivo animal studies, a microarray analysis of in vitro human macrophage culture treated with copper found up-regulation of seven cholesterolgenic genes, which include HMG-CoA synthase, squalene synthase, and LDLR (Svensson et al., 2003). These observations suggest that there may be tissue- and species-specific differences in cholesterol metabolism in response to copper. There is clearly a close and complex relationship between the copper and cholesterol metabolic pathways. The full impact of altered copper homeostasis with regard to cholesterol metabolism remains to be fully explored.
Figure 1.
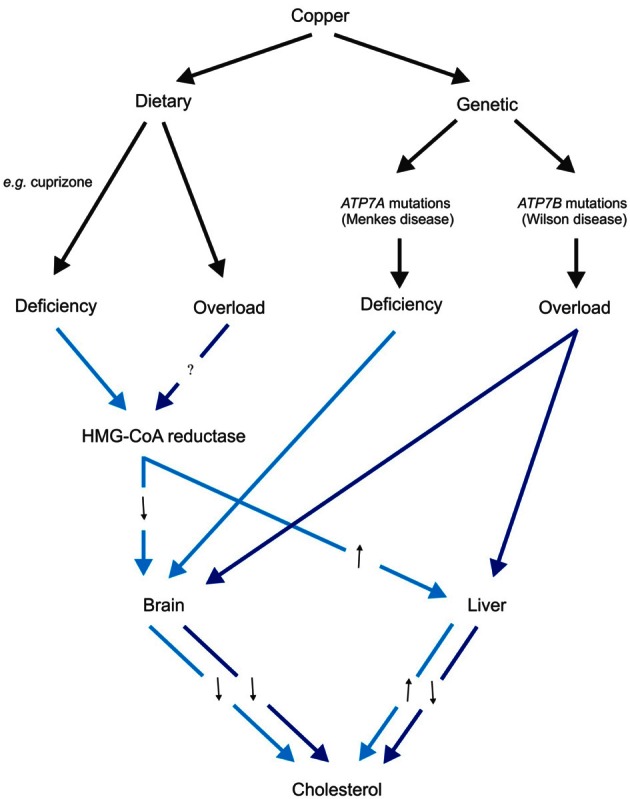
Schematic diagram of the effects of dietary and genetic copper manipulation on cholesterol levels, based on available data from animal models. Light blue and dark blue arrows represent pathways associated with copper deficiency and overload conditions, respectively. Black arrows denote either increased (↑) or decreased (↓) 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase expression or cholesterol levels as a consequence of altered copper levels.
Disintegration of copper and cholesterol metabolism in Alzheimer's disease
Late-onset AD is a complex multifactorial disorder, and to date, the exact etiology remains elusive. There are largely independent bodies of literature documenting the disruption to copper and cholesterol homeostasis and how they may contribute to the development and progression of AD. Observations from epidemiological studies of changes in plasma copper and cholesterol as discriminating markers for AD have been inconsistent (Jarvik et al., 1995; Notkola et al., 1998; Gonzalez et al., 1999; Squitti et al., 2002, 2003, 2005, 2006, 2007; Reitz et al., 2004, 2008; Mielke et al., 2005; Pajonk et al., 2005; Kessler et al., 2006; Kivipelto and Solomon, 2006; Solomon et al., 2007; Zambon et al., 2010; Rembach et al., 2013). The inherent genetic and environmental heterogeneity in different human populations may contribute to the ambiguous observations. Nevertheless, epidemiological and animal studies found that a high fat diet is associated with cognitive decline and increased risk for AD (Morris et al., 2003; Sparks, 2008). High copper consumption with a high saturated and trans fat diet further accelerates the cognitive decline (Morris et al., 2006). The latter study suggests that the association is independent of dietary cholesterol, zinc and iron; but this study did not account for potential effects due to stimulation of endogenous cholesterol synthesis. An AD cholesterol-lowering treatment trial involving atorvastatin reported an increase in plasma CP as a measure of copper (Sparks et al., 2005), further suggesting an interaction between copper and cholesterol homeostasis in AD. Collectively, these studies, which mostly document peripheral copper and cholesterol changes, indicate a synergistic failure of copper and cholesterol metabolic pathways that contributes to cognitive decline and pathogenesis of AD.
Interactions of copper and cholesterol with APP and Aβ
Amyloid plaques that are characteristic of AD brains contain high concentrations of the Aβ peptide, cholesterol and transition metals (copper, iron and zinc) (Glenner and Wong, 1984; Masters et al., 1985; Lovell et al., 1998; Dong et al., 2003; Miller et al., 2006; Panchal et al., 2010). Central to the formation of amyloid plaques is the processing of APP, a glycosylated type I transmembrane protein. There are two alternative APP processing pathways, commonly referred to as the amyloidogenic or non-amyloidogenic pathway (Thinakaran and Koo, 2008). The release of Aβ by sequential β- and γ-secretase cleavage of APP in the amyloidogenic pathway is most relevant to AD pathogenesis. Conversely, non-amyloidogenic processing of APP involves sequential α- and γ-secretase cleavages to release a non-neurotoxic p3 peptide. Cleavage by γ-secretase in both pathways liberates a common C-terminal fragment known as APP intracellular domain (AICD), which can translocate to the nucleus and function as a transcription factor.
Cholesterol is a widely recognized key regulator of APP processing. Amyloidogenic processing of APP preferentially occurs in cholesterol- and glycosphingolipid (GSL)-rich lipid raft microdomains (Simons and Ikonen, 1997; Ehehalt et al., 2003; Kawarabayashi et al., 2004; Vetrivel et al., 2004; Hooper, 2005; Cordy et al., 2006; Cheng et al., 2007; Schneider et al., 2008; Thinakaran and Koo, 2008; Hung et al., 2009; Vetrivel and Thinakaran, 2010). APP interactions at the cell surface with flotillin-2, a lipid raft protein (Schneider et al., 2008), and cholesterol via its C-terminal domain (Beel et al., 2008) are critical to its partitioning into lipid rafts and trafficking (Figure 2). Internalization of APP from the cell surface to BACE1 (β-secretase)-rich acidic endosomes is cholesterol-sensitive and pro-amyloidogenic (Refolo et al., 1995; Vassar et al., 1999; Schneider et al., 2008; Cossec et al., 2010; Marquer et al., 2011). Alternatively, cholesterol depletion triggers destabilization of lipid rafts, decreased APP internalization and lipid raft localization, inhibition of β- and γ-secretase activities, and reduced Aβ production (Simons et al., 1998; Fassbender et al., 2001; Riddell et al., 2001; Ehehalt et al., 2003; Vetrivel et al., 2004, 2005; Urano et al., 2005; Cordy et al., 2006; Schneider et al., 2008; Won et al., 2008; Cossec et al., 2010; Marquer et al., 2011). Two common agents used to deplete cholesterol are methyl-β-cyclodextrin (MβCD) and statins. MβCD extracts cholesterol from cellular membranes. Statins are inhibitors of HMG-CoA reductase, thereby achieving cholesterol reduction by interfering with endogenous cholesterol synthesis via the mevalonate pathway. Given the pro-amyloidogenic nature of cholesterol, it has been considered as a potential therapeutic target for treatment of AD. A number of clinical trials have investigated the beneficial effects of statins [reviewed in Shepardson et al. (2011a,b)]. The outcomes are variable due to the pleiotropic nature of statins, which differ in their BBB permeability and additional off-target effects on the isoprenylation pathway.
Figure 2.

Schematic models illustrating the interactions between copper, cholesterol and APP metabolism. (A) Under normal and/or elevated cellular copper conditions, there is an increased translocation of APP and ATP7A from the trans-Golgi network (TGN) to the plasma membrane (PM), where they participate in copper efflux. The majority of the APP molecules undergo non-amyloidogenic processing via α-secretase cleavage at the cell surface in non-lipid raft membrane domains, which produces a non-toxic p3 fragment. It is expected that a basal level of β-cleavage of APP occurs in cholesterol-rich lipid raft domains of acidic endosomes enriched in BACE1, the β-secretase. Aβ40, the product of the sequential cleavage of APP by β- and γ-secretases, is proposed to inhibit 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase activity as a part of a feedback mechanism by which cholesterol-dependent regulation of APP occurs (Grimm et al., 2005). (B) In contrast, under conditions of copper deficiency, such as that present in Alzheimer's disease (AD), there may be increased APP interactions with flotillin-2 at the PM, increased APP partitioning into lipid rafts, enhanced internalization of APP to BACE1-rich endosomes for amyloidogenic processing and generation of Aβ in lipid rafts. The combination of Aβ, cholesterol and copper in lipid rafts fosters a favorable environment for the formation of redox-active Aβ:Cu2+ complexes, which can catalytically oxidize cholesterol, and may potentiate neurotoxicity by generating H2O2 and toxic oxysterol species. Although there may be an increased production of Aβ, the formation of Aβ:Cu2+ oligomers may result in reduced availability of Aβ to inhibit HMG-CoA reductase activity and consequently, increased cholesterol synthesis.
Copper also modulates APP processing (Borchardt et al., 1999; Cater et al., 2008; Hung et al., 2009). Conditions of copper deficiency, such as that found in brain tissues from AD patients and transgenic mouse models promote amyloidogenic processing of APP (Deibel et al., 1996; Maynard et al., 2002, 2006; Bayer et al., 2003; Phinney et al., 2003; Magaki et al., 2007; Cater et al., 2008) in cholesterol-rich lipid rafts (Hung et al., 2009) (Figure 2). Conversely, increasing cellular copper levels via dietary, pharmacological or genetic manipulations favors the non-amyloidogenic processing of APP and inhibits Aβ production (Borchardt et al., 1999; Cherny et al., 2001; Bayer et al., 2003; Phinney et al., 2003; White et al., 2006; Adlard et al., 2008; Cater et al., 2008; Donnelly et al., 2008; Hung et al., 2009). Analogous to cholesterol depletion, increased cellular copper reduces the association of flotillin-2 with lipid rafts (Hung et al., 2009), retards APP internalization and attenuates Aβ production (Hung et al., 2009; Acevedo et al., 2011). It can be envisaged that the copper-dependent effect on APP processing may be achieved via regulation of cholesterol synthesis (see Section Crosstalk between copper and cholesterol metabolism). The alteration in copper-induced APP redistribution and processing may stimulate a compensatory response in the up-regulation of APP mRNA and protein expression (Armendariz et al., 2004; Bellingham et al., 2004b; Cater et al., 2008).
In a reciprocal manner, APP participates in the regulation of copper and cholesterol metabolism. A dual mechanism is involved in APP regulation of cholesterol metabolism. The binding of cholesterol to APP C-terminal domain promotes its amyloidogenic processing, and the release of Aβ and AICD. Experimental evidence demonstrates that Aβ40 inhibits the activity of HMG-CoA reductase, the rate limiting enzyme in the cholesterol synthesis pathway, so that APP deficiency results in cholesterol accumulation (Grimm et al., 2005). An additional mechanism of cellular cholesterol reduction is achieved by translocation of AICD into the nucleus, where it down-regulates LRP1 expression by transcription suppression (Liu et al., 2007). Thus, APP is proposed as a cholesterol sensor to control cellular cholesterol levels and is own processing (Beel et al., 2008). Additionally, APP is proposed to regulate copper efflux. This hypothesis evolved from evidence that APP expression inversely correlates with copper levels in cell and animal models (White et al., 1999b,c; Maynard et al., 2002; Phinney et al., 2003; Bellingham et al., 2004a). Over-expression of APP or its C-terminal domain (C100) that contains the Aβ sequence is associated with copper deficiency in transgenic mouse models of AD (Maynard et al., 2002; Bayer et al., 2003; Phinney et al., 2003). Conversely, APP deficiency causes copper accumulation (White et al., 1999c; Bellingham et al., 2004a). The concomitant rise in copper and cholesterol in APP knockout animals suggests an interaction between these pathways that becomes uncoupled in its absence.
Oxidative stress is a pathological feature of AD. It is noteworthy that the inverse nature of copper distribution to cholesterol-rich lipid rafts relative to cellular copper concentrations fosters an environment conducive to oxidative stress (Hung et al., 2009) (Figure 2). Lipid raft co-enrichment of Aβ and copper favors the formation of redox-active Aβ:Cu2+ complexes that catalytically generate H2O2 via reduction of Cu2+ to Cu+ (Atwood et al., 1998, 2004; Huang et al., 1999; Cuajungco et al., 2000; Hung et al., 2009). Cholesterol and long-chain fatty acids in lipid rafts are obvious electron donors that participate in this reaction (Opazo et al., 2002; Haeffner et al., 2005; Murray et al., 2005, 2007), and consequently are converted to oxysterols and lipid peroxidation products, such as 7β-hydroxycholesterol and 4-hydroxynonenal (HNE), which are present in abundance in the brains of human AD cases and mouse models (Opazo et al., 2002; Murray et al., 2005, 2007; Nelson and Alkon, 2005; Puglielli et al., 2005; Yoshimoto et al., 2005; Jiang et al., 2007). A neurotoxic vicious cycle then ensues, where oxidatively modified lipids stimulate the formation of Aβ oligomers complexed with copper and enter a continual pathological redox cycle oxidizing cholesterol and other membrane lipids (Koppaka and Axelsen, 2000; Barnham et al., 2003; Ciccotosto et al., 2004; Murray et al., 2005, 2007; Tickler et al., 2005; Liu et al., 2008). Furthermore, in vitro, oligomers of Aβ:Cu2+ can penetrate artificial membrane systems to form cation channels (Bhatia et al., 2000; Curtain et al., 2001, 2003; Lin et al., 2001). Collectively, these observations suggest that a combination of copper, soluble Aβ oligomers and cholesterol may potentiate neurotoxicity via lipid oxidation and altered membrane integrity.
Sporadic AD is often simulated by cholesterol feeding of animal models (rabbits, beagles, and mice) to induce AD-like pathologies and cognitive deficits (Ghribi et al., 2006; Woodruff-Pak et al., 2007; Ghribi, 2008; Sparks, 2008; Lu et al., 2009, 2006; Sparks et al., 2011). In rabbit brains, amyloid plaque-like structures are detected in the hippocampus and temporal lobe, similar to that in human AD brains (Sparks and Schreurs, 2003; Sparks, 2004; Sparks et al., 2006; Woodruff-Pak et al., 2007). Additional consumption of trace amounts of copper (0.12 ppm) in drinking water, but not zinc or aluminum, accelerated the cognitive decline with exacerbated amyloid pathology in cholesterol-fed rabbits suggestive of an interaction between dietary cholesterol and copper in this event (Sparks et al., 2006). In contrast, a normal diet with copper only treatment had no significant effect on amyloid pathology and cognition (Sparks and Schreurs, 2003; Lu et al., 2006). However, more recent studies showed conflicting data that copper treatment alone at a higher concentration of 0.24 ppm resulted in increased Aβ-positive neurons in the rabbit model (Deci et al., 2012); and chronic treatment of rodent models at a high dosage (250 ppm) also enhanced the amyloid pathology (Kitazawa et al., 2009; Mao et al., 2012). The impact of higher copper intake on cholesterol homeostasis in these studies is unclear. Potential mechanisms of dietary cholesterol in causing brain Aβ accumulation include increased circulating and brain concentrations of 27-OHC, and up-regulation of APP and BACE1 expression (Ghribi, 2008; Prasanthi et al., 2009; Shafaati et al., 2011; Popp et al., 2012). Copper may contribute to augmentation of dietary cholesterol-induced amyloid pathology and neurotoxicity by further up-regulating APP and BACE1 expression, increasing oxidative stress and activation of inflammatory pathways (Lu et al., 2006, 2009; Lin et al., 2008). An alternative hypothesis for copper-induced aggravation of amyloid pathology in cholesterol-fed animals is by impeding the clearance of Aβ complexed to ApoE-cholesterol from the brain for removal from the body by the liver (Sparks, 2007). However, the exact biochemical mechanism remains to be elucidated.
Link between copper and ApoE
There is an emerging literature linking the main extracellular cholesterol transporter ApoE and copper, alluding to a potential protective role for ApoE against copper-mediated toxicity and/or copper regulation. The antioxidant activity of ApoE is isoform-specific, in the order of E2>E3>E4, with ApoE2 demonstrating a >2-fold higher activity relative to ApoE4 (Miyata and Smith, 1996). The same study showed that ApoE binds metals including copper, zinc and iron, with highest affinity for copper, and it inhibits copper-mediated lipoprotein oxidation. The authors proposed that copper sequestration may account for the antioxidant activity of ApoE. Moreover, copper-induced Aβ accumulation and aggregation is most pronounced in the presence of ApoE4 compared with ApoE2 and ApoE3 (Moir et al., 1999). Hence, the ability of ApoE to bind to copper with high affinity is a potential mechanism for mitigating copper-mediated lipoprotein oxidation and copper-induced Aβ aggregation (Miyata and Smith, 1996; Moir et al., 1999). The precise ApoE copper-binding mechanism is unknown. Potential sites that may coordinate copper include the methionine residues within the N-terminal four-helix bundle (Miyata and Smith, 1996). The cysteines of ApoE2 at amino acid positions 112 and 158 have the potential to bind copper, while ApoE4, which lacks the cysteine residues, is proposed to have a reduced copper binding capacity and thus, decreased ability to clear Aβ from the brain (Brewer, 2009). However, this may not be feasible based on apoE structural studies [reviewed in Hatters et al. (2006)], which suggest that these cysteine residues are positioned in an opposing orientation.
An ApoE-copper connection was further supported by the finding that in Wilson disease patients with the common H1069Q mutation in the copper transporter ATP7B, an earlier onset of symptoms is associated with the APOE-ε4 genotype (Litwin et al., 2012). In comparison, APOE-ε3 genotype is associated with a significant delay in the onset of Wilson disease symptoms (Schiefermeier et al., 2000; Wang et al., 2003). The authors proposed that ApoE4 is less effective than ApoE2/ApoE3 as an antioxidant so that ApoE4 patients are more susceptible to copper toxicity. These studies further suggest a role for ApoE in copper regulation and in influencing Wilson disease phenotypes, but the exact mechanism remains unclear.
It is noteworthy that ApoJ also interacts with the ATP7A and ATP7B copper transporters that are defective in Menkes and Wilson diseases, respectively (Materia et al., 2011, 2012). ApoJ facilitates their degradation and thus can influence cellular copper levels. However, the integration of these apolipoproteins' lipid transport and Aβ clearance activities with their role in copper regulation remains to be further clarified.
Copper, cholesterol, and tau
NFTs, the other distinguishing pathological hallmark of AD, are mainly formed from aggregates of hyperphosphorylated tau protein [reviewed in Avila et al. (2004), Morris et al. (2011), Spires-Jones et al. (2009)]. Tau is best characterized for its role in polymerization and stabilization of neuronal microtubules, and regulation of axonal transport of synaptic vesicles in neurotransmission (Thies and Mandelkow, 2007; Dixit et al., 2008). However, it is increasingly recognized as a multi-functional protein. Recent findings report the release of tau with synaptic vesicles as a part of neurotransmission (Pooler et al., 2013) and it has been ascribed a regulatory role in neuronal signaling pathways (Ittner et al., 2010, 2011). Phosphorylation of tau is well-established as a modification that regulates its activity, particularly with respect to its interactions with microtubules. An imbalance of tau interacting kinase and phosphatase activities is believed to contribute to the genesis of hyperphosphorylated tau abundant in NFTs.
Tau has a peripheral membrane association via its N-terminal projection domain, implicating a role for tau in neurite outgrowth (Brandt et al., 1995). However, there is limited information regarding a direct interaction between tau and cholesterol or other lipids. Lipid raft studies have revealed an AD- and age-dependent enrichment of tau phosphorylated at NFT-associated epitopes in brain tissues from human AD cases and the Tg2576 mouse model (Kawarabayashi et al., 2004). Indeed, there is a co-localization of cholesterol and NFTs in the same neuronal populations in AD and NP-C brains (Distl et al., 2001). Pharmacological cholesterol depletion by statin-mediated inhibition of cholesterol synthesis resulted in enhanced tau phosphorylation at NFT-associated epitopes (Fan et al., 2001). These data are suggestive of a role for cholesterol in tau phosphorylation and development of NFTs, although the mechanism involved remains to be elucidated.
Literature on the interaction between copper and tau is also limited. The promoter of the tau (MAPT) gene has the potential to be regulated by Sp1, a copper-responsive transcription factor (Heicklen-Klein and Ginzburg, 2000; Song et al., 2008; Bellingham et al., 2009). Over-expression of tau inhibits trafficking of APP to the cells surface (Stamer et al., 2002); and ATP7A traffics to the cell surface along the microtubule network (Cobbold et al., 2004). Therefore, it is plausible that copper-regulated tau expression may be a regulatory mechanism to control copper-responsive trafficking of APP and ATP7A (Stamer et al., 2002; Cobbold et al., 2004; Hung et al., 2009; Acevedo et al., 2011). The functional implications of this regulatory control include NMDA receptor-mediated excitotoxicity, ATP7A-mediated synaptic release of copper and APP metabolism.
Copper is associated with NFTs (Sayre et al., 2000). In vitro experiments indicate that adventitious binding of copper to tau is redox-competent and contributes to neuronal oxidative stress through the generation of hydrogen peroxide. Detailed structural and biophysical characterization of the longest human tau isoform established that tau binds to one Cu2+ ion per monomer under mild acidic conditions with high affinity (Kd = 0.5 μM) (Soragni et al., 2008). Copper coordination involves the cysteine residues 291 and 322 within the microtubule-binding domain. It is notable that evidence from in vitro experiments suggests that copper-mediated tau aggregation is phosphorylation independent; but the manner in which copper may catalyze tau aggregation in vivo is unknown.
The effects of copper on tau pathology in vivo in AD models also reveal conflicting results. Copper modulation by treating a double transgenic AD mouse model with CuII(gtsm), a bis(thiosemicarbazone) or PBT2, a copper/zinc ionophore, improved tau phosphorylation, Aβ oligomerization and cognition (Adlard et al., 2008; Crouch et al., 2009). However, chronic administration of CuSO4 to a triple transgenic AD mouse model caused an increase in cdk5/p25 phosphorylated tau species (Kitazawa et al., 2009). These apparent discrepancies remain to be resolved.
In general, studies that investigate the interaction between copper, cholesterol and tau are lacking. In cholesterol-fed rabbits, copper appears to ameliorate tau pathology in the hippocampus and frontal cortex of cholesterol-fed rabbits, but induces a concomitant rise in plasma tau concentration (Sparks et al., 2011). The mechanism that underlies this observation awaits further studies. Interestingly, in the cholesterol-fed rabbits, the apparent normalization effect of copper on brain tau pathology opposes the negative impact of copper on the amyloid pathology. There is no reasonable explanation for this apparent contradiction at present.
Conclusions
AD is a complex and multifactorial neurodegenerative disorder, and failures in many biochemical pathways are implicated in the pathogenesis of sporadic late-onset AD. Disruption of copper and cholesterol homeostasis are two major pathological features of AD, but whether abnormalities in copper and cholesterol are a cause or consequence of the disease process is not clear. The finding of impaired copper and cholesterol metabolism in other neurodegenerative diseases such as PD and NP-C highlights the possibility that there are fundamental mechanisms linking the biochemistry of these essential cellular constituents. This review has highlighted the essential requirements for copper and cholesterol for normal brain functions, and has summarized the growing body of evidence documenting the close and complex relationship between copper and cholesterol metabolism. Clearly disturbance to one is associated with dysregulation of the other. The complexity is further enhanced by the interactions of copper and cholesterol with key proteins that are dysregulated or malfunction in AD such as, APP, Aβ and tau; and the heterogeneity of genetic and environmental influences between individuals and populations. For example, there is an intimate interaction between copper and cholesterol in APP metabolism that may explain some of the neurodegeneration evident in AD. Key issues that could be addressed to provide further insight are: (1) the relationship between copper, cholesterol and tau; (2) whether the major risk associated factors ApoE and ApoJ are associated with copper in vivo and whether copper is necessary for their function; (3) whether copper is associated with HDL-like particles containing cholesterol-bound ApoE and if so, the functional significance of this in both copper and cholesterol transport and AD. Further research is required for an integrated understanding of the molecular mechanisms connecting copper and cholesterol homeostasis with various players in AD, and how their disintegration may contribute to the development and progression of AD. Such knowledge is critical to the design of effective disease-modifying therapeutic targets.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funding from the National Health and Medical Research Council of Australia (Ashley I. Bush. and Sharon La Fontaine.), Cooperative Research Centre for Mental Health (Ashley I. Bush), the Addi and Cassi Fund (Ashley I. Bush and Ya Hui Hung), Perpetual Trustees (Harry Secomb Foundation and Payne L & Heather Medical Research Charitable Foundation; Ashley I. Bush). The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
References
- Acevedo K. M., Hung Y. H., Dalziel A. H., Li Q. X., Laughton K., Wikhe K., et al. (2011). Copper promotes the trafficking of the amyloid precursor protein. J. Biol. Chem. 286, 8252–8262 10.1074/jbc.M110.128512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlard P. A., Cherny R. A., Finkelstein D. I., Gautier E., Robb E., Cortes M., et al. (2008). Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59, 43–55 10.1016/j.neuron.2008.06.018 [DOI] [PubMed] [Google Scholar]
- Allain P., Krari N., Chaleil D., Balanant Y., Bled F., Girault M. (1989). The distribution of elements in the tissues of Watanabe Heritable Hyperlipidemic Rabbits. Biol. Trace Elem. Res. 19, 153–160 [DOI] [PubMed] [Google Scholar]
- al-Othman A. A., Rosenstein F., Lei K. Y. (1994). Pool size and concentration of plasma cholesterol are increased and tissue copper levels are reduced during early stages of copper deficiency in rats. J. Nutr. 124, 628–635 [DOI] [PubMed] [Google Scholar]
- Alzheimer A. (1907). Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie und psychisch-gerichtliche Medizin 64, 146–148 [Google Scholar]
- Amaravadi R., Glerum D. M., Tzagoloff A. (1997). Isolation of a cDNA encoding the human homolog of COX17, a yeast gene essential for mitochondrial copper recruitment. Hum. Genet. 99, 329–333 [DOI] [PubMed] [Google Scholar]
- Andersson M., Elmberger P. G., Edlund C., Kristensson K., Dallner G. (1990). Rates of cholesterol, ubiquinone, dolichol and dolichyl-P biosynthesis in rat brain slices. FEBS Lett. 269, 15–18 [DOI] [PubMed] [Google Scholar]
- Angeletti B., Waldron K. J., Freeman K. B., Bawagan H., Hussain I., Miller C. C., et al. (2005). BACE1 cytoplasmic domain interacts with the copper chaperone for superoxide dismutase-1 and binds copper. J. Biol. Chem. 280, 17930–17937 10.1074/jbc.M412034200 [DOI] [PubMed] [Google Scholar]
- Aoki K., Uchihara T., Sanjo N., Nakamura A., Ikeda K., Tsuchiya K., et al. (2003). Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke 34, 875–880 10.1161/01.STR.0000064320.73388.C6 [DOI] [PubMed] [Google Scholar]
- Armendariz A. D., Gonzalez M., Loguinov A. V., Vulpe C. D. (2004). Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App. Physiol. Genomics 20, 45–54 10.1152/physiolgenomics.00196.2003 [DOI] [PubMed] [Google Scholar]
- Arredondo M., Munoz P., Mura C. V., Nunez M. T. (2003). DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am. J. Physiol. Cell Physiol. 284, C1525–C1530 10.1152/ajpcell.00480.2002 [DOI] [PubMed] [Google Scholar]
- Atwood C. S., Moir R. D., Huang X., Scarpa R. C., Bacarra N. M., Romano D. M., et al. (1998). Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273, 12817–12826 10.1074/jbc.273.21.12817 [DOI] [PubMed] [Google Scholar]
- Atwood C. S., Perry G., Zeng H., Kato Y., Jones W. D., Ling K. Q., et al. (2004). Copper mediates dityrosine cross-linking of Alzheimer's amyloid-beta. Biochemistry 43, 560–568 10.1021/bi0358824 [DOI] [PubMed] [Google Scholar]
- Avila J., Lucas J. J., Perez M., Hernandez F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384 10.1152/physrev.00024.2003 [DOI] [PubMed] [Google Scholar]
- Barnea A., Hartter D. E., Cho G., Bhasker K. R., Katz B. M., Edwards M. D. (1990). Further characterization of the process of in vitro uptake of radiolabeled copper by the rat brain. J. Inorg. Biochem. 40, 103–110 10.1016/0162-0134(90)80043-W [DOI] [PubMed] [Google Scholar]
- Barnes N., Tsivkovskii R., Tsivkovskaia N., Lutsenko S. (2005). The Copper-transporting ATPases, menkes and wilson disease proteins, have distinct roles in adult and developing cerebellum. J. Biol. Chem. 280, 9640–9645 10.1074/jbc.M413840200 [DOI] [PubMed] [Google Scholar]
- Barnham K. J., Ciccotosto G. D., Tickler A. K., Ali F. E., Smith D. G., Williamson N. A., et al. (2003). Neurotoxic, redox-competent Alzheimer's beta-amyloid is released from lipid membrane by methionine oxidation. J. Biol. Chem. 278, 42959–42965 10.1074/jbc.M305494200 [DOI] [PubMed] [Google Scholar]
- Bayer T. A., Schafer S., Simons A., Kemmling A., Kamer T., Tepest R., et al. (2003). Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 14187–14192 10.1073/pnas.2332818100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearn A. G., Kunkel H. G. (1954). Localization of Cu64 in serum fractions following oral administration: an alteration in Wilson's disease. Proc. Soc. Exp. Biol. Med. 85, 44–48 [DOI] [PubMed] [Google Scholar]
- Beel A. J., Mobley C. K., Kim H. J., Tian F., Hadziselimovic A., Jap B., et al. (2008). Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): does APP function as a cholesterol sensor? Biochemistry 47, 9428–9446 10.1021/bi800993c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers J., Glerum D. M., Tzagoloff A. (1997). Purification, characterization, and localization of yeast Cox17p, a mitochondrial copper shuttle. J. Biol. Chem. 272, 33191–33196 10.1074/jbc.272.52.33191 [DOI] [PubMed] [Google Scholar]
- Bekersky I., Fielding R. M., Dressler D. E., Kline S., Buell D. N., Walsh T. J. (2001). Pharmacokinetics, excretion, and mass balance of 14C after administration of 14C-cholesterol-labeled AmBisome to healthy volunteers. J. Clin. Pharmacol. 41, 963–971 10.1177/009127000104100906 [DOI] [PubMed] [Google Scholar]
- Bell R. D., Winkler E. A., Singh I., Sagare A. P., Deane R., Wu Z., et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 10.1038/nature11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham S. A., Ciccotosto G. D., Needham B. E., Fodero L. R., White A. R., Masters C. L., et al. (2004a). Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J. Neurochem. 91, 423–428 10.1111/j.1471-4159.2004.02731.x [DOI] [PubMed] [Google Scholar]
- Bellingham S. A., Lahiri D. K., Maloney B., La Fontaine S., Multhaup G., Camakaris J. (2004b). Copper depletion down-regulates expression of the Alzheimer's disease amyloid-beta precursor protein gene. J. Biol. Chem. 279, 20378–20386 10.1074/jbc.M400805200 [DOI] [PubMed] [Google Scholar]
- Bellingham S. A., Coleman L. A., Masters C. L., Camakaris J., Hill A. F. (2009). Regulation of prion gene expression by transcription factors SP1 and metal transcription factor-1. J. Biol. Chem. 284, 1291–1301 10.1074/jbc.M804755200 [DOI] [PubMed] [Google Scholar]
- Bertram L., Tanzi R. E. (2009). Genome-wide association studies in Alzheimer's disease. Hum. Mol. Genet. 18, R137–R145 10.1093/hmg/ddp406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L., Tanzi R. E. (2012). The genetics of Alzheimer's disease. Prog. Mol. Biol. Trans. Sci. 107, 79–100 10.1016/B978-0-12-385883-2.00008-4 [DOI] [PubMed] [Google Scholar]
- Bhatia R., Lin H., Lal R. (2000). Fresh and globular amyloid beta protein (1-42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. FASEB J. 14, 1233–1243 [DOI] [PubMed] [Google Scholar]
- Björkhem I. (2013). Five decades with oxysterols. Biochimie 95, 448–454 10.1016/j.biochi.2012.02.029 [DOI] [PubMed] [Google Scholar]
- Björkhem I., Andersson U., Ellis E., Alvelius G., Ellegard L., Diczfalusy U., et al. (2001). From brain to bile. Evidence that conjugation and omega-hydroxylation are important for elimination of 24S-hydroxycholesterol (cerebrosterol) in humans. J. Biol. Chem. 276, 37004–37010 10.1074/jbc.M103828200 [DOI] [PubMed] [Google Scholar]
- Björkhem I., Cedazo-Minguez A., Leoni V., Meaney S. (2009). Oxysterols and neurodegenerative diseases. Mol. Aspects Med. 30, 171–179 10.1016/j.mam.2009.02.001 [DOI] [PubMed] [Google Scholar]
- Björkhem I., Leoni V., Meaney S. (2010). Genetic connections between neurological disorders and cholesterol metabolism. J. Lipid Res. 51, 2489–2503 10.1194/jlr.R006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkhem I., Lutjohann D., Breuer O., Sakinis A., Wennmalm A. (1997). Importance of a novel oxidative mechanism for elimination of brain cholesterol. Turnover of cholesterol and 24(S)-hydroxycholesterol in rat brain as measured with 18O2 techniques in vivo and in vitro. J. Biol. Chem. 272, 30178–30184 10.1074/jbc.272.48.30178 [DOI] [PubMed] [Google Scholar]
- Björkhem I., Lutjohann D., Diczfalusy U., Stahle L., Ahlborg G., Wahren J. (1998). Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39, 1594–1600 [PubMed] [Google Scholar]
- Björkhem I., Meaney S. (2004). Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24, 806–815 10.1161/01.ATV.0000120374.59826.1b [DOI] [PubMed] [Google Scholar]
- Borchardt T., Camakaris J., Cappai R., Masters C. L., Beyreuther K., Multhaup G. (1999). Copper inhibits beta-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion. Biochem. J. 344(Pt 2), 461–467 [PMC free article] [PubMed] [Google Scholar]
- Borghini I., Barja F., Pometta D., James R. W. (1995). Characterization of subpopulations of lipoprotein particles isolated from human cerebrospinal fluid. Biochim. Biophys. Acta 1255, 192–200 [DOI] [PubMed] [Google Scholar]
- Boyles J. K., Pitas R. E., Wilson E., Mahley R. W., Taylor J. M. (1985). Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest. 76, 1501–1513 10.1172/JCI112130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R., Leger J., Lee G. (1995). Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J. Cell Biol. 131, 1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretillon L., Lutjohann D., Stahle L., Widhe T., Bindl L., Eggertsen G., et al. (2000). Plasma levels of 24S-hydroxycholesterol reflect the balance between cerebral production and hepatic metabolism and are inversely related to body surface. J. Lipid Res. 41, 840–845 [PubMed] [Google Scholar]
- Brewer G. J. (2009). The risks of copper toxicity contributing to cognitive decline in the aging population and to Alzheimer's disease. J. Am. Coll. Nutr. 28, 238–242 [DOI] [PubMed] [Google Scholar]
- Brookmeyer R., Johnson E., Ziegler-Graham K., Arrighi H. M. (2007). Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 3, 186–191 10.1016/j.jalz.2007.04.381 [DOI] [PubMed] [Google Scholar]
- Buhaescu I., Izzedine H. (2007). Mevalonate pathway: a review of clinical and therapeutical implications. Clin. Biochem. 40, 575–584 10.1016/j.clinbiochem.2007.03.016 [DOI] [PubMed] [Google Scholar]
- Burkhead J. L., Gray L. W., Lutsenko S. (2011). Systems biology approach to Wilson's disease. Biometals 24, 455–466 10.1007/s10534-011-9430-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C. H., Brown R., Linder M. C. (1981). Circulating ceruloplasmin is an important source of copper for normal and malignant animal cells. Biochim. Biophys. Acta 678, 27–38 [DOI] [PubMed] [Google Scholar]
- Camponeschi F., Valensin D., Tessari I., Bubacco L., Dell'Acqua S., Casella L., et al. (2013). Copper(I)-alpha-synuclein interaction: structural description of two independent and competing metal binding sites. Inorg. Chem. 52, 1358–1367 10.1021/ic302050m [DOI] [PubMed] [Google Scholar]
- Cannon J. R., Greenamyre J. T. (2011). The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 124, 225–250 10.1093/toxsci/kfr239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr T. P., Lei K. Y. (1989). In vivo apoprotein catabolism of high density lipoproteins in copper-deficient, hypercholesterolemic rats. Proc. Soc. Exp. Biol. Med. 191, 370–376 [DOI] [PubMed] [Google Scholar]
- Cartwright G. E., Wintrobe M. M. (1964). Copper metabolism in normal subjects. Am. J. Clin. Nutr. 14, 224–232 [DOI] [PubMed] [Google Scholar]
- Cater M. A., McInnes K. T., Li Q. X., Volitakis I., La Fontaine S., Mercer J. F., et al. (2008). Intracellular copper deficiency increases amyloid-beta secretion by diverse mechanisms. Biochem. J. 412, 141–152 10.1042/BJ20080103 [DOI] [PubMed] [Google Scholar]
- Cheng H., Vetrivel K. S., Gong P., Meckler X., Parent A., Thinakaran G. (2007). Mechanisms of disease: new therapeutic strategies for Alzheimer's disease–targeting APP processing in lipid rafts. Nat. Clin. Pract. Neurol. 3, 374–382 10.1038/ncpneuro0549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny R. A., Atwood C. S., Xilinas M. E., Gray D. N., Jones W. D., McLean C. A., et al. (2001). Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30, 665–676 [DOI] [PubMed] [Google Scholar]
- Chobanian A. V., Hollander W. (1962). Body cholesterol metabolism in man. I. The equilibration of serum and tissue cholesterol. J. Clin. Invest. 41, 1732–1737 10.1172/JCI104631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi B. S., Zheng W. (2009). Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 1248, 14–21 10.1016/j.brainres.2008.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccotosto G. D., Tew D., Curtain C. C., Smith D., Carrington D., Masters C. L., et al. (2004). Enhanced toxicity and cellular binding of a modified amyloid beta peptide with a methionine to valine substitution. J. Biol. Chem. 279, 42528–42534 10.1074/jbc.M406465200 [DOI] [PubMed] [Google Scholar]
- Cobbold C., Coventry J., Ponnambalam S., Monaco A. P. (2004). Actin and microtubule regulation of trans-Golgi network architecture, and copper-dependent protein transport to the cell surface. Mol. Membr. Biol. 21, 59–66 10.1080/096870310001607350 [DOI] [PubMed] [Google Scholar]
- Cordy J. M., Hooper N. M., Turner A. J. (2006). The involvement of lipid rafts in Alzheimer's disease. Mol. Membr. Biol. 23, 111–122 10.1080/09687860500496417 [DOI] [PubMed] [Google Scholar]
- Cossec J. C., Simon A., Marquer C., Moldrich R. X., Leterrier C., Rossier J., et al. (2010). Clathrin-dependent APP endocytosis and Abeta secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim. Biophys. Acta 1801, 846–852 10.1016/j.bbalip.2010.05.010 [DOI] [PubMed] [Google Scholar]
- Crouch P. J., Hung L. W., Adlard P. A., Cortes M., Lal V., Filiz G., et al. (2009). Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 381–386 10.1073/pnas.0809057106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuajungco M. P., Goldstein L. E., Nunomura A., Smith M. A., Lim J. T., Atwood C. S., et al. (2000). Evidence that the beta-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of abeta by zinc. J. Biol. Chem. 275, 19439–19442 10.1074/jbc.C000165200 [DOI] [PubMed] [Google Scholar]
- Culotta V. C., Klomp L. W., Strain J., Casareno R. L., Krems B., Gitlin J. D. (1997). The copper chaperone for superoxide dismutase. J. Biol. Chem. 272, 23469–23472 10.1074/jbc.272.38.23469 [DOI] [PubMed] [Google Scholar]
- Curtain C. C., Ali F., Volitakis I., Cherny R. A., Norton R. S., Beyreuther K., et al. (2001). Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276, 20466–20473 10.1074/jbc.M100175200 [DOI] [PubMed] [Google Scholar]
- Curtain C. C., Ali F. E., Smith D. G., Bush A. I., Masters C. L., Barnham K. J. (2003). Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-beta peptide with membrane lipid. J. Biol. Chem. 278, 2977–2982 10.1074/jbc.M205455200 [DOI] [PubMed] [Google Scholar]
- Davies K. M., Hare D. J., Cottam V., Chen N., Hilgers L., Halliday G., et al. (2013). Localization of copper and copper transporters in the human brain. Metallomics 5, 43–51 10.1039/c2mt20151h [DOI] [PubMed] [Google Scholar]
- de Chaves E. I., Rusinol A. E., Vance D. E., Campenot R. B., Vance J. E. (1997). Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J. Biol. Chem. 272, 30766–30773 10.1074/jbc.272.49.30766 [DOI] [PubMed] [Google Scholar]
- Deci S., Lemieux S. K., Smith-Bell C. A., Sparks D. L., Schreurs B. G. (2012). Cholesterol increases ventricular volume in a rabbit model of Alzheimer's disease. J. Alzheimers Dis. 29, 283–292 10.3233/JAD-2011-111415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deibel M. A., Ehmann W. D., Markesbery W. R. (1996). Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer's disease: possible relation to oxidative stress. J. Neurol. Sci. 143, 137–142 [DOI] [PubMed] [Google Scholar]
- de Wolf I., Fielmich-Bouman X., Lankhorst A., van Oost B., Beynen A., Kren V., et al. (2003a). Liver copper content of rats hypo- or hyperresponsive to dietary cholesterol. J. Trace Elem. Med. Biol. 17, 177–182 10.1016/S0946-672X(03)80023-8 [DOI] [PubMed] [Google Scholar]
- de Wolf I. D., Fielmich-Bouman X. M., Lankhorst A., den Bieman M., van Oost B. A., Beynen A. C., et al. (2003b). Cholesterol and copper in the liver of rabbit inbred strains with differences in dietary cholesterol response. J. Nutr. Biochem. 14, 459–465 10.1016/S0955-2863(03)00059-7 [DOI] [PubMed] [Google Scholar]
- Dietschy J. M., Turley S. D. (2004). Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid. Res. 45, 1375–1397 10.1194/jlr.R400004-JLR200 [DOI] [PubMed] [Google Scholar]
- Dingwall C. (2007). A copper-binding site in the cytoplasmic domain of BACE1 identifies a possible link to metal homoeostasis and oxidative stress in Alzheimer's disease. Biochem. Soc. Trans. 35, 571–573 10.1042/BST0350571 [DOI] [PubMed] [Google Scholar]
- Distl R., Meske V., Ohm T. G. (2001). Tangle-bearing neurons contain more free cholesterol than adjacent tangle-free neurons. Acta Neuropathol. 101, 547–554 10.1007/s004010000314 [DOI] [PubMed] [Google Scholar]
- Distl R., Treiber-Held S., Albert F., Meske V., Harzer K., Ohm T. G. (2003). Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain. J. Pathol. 200, 104–111 10.1002/path.1320 [DOI] [PubMed] [Google Scholar]
- Dixit R., Ross J. L., Goldman Y. E., Holzbaur E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089 10.1126/science.1152993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolska J., Dehnhardt M., Matusch A., Zoriy M., Palomero-Gallagher N., Koscielniak P., et al. (2008). Quantitative imaging of zinc, copper and lead in three distinct regions of the human brain by laser ablation inductively coupled plasma mass spectrometry. Talanta 74, 717–723 10.1016/j.talanta.2007.06.051 [DOI] [PubMed] [Google Scholar]
- Dong J., Atwood C. S., Anderson V. E., Siedlak S. L., Smith M. A., Perry G., et al. (2003). Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: raman microscopic evidence. Biochemistry 42, 2768–2773 10.1021/bi0272151 [DOI] [PubMed] [Google Scholar]
- Dong L. M., Parkin S., Trakhanov S. D., Rupp B., Simmons T., Arnold K. S., et al. (1996). Novel mechanism for defective receptor binding of apolipoprotein E2 in type III hyperlipoproteinemia. Nat. Struct. Biol. 3, 718–722 [DOI] [PubMed] [Google Scholar]
- Donnelly P. S., Caragounis A., Du T., Laughton K. M., Volitakis I., Cherny R. A., et al. (2008). Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J. Biol. Chem. 283, 4568–4577 10.1074/jbc.M705957200 [DOI] [PubMed] [Google Scholar]
- Donsante A., Johnson P., Jansen L. A., Kaler S. G. (2010). Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am. J. Med. Genet. A 152A, 2529–2534 10.1002/ajmg.a.33632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doreulee N., Yanovsky Y., Haas H. L. (1997). Suppression of long-term potentiation in hippocampal slices by copper. Hippocampus 7, 666–669 [DOI] [PubMed] [Google Scholar]
- Drew S. C., Leong S. L., Pham C. L., Tew D. J., Masters C. L., Miles L. A., et al. (2008). Cu2+ binding modes of recombinant alpha-synuclein–insights from EPR spectroscopy. J. Am. Chem. Soc. 130, 7766–7773 10.1021/ja800708x [DOI] [PubMed] [Google Scholar]
- Ehehalt R., Keller P., Haass C., Thiele C., Simons K. (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123 10.1083/jcb.200207113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Meskini R., Cline L. B., Eipper B. A., Ronnett G. V. (2005). The developmentally regulated expression of Menkes protein ATP7A suggests a role in axon extension and synaptogenesis. Dev. Neurosci. 27, 333–348 10.1159/000086713 [DOI] [PubMed] [Google Scholar]
- El Meskini R., Crabtree K. L., Cline L. B., Mains R. E., Eipper B. A., Ronnett G. V. (2007). ATP7A (Menkes protein) functions in axonal targeting and synaptogenesis. Mol. Cell. Neurosci. 34, 409–421 10.1016/j.mcn.2006.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q. W., Yu W., Senda T., Yanagisawa K., Michikawa M. (2001). Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J. Neurochem. 76, 391–400 10.1046/j.1471-4159.2001.00063.x [DOI] [PubMed] [Google Scholar]
- Fassbender K., Simons M., Bergmann C., Stroick M., Lutjohann D., Keller P., et al. (2001). Simvastatin strongly reduces levels of Alzheimer's disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 5856–5861 10.1073/pnas.081620098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd R. A. (1999). Antioxidants, oxidative stress, and degenerative neurological disorders. Proc. Soc. Exp. Biol. Med. 222, 236–245 [DOI] [PubMed] [Google Scholar]
- Freedman J. H., Ciriolo M. R., Peisach J. (1989). The role of glutathione in copper metabolism and toxicity. J. Biol. Chem. 264, 5598–5605 [PubMed] [Google Scholar]
- Fünfschilling U., Jockusch W. J., Sivakumar N., Mobius W., Corthals K., Li S., et al. (2012). Critical time window of neuronal cholesterol synthesis during neurite outgrowth. J. Neurosci. 32, 7632–7645 10.1523/JNEUROSCI.1352-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U., Saher G., Xiao L., Mobius W., Nave K. A. (2007). Survival of adult neurons lacking cholesterol synthesis in vivo. BMC Neurosci. 8:1 10.1186/1471-2202-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghribi O. (2008). Potential mechanisms linking cholesterol to Alzheimer's disease-like pathology in rabbit brain, hippocampal organotypic slices, and skeletal muscle. J. Alzheimers Dis. 15, 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ghribi O., Larsen B., Schrag M., Herman M. M. (2006). High cholesterol content in neurons increases BACE, beta-amyloid, and phosphorylated tau levels in rabbit hippocampus. Exp. Neurol. 200, 460–467 10.1016/j.expneurol.2006.03.019 [DOI] [PubMed] [Google Scholar]
- Glenner G. G., Wong C. W. (1984). Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890 10.1016/S0006-291X(84)80190-4 [DOI] [PubMed] [Google Scholar]
- Glerum D. M., Shtanko A., Tzagoloff A. (1996a). Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 271, 14504–14509 10.1074/jbc.271.24.14504 [DOI] [PubMed] [Google Scholar]
- Glerum D. M., Shtanko A., Tzagoloff A. (1996b). SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J. Biol. Chem. 271, 20531–20535 10.1074/jbc.271.34.20531 [DOI] [PubMed] [Google Scholar]
- Goldstein J. L., Brown M. S. (1990). Regulation of the mevalonate pathway. Nature 343, 425–430 10.1038/343425a0 [DOI] [PubMed] [Google Scholar]
- Goldstein J. L., Brown M. S. (2009). The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 29, 431–438 10.1161/ATVBAHA.108.179564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C., Martin T., Cacho J., Brenas M. T., Arroyo T., Garcia-Berrocal B., et al. (1999). Serum zinc, copper, insulin and lipids in Alzheimer's disease epsilon 4 apolipoprotein E allele carriers. Eur. J. Clin. Invest. 29, 637–642 10.1046/j.1365-2362.1999.00471.x [DOI] [PubMed] [Google Scholar]
- Goritz C., Mauch D. H., Pfrieger F. W. (2005). Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell. Neurosci. 29, 190–201 10.1016/j.mcn.2005.02.006 [DOI] [PubMed] [Google Scholar]
- Gray E. H., De Vos K. J., Dingwall C., Perkinton M. S., Miller C. C. (2010). Deficiency of the copper chaperone for superoxide dismutase increases amyloid-beta production. J. Alzheimers Dis. 21, 1101–1105 10.3233/JAD-2010-100717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M. O., Grimm H. S., Patzold A. J., Zinser E. G., Halonen R., Duering M., et al. (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 7, 1118–1123 10.1038/ncb1313 [DOI] [PubMed] [Google Scholar]
- Guirland C., Zheng J. Q. (2007). Membrane lipid rafts and their role in axon guidance. Adv. Exp. Med. Biol. 621, 144–155 10.1007/978-0-387-76715-4_11 [DOI] [PubMed] [Google Scholar]
- Gunn A. P., Roberts B. R., Bush A. I. (2012). Rapid generation of dityrosine cross-linked Abeta oligomers via Cu-redox cycling. Methods Mol. Biol. 849, 3–10 10.1007/978-1-61779-551-0_1 [DOI] [PubMed] [Google Scholar]
- Gybina A. A., Prohaska J. R. (2006). Variable response of selected cuproproteins in rat choroid plexus and cerebellum following perinatal copper deficiency. Genes Nutr. 1, 51–59 10.1007/BF02829936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeffner F., Smith D. G., Barnham K. J., Bush A. I. (2005). Model studies of cholesterol and ascorbate oxidation by copper complexes: relevance to Alzheimer's disease beta-amyloid metallochemistry. J. Inorg. Biochem. 99, 2403–2422 10.1016/j.jinorgbio.2005.09.011 [DOI] [PubMed] [Google Scholar]
- Halliwell B., Gutteridge J. M. C. (1984). Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 219, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza I., Prohaska J., Gitlin J. D. (2003). Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc. Natl. Acad. Sci. U.S.A. 100, 1215–1220 10.1073/pnas.0336230100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara A., Taketomi T. (1986). Cerebral lipid and protein abnormalities in Menkes' steely-hair disease. Jpn. J. Exp. Med. 56, 277–284 [PubMed] [Google Scholar]
- Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 41, 1088–1093 10.1038/ng.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartter D. E., Barnea A. (1988). Evidence for release of copper in the brain: depolarization-induced release of newly taken-up 67copper. Synapse 2, 412–415 10.1002/syn.890020408 [DOI] [PubMed] [Google Scholar]
- Hatters D. M., Peters-Libeu C. A., Weisgraber K. H. (2006). Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 31, 445–454 10.1016/j.tibs.2006.06.008 [DOI] [PubMed] [Google Scholar]
- Hauser P. S., Narayanaswami V., Ryan R. O. (2011). Apolipoprotein E: from lipid transport to neurobiology. Prog. Lipid Res. 50, 62–74 10.1016/j.plipres.2010.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heicklen-Klein A., Ginzburg I. (2000). Tau promoter confers neuronal specificity and binds Sp1 and AP-2. J. Neurochem. 75, 1408–1418 10.1046/j.1471-4159.2000.0751408.x [DOI] [PubMed] [Google Scholar]
- Hering H., Lin C. C., Sheng M. (2003). Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 23, 3262–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heverin M., Meaney S., Lutjohann D., Diczfalusy U., Wahren J., Björkhem I. (2005). Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. J. Lipid Res. 46, 1047–1052 10.1194/jlr.M500024-JLR200 [DOI] [PubMed] [Google Scholar]
- Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J. C., Carrasquillo M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 10.1038/ng.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman D. M., Herz J., Bu G. (2012). Apolipoprotein e and apolipoprotein e receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Pers. Med. 2:a006312 10.1101/cshperspect.a006312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper N. M. (2005). Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein. Biochem. Soc. Trans. 33, 335–338 10.1042/BST0330335 [DOI] [PubMed] [Google Scholar]
- Hopt A., Korte S., Fink H., Panne U., Niessner R., Jahn R., et al. (2003). Methods for studying synaptosomal copper release. J. Neurosci. Methods 128, 159–172 10.1016/S0165-0270(03)00173-0 [DOI] [PubMed] [Google Scholar]
- Horng Y. C., Cobine P. A., Maxfield A. B., Carr H. S., Winge D. R. (2004). Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem. 279, 35334–35340 10.1074/jbc.M404747200 [DOI] [PubMed] [Google Scholar]
- Horvath R., Lochmuller H., Stucka R., Yao J., Shoubridge E. A., Kim S. H., et al. (2000). Characterization of human SCO1 and COX17 genes in mitochondrial cytochrome-c-oxidase deficiency. Biochem. Biophys. Res. Commun. 276, 530–533 10.1006/bbrc.2000.3495 [DOI] [PubMed] [Google Scholar]
- Huang X., Cuajungco M. P., Atwood C. S., Hartshorn M. A., Tyndall J. D., Hanson G. R., et al. (1999). Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274, 37111–37116 10.1074/jbc.274.52.37111 [DOI] [PubMed] [Google Scholar]
- Hung Y. H., Layton M. J., Voskoboinik I., Mercer J. F., Camakaris J. (2007). Purification and membrane reconstitution of catalytically active Menkes copper-transporting P-type ATPase (MNK; ATP7A). Biochem. J. 401, 569–579 10.1042/BJ20060924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung Y. H., Robb E. L., Volitakis I., Ho M., Evin G., Li Q. X., et al. (2009). Paradoxical condensation of copper with elevated beta-amyloid in lipid rafts under cellular copper deficiency conditions: implications for Alzheimer disease. J. Biol. Chem. 284, 21899–21907 10.1074/jbc.M109.019521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huster D., Purnat T. D., Burkhead J. L., Ralle M., Fiehn O., Stuckert F., et al. (2007). High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 282, 8343–8355 10.1074/jbc.M607496200 [DOI] [PubMed] [Google Scholar]
- Ikonen E. (2006). Mechanisms for cellular cholesterol transport: defects and human disease. Physiol. Rev. 86, 1237–1261 10.1152/physrev.00022.2005 [DOI] [PubMed] [Google Scholar]
- Illing A. C., Shawki A., Cunningham C. L., Mackenzie B. (2012). Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J. Biol. Chem. 287, 30485–30496 10.1074/jbc.M112.364208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante R. E., Abi-Mosleh L., Radhakrishnan A., Dale J. D., Brown M. S., Goldstein J. L. (2008a). Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 283, 1052–1063 10.1074/jbc.M707943200 [DOI] [PubMed] [Google Scholar]
- Infante R. E., Radhakrishnan A., Abi-Mosleh L., Kinch L. N., Wang M. L., Grishin N. V., et al. (2008b). Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283, 1064–1075 10.1074/jbc.M707944200 [DOI] [PubMed] [Google Scholar]
- Infante R. E., Wang M. L., Radhakrishnan A., Kwon H. J., Brown M. S., Goldstein J. L. (2008c). NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. U.S.A. 105, 15287–15292 10.1073/pnas.0807328105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innerarity T. L., Weisgraber K. H., Arnold K. S., Rall S. C., Jr., Mahley R. W. (1984). Normalization of receptor binding of apolipoprotein E2. Evidence for modulation of the binding site conformation. J. Biol. Chem. 259, 7261–7267 [PubMed] [Google Scholar]
- Ittner A., Ke Y. D., Eersel J., Gladbach A., Gotz J., Ittner L. M. (2011). Brief update on different roles of tau in neurodegeneration. IUBMB Life 63, 495–502 10.1002/iub.467 [DOI] [PubMed] [Google Scholar]
- Ittner L. M., Ke Y. D., Delerue F., Bi M., Gladbach A., van Eersel J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397 10.1016/j.cell.2010.06.036 [DOI] [PubMed] [Google Scholar]
- Iwase T., Nishimura M., Sugimura H., Igarashi H., Ozawa F., Shinmura K., et al. (1996). Localization of Menkes gene expression in the mouse brain; its association with neurological manifestations in Menkes model mice. Acta Neuropathol. 91, 482–488 [DOI] [PubMed] [Google Scholar]
- Jarvik G. P., Wijsman E. M., Kukull W. A., Schellenberg G. D., Yu C., Larson E. B. (1995). Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer's disease: a case-control study. Neurology 45, 1092–1096 [DOI] [PubMed] [Google Scholar]
- Jiang D., Men L., Wang J., Zhang Y., Chickenyen S., Wang Y., et al. (2007). Redox reactions of copper complexes formed with different beta-amyloid peptides and their neuropathological [correction of neuropathalogical] relevance. Biochemistry 46, 9270–9282 10.1021/bi700508n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevics H., Hostettler J., Muse E. D., Sammond D. W., Matsushima G. K., Toews A. D., et al. (2001). Cerebroside synthesis as a measure of the rate of remyelination following cuprizone-induced demyelination in brain. J. Neurochem. 77, 1067–1076 10.1046/j.1471-4159.2001.00310.x [DOI] [PubMed] [Google Scholar]
- Jurevics H., Largent C., Hostettler J., Sammond D. W., Matsushima G. K., Kleindienst A., et al. (2002). Alterations in metabolism and gene expression in brain regions during cuprizone-induced demyelination and remyelination. J. Neurochem. 82, 126–136 10.1046/j.1471-4159.2002.00954.x [DOI] [PubMed] [Google Scholar]
- Kako K., Tsumori K., Ohmasa Y., Takahashi Y., Munekata E. (2000). The expression of Cox17p in rodent tissues and cells. Eur. J. Biochem. 267, 6699–6707 10.1046/j.1432-1327.2000.01771.x [DOI] [PubMed] [Google Scholar]
- Kaler S. G. (2011). ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 7, 15–29 10.1038/nrneurol.2010.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos J., Kovacs I., Hajos F., Kalman M., Simonyi M. (1989). Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 103, 139–144 10.1016/0304-3940(89)90565-X [DOI] [PubMed] [Google Scholar]
- Karten B., Vance D. E., Campenot R. B., Vance J. E. (2002). Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J. Neurochem. 83, 1154–1163 10.1046/j.1471-4159.2002.01220.x [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T., Shoji M., Younkin L. H., Wen-Lang L., Dickson D. W., Murakami T., et al. (2004). Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 24, 3801–3809 10.1523/JNEUROSCI.5543-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelner G. S., Lee M., Clark M. E., Maciejewski D., McGrath D., Rabizadeh S., et al. (2000). The copper transport protein Atox1 promotes neuronal survival. J. Biol. Chem. 275, 580–584 10.1074/jbc.275.1.580 [DOI] [PubMed] [Google Scholar]
- Kessler H., Pajonk F. G., Meisser P., Schneider-Axmann T., Hoffmann K. H., Supprian T., et al. (2006). Cerebrospinal fluid diagnostic markers correlate with lower plasma copper and ceruloplasmin in patients with Alzheimer's disease. J. Neural Transm. 113, 1763–1769 10.1007/s00702-006-0485-7 [DOI] [PubMed] [Google Scholar]
- Kim W. S., Rahmanto A. S., Kamili A., Rye K. A., Guillemin G. J., Gelissen I. C., et al. (2007). Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 282, 2851–2861 10.1074/jbc.M607831200 [DOI] [PubMed] [Google Scholar]
- Kim W. S., Weickert C. S., Garner B. (2008). Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J. Neurochem. 104, 1145–1166 10.1111/j.1471-4159.2007.05099.x [DOI] [PubMed] [Google Scholar]
- Kitazawa M., Cheng D., Laferla F. M. (2009). Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J. Neurochem. 108, 1550–1560 10.1111/j.1471-4159.2009.05901.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivipelto M., Solomon A. (2006). Cholesterol as a risk factor for Alzheimer's disease - epidemiological evidence. Acta Neurol. Scand. Suppl. 185, 50–57 10.1111/j.1600-0404.2006.00685.x [DOI] [PubMed] [Google Scholar]
- Kjellin K. (1963). Determination of copper in cerebrospinal fluid by activation analysis. J. Neurochem. 10, 89–93 [DOI] [PubMed] [Google Scholar]
- Klevay L. M. (1988). Dietary cholesterol lowers liver copper in rabbits. Biol. Trace Elem. Res. 16, 51–57 10.1007/BF02795333 [DOI] [PubMed] [Google Scholar]
- Klomp A. E., Tops B. B., Van Denberg I. E., Berger R., Klomp L. W. (2002). Biochemical characterization and subcellular localization of human copper transporter 1 (hCTR1). Biochem. J. 364, 497–505 10.1042/BJ20011803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klomp L. W., Lin S. J., Yuan D. S., Klausner R. D., Culotta V. C., Gitlin J. D. (1997). Identification and functional expression of HAH1, a novel human gene involved in copper homeostasis. J. Biol. Chem. 272, 9221–9226 10.1074/jbc.272.14.9221 [DOI] [PubMed] [Google Scholar]
- Ko M., Zou K., Minagawa H., Yu W., Gong J. S., Yanagisawa K., et al. (2005). Cholesterol-mediated neurite outgrowth is differently regulated between cortical and hippocampal neurons. J. Biol. Chem. 280, 42759–42765 10.1074/jbc.M509164200 [DOI] [PubMed] [Google Scholar]
- Kolovou G. D., Mikhailidis D. P., Anagnostopoulou K. K., Daskalopoulou S. S., Cokkinos D. V. (2006). Tangier disease four decades of research: a reflection of the importance of HDL. Curr. Med. Chem. 13, 771–782 10.2174/092986706776055580 [DOI] [PubMed] [Google Scholar]
- Koppaka V., Axelsen P. H. (2000). Accelerated accumulation of amyloid beta proteins on oxidatively damaged lipid membranes. Biochemistry 39, 10011–10016 10.1021/bi000619d [DOI] [PubMed] [Google Scholar]
- Korade Z., Mi Z., Portugal C., Schor N. F. (2007). Expression and p75 neurotrophin receptor dependence of cholesterol synthetic enzymes in adult mouse brain. Neurobiol. Aging 28, 1522–1531 10.1016/j.neurobiolaging.2006.06.026 [DOI] [PubMed] [Google Scholar]
- Koudinov A. R., Koudinova N. V. (2001). Essential role for cholesterol in synaptic plasticity and neuronal degeneration. FASEB J. 15, 1858–1860 10.1096/fj.00-0815fje [DOI] [PubMed] [Google Scholar]
- Koudinov A. R., Koudinova N. V. (2005). Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. J. Neurol. Sci. 229–230, 233–240 10.1016/j.jns.2004.11.036 [DOI] [PubMed] [Google Scholar]
- Kruth H. S., Comly M. E., Butler J. D., Vanier M. T., Fink J. K., Wenger D. A., et al. (1986). Type C Niemann-Pick disease. Abnormal metabolism of low density lipoprotein in homozygous and heterozygous fibroblasts. J. Biol. Chem. 261, 16769–16774 [PubMed] [Google Scholar]
- Kuo Y. M., Gybina A. A., Pyatskowit J. W., Gitschier J., Prohaska J. R. (2006). Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. J. Nutr. 136, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y. M., Zhou B., Cosco D., Gitschier J. (2001). The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc. Natl. Acad. Sci. U.S.A. 98, 6836–6841 10.1073/pnas.111057298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H. J., Abi-Mosleh L., Wang M. L., Deisenhofer J., Goldstein J. L., Brown M. S., et al. (2009). Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137, 1213–1224 10.1016/j.cell.2009.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc V., Jasmin-Belanger S., Poirier J. (2010). APOE and cholesterol homeostasis in Alzheimer's disease. Trends Mol. Med. 16, 469–477 10.1016/j.molmed.2010.07.008 [DOI] [PubMed] [Google Scholar]
- Lee J., Prohaska J. R., Thiele D. J. (2001). Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc. Natl. Acad. Sci. U.S.A. 98, 6842–6847 10.1073/pnas.111058698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H., Lancey R., Montaser A., Madani N., Linder M. C. (1993). Ceruloplasmin and copper transport during the latter part of gestation in the rat. Proc. Soc. Exp. Biol. Med. 203, 428–439 [DOI] [PubMed] [Google Scholar]
- Lei K. Y. (1991). Dietary copper: cholesterol and lipoprotein metabolism. Annu. Rev. Nutr. 11, 265–283 10.1146/annurev.nu.11.070191.001405 [DOI] [PubMed] [Google Scholar]
- Lentner C. (Ed.) (1981). Geigy Scientific Tables: Units of Measurement, Body Fluids, Composition of the Body, Nutrition, 8th Edn, Ciba-Geigy Corporation Medical Education Division, (Basel). [Google Scholar]
- Lin H., Bhatia R., Lal R. (2001). Amyloid beta protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 15, 2433–2444 10.1096/fj.01-0377com [DOI] [PubMed] [Google Scholar]
- Lin R., Chen X., Li W., Han Y., Liu P., Pi R. (2008). Exposure to metal ions regulates mRNA levels of APP and BACE1 in PC12 cells: blockage by curcumin. Neurosci. Lett. 440, 344–347 10.1016/j.neulet.2008.05.070 [DOI] [PubMed] [Google Scholar]
- Lin S. J., Culotta V. C. (1995). The ATX1 gene of Saccharomyces cerevisiae encodes a small metal homeostasis factor that protects cells against reactive oxygen toxicity. Proc. Natl. Acad. Sci. U.S.A. 92, 3784–3788 10.1073/pnas.92.9.3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder M. C. (1991). Biochemistry of Copper. New York, NY: Plenum Press [Google Scholar]
- Linder M. C., Hazegh-Azam M. (1996). Copper biochemistry and molecular biology. Am. J. Clin. Nutr. 63, 797S–811S [DOI] [PubMed] [Google Scholar]
- Linton M. F., Gish R., Hubl S. T., Butler E., Esquivel C., Bry W. I., et al. (1991). Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J. Clin. Invest. 88, 270–281 10.1172/JCI115288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin T., Gromadzka G., Czlonkowska A. (2012). Apolipoprotein E gene (APOE) genotype in Wilson's disease: impact on clinical presentation. Parkinsonism Relat. Disord. 18, 367–369 10.1016/j.parkreldis.2011.12.005 [DOI] [PubMed] [Google Scholar]
- Liu L., Komatsu H., Murray I. V., Axelsen P. H. (2008). Promotion of amyloid beta protein misfolding and fibrillogenesis by a lipid oxidation product. J. Mol. Biol. 377, 1236–1250 10.1016/j.jmb.2008.01.057 [DOI] [PubMed] [Google Scholar]
- Liu Q., Zerbinatti C. V., Zhang J., Hoe H. S., Wang B., Cole S. L., et al. (2007). Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78 10.1016/j.neuron.2007.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. (1998). Copper, iron and zinc in Alzheimer's disease senile plaques. J. Neurol. Sci. 158, 47–52 [DOI] [PubMed] [Google Scholar]
- Lu J., Wu D. M., Zheng Y. L., Sun D. X., Hu B., Shan Q., et al. (2009). Trace amounts of copper exacerbate beta amyloid-induced neurotoxicity in the cholesterol-fed mice through TNF-mediated inflammatory pathway. Brain Behav. Immun. 23, 193–203 10.1016/j.bbi.2008.09.003 [DOI] [PubMed] [Google Scholar]
- Lu J., Zheng Y. L., Wu D. M., Sun D. X., Shan Q., Fan S. H. (2006). Trace amounts of copper induce neurotoxicity in the cholesterol-fed mice through apoptosis. FEBS Lett. 580, 6730–6740 10.1016/j.febslet.2006.10.072 [DOI] [PubMed] [Google Scholar]
- Lund-Katz S., Wehrli S., Zaiou M., Newhouse Y., Weisgraber K. H., Phillips M. C. (2001). Effects of polymorphism on the microenvironment of the LDL receptor-binding region of human apoE. J. Lipid Res. 42, 894–901 [PubMed] [Google Scholar]
- Lund E. G., Guileyardo J. M., Russell D. W. (1999). cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U.S.A. 96, 7238–7243 10.1073/pnas.96.13.7238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E. G., Xie C., Kotti T., Turley S. D., Dietschy J. M., Russell D. W. (2003). Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 278, 22980–22988 10.1074/jbc.M303415200 [DOI] [PubMed] [Google Scholar]
- Lutjohann D., Breuer O., Ahlborg G., Nennesmo I., Siden A., Diczfalusy U., et al. (1996). Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. U.S.A. 93, 9799–9804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutjohann D., von Bergmann K. (2003). 24S-hydroxycholesterol: a marker of brain cholesterol metabolism. Pharmacopsychiatry 36(Suppl. 2), S102–S106 10.1055/s-2003-43053 [DOI] [PubMed] [Google Scholar]
- Magaki S., Raghavan R., Mueller C., Oberg K. C., Vinters H. V., Kirsch W. M. (2007). Iron, copper, and iron regulatory protein 2 in Alzheimer's disease and related dementias. Neurosci. Lett. 418, 72–76 10.1016/j.neulet.2007.02.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630 10.1126/science.3283935 [DOI] [PubMed] [Google Scholar]
- Mailman T., Hariharan M., Karten B. (2011). Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J. Neurochem. 119, 1002–1015 10.1111/j.1471-4159.2011.07474.x [DOI] [PubMed] [Google Scholar]
- Mao X., Ye J., Zhou S., Pi R., Dou J., Zang L., et al. (2012). The effects of chronic copper exposure on the amyloid protein metabolisim associated genes' expression in chronic cerebral hypoperfused rats. Neurosci. Lett. 518, 14–18 10.1016/j.neulet.2012.04.030 [DOI] [PubMed] [Google Scholar]
- Marquer C., Devauges V., Cossec J. C., Liot G., Lecart S., Saudou F., et al. (2011). Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 25, 1295–1305 10.1096/fj.10-168633 [DOI] [PubMed] [Google Scholar]
- Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materia S., Cater M. A., Klomp L. W., Mercer J. F., La Fontaine S. (2011). Clusterin (apolipoprotein J), a molecular chaperone that facilitates degradation of the copper-ATPases ATP7A and ATP7B. J. Biol. Chem. 286, 10073–10083 10.1074/jbc.M110.190546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materia S., Cater M. A., Klomp L. W., Mercer J. F., La Fontaine S. (2012). Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem. 287, 2485–2499 10.1074/jbc.M111.302216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. (2004). Pathways towards and away from Alzheimer's disease. Nature 430, 631–639 10.1038/nature02621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch D. H., Nagler K., Schumacher S., Goritz C., Muller E. C., Otto A., et al. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357 10.1126/science.294.5545.1354 [DOI] [PubMed] [Google Scholar]
- Maynard C. J., Cappai R., Volitakis I., Cherny R. A., Masters C. L., Li Q. X., et al. (2006). Gender and genetic background effects on brain metal levels in APP transgenic and normal mice: implications for Alzheimer beta-amyloid pathology. J. Inorg. Biochem. 100, 952–962 10.1016/j.jinorgbio.2006.02.010 [DOI] [PubMed] [Google Scholar]
- Maynard C. J., Cappai R., Volitakis I., Cherny R. A., White A. R., Beyreuther K., et al. (2002). Overexpression of Alzheimer's disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277, 44670–44676 10.1074/jbc.M204379200 [DOI] [PubMed] [Google Scholar]
- Meaney S., Lutjohann D., Diczfalusy U., Björkhem I. (2000). Formation of oxysterols from different pools of cholesterol as studied by stable isotope technique: cerebral origin of most circulating 24S-hydroxycholesterol in rats, but not in mice. Biochim. Biophys. Acta 1486, 293–298 10.1016/S1388-1981(00)00070-6 [DOI] [PubMed] [Google Scholar]
- Meloni G., Sonois V., Delaine T., Guilloreau L., Gillet A., Teissie J., et al. (2008). Metal swap between Zn7-metallothionein-3 and amyloid-beta-Cu protects against amyloid-beta toxicity. Nat. Chem. Biol. 4, 366–372 10.1038/nchembio.89 [DOI] [PubMed] [Google Scholar]
- Methia N., Andre P., Hafezi-Moghadam A., Economopoulos M., Thomas K. L., Wagner D. D. (2001). ApoE deficiency compromises the blood brain barrier especially after injury. Mol. Med. 7, 810–815 [PMC free article] [PubMed] [Google Scholar]
- Michikawa M., Fan Q. W., Isobe I., Yanagisawa K. (2000). Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem. 74, 1008–1016 10.1046/j.1471-4159.2000.0741008.x [DOI] [PubMed] [Google Scholar]
- Mielke M. M., Zandi P. P., Sjogren M., Gustafson D., Ostling S., Steen B., et al. (2005). High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology 64, 1689–1695 10.1212/01.WNL.0000161870.78572.A5 [DOI] [PubMed] [Google Scholar]
- Miller L. M., Wang Q., Telivala T. P., Smith R. J., Lanzirotti A., Miklossy J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer's disease. J. Struct. Biol. 155, 30–37 10.1016/j.jsb.2005.09.004 [DOI] [PubMed] [Google Scholar]
- Minagawa H., Gong J. S., Jung C. G., Watanabe A., Lund-Katz S., Phillips M. C., et al. (2009). Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J. Neurosci. Res. 87, 2498–2508 10.1002/jnr.22073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M., Smith J. D. (1996). Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 14, 55–61 10.1038/ng0996-55 [DOI] [PubMed] [Google Scholar]
- Moir R. D., Atwood C. S., Romano D. M., Laurans M. H., Huang X., Bush A. I., et al. (1999). Differential effects of apolipoprotein E isoforms on metal-induced aggregation of A beta using physiological concentrations. Biochemistry 38, 4595–4603 10.1021/bi982437d [DOI] [PubMed] [Google Scholar]
- Morris M., Maeda S., Vossel K., Mucke L. (2011). The many faces of tau. Neuron 70, 410–426 10.1016/j.neuron.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M. C., Evans D. A., Bienias J. L., Tangney C. C., Bennett D. A., Aggarwal N., et al. (2003). Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol. 60, 194–200 10.1001/archneur.60.2.194 [DOI] [PubMed] [Google Scholar]
- Morris M. C., Evans D. A., Tangney C. C., Bienias J. L., Schneider J. A., Wilson R. S., et al. (2006). Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch. Neurol. 63, 1085–1088 10.1001/archneur.63.8.1085 [DOI] [PubMed] [Google Scholar]
- Murray I. V., Liu L., Komatsu H., Uryu K., Xiao G., Lawson J. A., et al. (2007). Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. J. Biol. Chem. 282, 9335–9345 10.1074/jbc.M608589200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray I. V., Sindoni M. E., Axelsen P. H. (2005). Promotion of oxidative lipid membrane damage by amyloid beta proteins. Biochemistry 44, 12606–12613 10.1021/bi050926p [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeve G. S., Vana A. M., Eggold J. R., Kelner G. S., Maki R., Desouza E. B., et al. (1999). Expression profile of the copper homeostasis gene, rAtox1, in the rat brain. Neuroscience 93, 1179–1187 10.1016/S0306-4522(99)00175-X [DOI] [PubMed] [Google Scholar]
- Naj A. C., Jun G., Beecham G. W., Wang L. S., Vardarajan B. N., Buros J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 43, 436–441 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negi S. I., Brautbar A., Virani S. S., Anand A., Polisecki E., Asztalos B. F., et al. (2013). A novel mutation in the ABCA1 gene causing an atypical phenotype of Tangier disease. J. Clin. Lipidol. 7, 82–87 10.1016/j.jacl.2012.09.004 [DOI] [PubMed] [Google Scholar]
- Nelson T. J., Alkon D. L. (2005). Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J. Biol. Chem. 280, 7377–7387 10.1074/jbc.M409071200 [DOI] [PubMed] [Google Scholar]
- Neumann P. Z., Sass-Kortsak A. (1967). The state of copper in human serum: evidence for an amino acid-bound fraction. J. Clin. Invest. 46, 646–658 10.1172/JCI105566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevitt T., Ohrvik H., Thiele D. J. (2012). Charting the travels of copper in eukaryotes from yeast to mammals. Biochim. Biophys. Acta 1823, 1580–1593 10.1016/j.bbamcr.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niciu M. J., Ma X. M., El Meskini R., Ronnett G. V., Mains R. E., Eipper B. A. (2006). Developmental changes in the expression of ATP7A during a critical period in postnatal neurodevelopment. Neuroscience 139, 947–964 10.1016/j.neuroscience.2006.01.044 [DOI] [PubMed] [Google Scholar]
- Nishitsuji K., Hosono T., Nakamura T., Bu G., Michikawa M. (2011). Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 286, 17536–17542 10.1074/jbc.M111.225532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notkola I. L., Sulkava R., Pekkanen J., Erkinjuntti T., Ehnholm C., Kivinen P., et al. (1998). Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology 17, 14–20 [DOI] [PubMed] [Google Scholar]
- Opazo C., Huang X., Cherny R. A., Moir R. D., Roher A. E., White A. R., et al. (2002). Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J. Biol. Chem. 277, 40302–40308 10.1074/jbc.M206428200 [DOI] [PubMed] [Google Scholar]
- Pajonk F. G., Kessler H., Supprian T., Hamzei P., Bach D., Schweickhardt J., et al. (2005). Cognitive decline correlates with low plasma concentrations of copper in patients with mild to moderate Alzheimer's disease. J. Alzheimers Dis. 8, 23–27 [DOI] [PubMed] [Google Scholar]
- Panchal M., Loeper J., Cossec J. C., Perruchini C., Lazar A., Pompon D., et al. (2010). Enrichment of cholesterol in microdissected Alzheimer's disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 51, 598–605 10.1194/jlr.M001859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappolla M. A., Bryant-Thomas T. K., Herbert D., Pacheco J., Fabra Garcia M., Manjon M., et al. (2003). Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 61, 199–205 [DOI] [PubMed] [Google Scholar]
- Pentchev P. G., Comly M. E., Kruth H. S., Vanier M. T., Wenger D. A., Patel S., et al. (1985). A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc. Natl. Acad. Sci. U.S.A. 82, 8247–8251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger F. W. (2003). Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? Bioessays 25, 72–78 10.1002/bies.10195 [DOI] [PubMed] [Google Scholar]
- Phinney A. L., Drisaldi B., Schmidt S. D., Lugowski S., Coronado V., Liang Y., et al. (2003). In vivo reduction of amyloid-beta by a mutant copper transporter. Proc. Natl. Acad. Sci. U.S.A. 100, 14193–14198 10.1073/pnas.2332851100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrini V., Pinna V., Milone F. F. (1990). Tangier disease: central nervous system impairment in a case of syringomyelia-like syndrome. J. Neurol. Sci. 98, 245–250 [DOI] [PubMed] [Google Scholar]
- Pitas R. E., Boyles J. K., Lee S. H., Foss D., Mahley R. W. (1987a). Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta 917, 148–161 10.1016/0005-2760(87)90295-5 [DOI] [PubMed] [Google Scholar]
- Pitas R. E., Boyles J. K., Lee S. H., Hui D., Weisgraber K. H. (1987b). Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B, E(LDL) receptors in the brain. J. Biol. Chem. 262, 14352–14360 [PubMed] [Google Scholar]
- Pooler A. M., Phillips E. C., Lau D. H., Noble W., Hanger D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394 10.1038/embor.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp J., Lewczuk P., Kolsch H., Meichsner S., Maier W., Kornhuber J., et al. (2012). Cholesterol metabolism is associated with soluble amyloid precursor protein production in Alzheimer's disease. J. Neurochem. 123, 310–316 10.1111/j.1471-4159.2012.07893.x [DOI] [PubMed] [Google Scholar]
- Posse De Chaves E. I., Vance D. E., Campenot R. B., Kiss R. S., Vance J. E. (2000). Uptake of lipoproteins for axonal growth of sympathetic neurons. J. Biol. Chem. 275, 19883–19890 10.1074/jbc.275.26.19883 [DOI] [PubMed] [Google Scholar]
- Prasanthi J. R., Huls A., Thomasson S., Thompson A., Schommer E., Ghribi O. (2009). Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on beta-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Mol. Neurodegener. 4:1 10.1186/1750-1326-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli L., Friedlich A. L., Setchell K. D., Nagano S., Opazo C., Cherny R. A., et al. (2005). Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J. Clin. Invest. 115, 2556–2563 10.1172/JCI23610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y., Tiffany-Castiglioni E., Welsh J., Harris E. D. (1998). Copper efflux from murine microvascular cells requires expression of the menkes disease Cu-ATPase. J. Nutr. 128, 1276–1282 [DOI] [PubMed] [Google Scholar]
- Rajan K. S., Colburn R. W., Davis J. M. (1976). Distribution of metal ions in the subcellular fractions of several rat brain areas. Life Sci. 18, 423–431 [DOI] [PubMed] [Google Scholar]
- Refolo L. M., Sambamurti K., Efthimiopoulos S., Pappolla M. A., Robakis N. K. (1995). Evidence that secretase cleavage of cell surface Alzheimer amyloid precursor occurs after normal endocytic internalization. J. Neurosci. Res. 40, 694–706 10.1002/jnr.490400515 [DOI] [PubMed] [Google Scholar]
- Reitz C., Tang M. X., Luchsinger J., Mayeux R. (2004). Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch. Neurol. 61, 705–714 10.1001/archneur.61.5.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C., Tang M. X., Manly J., Schupf N., Mayeux R., Luchsinger J. A. (2008). Plasma lipid levels in the elderly are not associated with the risk of mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 25, 232–237 10.1159/000115847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembach A., Doecke J. D., Roberts B. R., Watt A. D., Faux N. G., Volitakis I., et al. (2013). Longitudinal analysis of serum copper and ceruloplasmin in Alzheimer's disease. J. Alzheimers Dis. 34, 171–182 10.3233/JAD-121474 [DOI] [PubMed] [Google Scholar]
- Riddell D. R., Christie G., Hussain I., Dingwall C. (2001). Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 11, 1288–1293 10.1016/S0960-9822(01)00394-3 [DOI] [PubMed] [Google Scholar]
- Saggu H., Cooksey J., Dexter D., Wells F. R., Lees A., Jenner P., et al. (1989). A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J. Neurochem. 53, 692–697 [DOI] [PubMed] [Google Scholar]
- Saher G., Brugger B., Lappe-Siefke C., Mobius W., Tozawa R., Wehr M. C., et al. (2005). High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 8, 468–475 10.1038/nn1426 [DOI] [PubMed] [Google Scholar]
- Saito H., Dhanasekaran P., Baldwin F., Weisgraber K. H., Phillips M. C., Lund-Katz S. (2003). Effects of polymorphism on the lipid interaction of human apolipoprotein E. J. Biol. Chem. 278, 40723–40729 10.1074/jbc.M304814200 [DOI] [PubMed] [Google Scholar]
- Saito T., Okabe M., Hosokawa T., Kurasaki M., Hata A., Endo F., et al. (1999). Immunohistochemical determination of the Wilson Copper-transporting P- type ATPase in the brain tissues of the rat. Neurosci. Lett. 266, 13–16 10.1016/S0304-3940(99)00258-X [DOI] [PubMed] [Google Scholar]
- Sauer S. W., Merle U., Opp S., Haas D., Hoffmann G. F., Stremmel W., et al. (2011). Severe dysfunction of respiratory chain and cholesterol metabolism in Atp7b(-/-) mice as a model for Wilson disease. Biochim. Biophys. Acta 1812, 1607–1615 10.1016/j.bbadis.2011.08.011 [DOI] [PubMed] [Google Scholar]
- Sayre L. M., Perry G., Harris P. L., Liu Y., Schubert K. A., Smith M. A. (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J. Neurochem. 74, 270–279 10.1046/j.1471-4159.2000.0740270.x [DOI] [PubMed] [Google Scholar]
- Scheiber I. F., Mercer J. F., Dringen R. (2010). Copper accumulation by cultured astrocytes. Neurochem. Int. 56, 451–460 10.1016/j.neuint.2009.12.002 [DOI] [PubMed] [Google Scholar]
- Scheiber I. F., Schmidt M. M., Dringen R. (2012). Copper export from cultured astrocytes. Neurochem. Int. 60, 292–300 10.1016/j.neuint.2011.12.012 [DOI] [PubMed] [Google Scholar]
- Schiefermeier M., Kollegger H., Madl C., Polli C., Oder W., Kuhn H., et al. (2000). The impact of apolipoprotein E genotypes on age at onset of symptoms and phenotypic expression in Wilson's disease. Brain 123(Pt 3), 585–590 10.1093/brain/123.3.585 [DOI] [PubMed] [Google Scholar]
- Schlief M. L., Craig A. M., Gitlin J. D. (2005). NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 25, 239–246 10.1523/JNEUROSCI.3699-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlief M. L., West T., Craig A. M., Holtzman D. M., Gitlin J. D. (2006). Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 14919–14924 10.1073/pnas.0605390103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A., Rajendran L., Honsho M., Gralle M., Donnert G., Wouters F., et al. (2008). Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 28, 2874–2882 10.1523/JNEUROSCI.5345-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seessle J., Gohdes A., Gotthardt D. N., Pfeiffenberger J., Eckert N., Stremmel W., et al. (2011). Alterations of lipid metabolism in Wilson disease. Lipids Health Dis. 10:83 10.1186/1476-511X-10-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafaati M., Marutle A., Pettersson H., Lovgren-Sandblom A., Olin M., Pikuleva I., et al. (2011). Marked accumulation of 27-hydroxycholesterol in the brains of Alzheimer's patients with the Swedish APP 670/671 mutation. J. Lipid Res. 52, 1004–1010 10.1194/jlr.M014548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepardson N. E., Shankar G. M., Selkoe D. J. (2011a). Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch. Neurol. 68, 1239–1244 10.1001/archneurol.2011.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepardson N. E., Shankar G. M., Selkoe D. J. (2011b). Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch. Neurol. 68, 1385–1392 10.1001/archneurol.2011.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K., Ikonen E. (1997). Functional rafts in cell membranes. Nature 387, 569–572 10.1038/42408 [DOI] [PubMed] [Google Scholar]
- Simons M., Keller P., De Strooper B., Beyreuther K., Dotti C. G., Simons K. (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snipes G. J., Suter U. (1997). Cholesterol and myelin. Subcell. Biochem. 28, 173–204 [DOI] [PubMed] [Google Scholar]
- Solomon A., Kareholt I., Ngandu T., Winblad B., Nissinen A., Tuomilehto J., et al. (2007). Serum cholesterol changes after midlife and late-life cognition: twenty-one-year follow-up study. Neurology 68, 751–756 10.1212/01.wnl.0000256368.57375.b7 [DOI] [PubMed] [Google Scholar]
- Song I. S., Chen H. H., Aiba I., Hossain A., Liang Z. D., Klomp L. W., et al. (2008). Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol. Pharmacol. 74, 705–713 10.1124/mol.108.046771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soragni A., Zambelli B., Mukrasch M. D., Biernat J., Jeganathan S., Griesinger C., et al. (2008). Structural characterization of binding of Cu(II) to tau protein. Biochemistry 47, 10841–10851 10.1021/bi8008856 [DOI] [PubMed] [Google Scholar]
- Sparks D. L. (2004). Cholesterol, copper, and accumulation of thioflavine S-reactive Alzheimer's-like amyloid beta in rabbit brain. J. Mol. Neurosci. 24, 97–104 [DOI] [PubMed] [Google Scholar]
- Sparks D. L. (2007). Cholesterol metabolism and brain amyloidosis: evidence for a role of copper in the clearance of Abeta through the liver. Curr. Alzheimer Res. 4, 165–169 10.2174/156720507780362119 [DOI] [PubMed] [Google Scholar]
- Sparks D. L. (2008). The early and ongoing experience with the cholesterol-fed rabbit as a model of Alzheimer's disease: the old, the new and the pilot. J. Alzheimers Dis. 15, 641–656 [DOI] [PubMed] [Google Scholar]
- Sparks D. L., Friedland R., Petanceska S., Schreurs B. G., Shi J., Perry G., et al. (2006). Trace copper levels in the drinking water, but not zinc or aluminum influence CNS Alzheimer-like pathology. J. Nutr. Health Aging 10, 247–254 [PMC free article] [PubMed] [Google Scholar]
- Sparks D. L., Petanceska S., Sabbagh M., Connor D., Soares H., Adler C., et al. (2005). Cholesterol, copper and Abeta in controls, MCI, AD and the AD cholesterol-lowering treatment trial (ADCLT). Curr. Alzheimer Res. 2, 527–539 10.2174/156720505774932296 [DOI] [PubMed] [Google Scholar]
- Sparks D. L., Schreurs B. G. (2003). Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 11065–11069 10.1073/pnas.1832769100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks D. L., Ziolkowski C., Lawmaster T., Martin T. (2011). Influence of water quality on cholesterol-induced tau pathology: preliminary data. Int. J. Alzheimers Dis. 2011:987023 10.4061/2011/987023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones T. L., Stoothoff W. H., de Calignon A., Jones P. B., Hyman B. T. (2009). Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 32, 150–159 10.1016/j.tins.2008.11.007 [DOI] [PubMed] [Google Scholar]
- Squitti R., Barbati G., Rossi L., Ventriglia M., Dal Forno G., Cesaretti S., et al. (2006). Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF [beta]-amyloid, and h-tau. Neurology 67, 76–82 10.1212/01.wnl.0000223343.82809.cf [DOI] [PubMed] [Google Scholar]
- Squitti R., Lupoi D., Pasqualetti P., Dal Forno G., Vernieri F., Chiovenda P., et al. (2002). Elevation of serum copper levels in Alzheimer's disease. Neurology 59, 1153–1161 [DOI] [PubMed] [Google Scholar]
- Squitti R., Pasqualetti P., Cassetta E., Dal Forno G., Cesaretti S., Pedace F., et al. (2003). Elevation of serum copper levels discriminates Alzheimer's disease from vascular dementia. Neurology 60, 2013–2014 [DOI] [PubMed] [Google Scholar]
- Squitti R., Pasqualetti P., Dal Forno G., Moffa F., Cassetta E., Lupoi D., et al. (2005). Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology 64, 1040–1046 10.1212/01.WNL.0000154531.79362.23 [DOI] [PubMed] [Google Scholar]
- Squitti R., Ventriglia M., Barbati G., Cassetta E., Ferreri F., Dal Forno G., et al. (2007). ‘Free’ copper in serum of Alzheimer's disease patients correlates with markers of liver function. J. Neural. Transm. 114, 1589–1594 10.1007/s00702-007-0777-6 [DOI] [PubMed] [Google Scholar]
- Stamer K., Vogel R., Thies E., Mandelkow E., Mandelkow E. M. (2002). Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063 10.1083/jcb.200108057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottmann R. W., Turbe-Doan A., Tran P., Kratz L. E., Moran J. L., Kelley R. I., et al. (2011). Cholesterol metabolism is required for intracellular hedgehog signal transduction in vivo. PLoS Genet. 7:e1002224 10.1371/journal.pgen.1002224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson P. A., Englund M. C., Markstrom E., Ohlsson B. G., Jernas M., Billig H., et al. (2003). Copper induces the expression of cholesterogenic genes in human macrophages. Atherosclerosis 169, 71–76 10.1016/S0021-9150(03)00145-X [DOI] [PubMed] [Google Scholar]
- Tang Z., Gasperkova D., Xu J., Baillie R., Lee J. H., Clarke S. D. (2000). Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J. Nutr. 130, 2915–2921 [DOI] [PubMed] [Google Scholar]
- Thies E., Mandelkow E. M. (2007). Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 27, 2896–2907 10.1523/JNEUROSCI.4674-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G., Koo E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 10.1074/jbc.R800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickler A. K., Smith D. G., Ciccotosto G. D., Tew D. J., Curtain C. C., Carrington D., et al. (2005). Methylation of the imidazole side chains of the Alzheimer disease amyloid-beta peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. J. Biol. Chem. 280, 13355–13363 10.1074/jbc.M414178200 [DOI] [PubMed] [Google Scholar]
- Tietz N. W. (1987). Fundamentals of Clinical Chemistry, 3rd Edn, Philadelphia: Saunders [Google Scholar]
- Treiber-Held S., Distl R., Meske V., Albert F., Ohm T. G. (2003). Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. J. Pathol. 200, 95–103 10.1002/path.1345 [DOI] [PubMed] [Google Scholar]
- Tsivkovskii R., Eisses J. F., Kaplan J. H., Lutsenko S. (2002). Functional properties of the copper-transporting ATPase ATP7B (the Wilson's disease protein) expressed in insect cells. J. Biol. Chem. 277, 976–983 10.1074/jbc.M109368200 [DOI] [PubMed] [Google Scholar]
- Uchida Y., Gomi F., Masumizu T., Miura Y. (2002). Growth inhibitory factor prevents neurite extension and the death of cortical neurons caused by high oxygen exposure through hydroxyl radical scavenging. J. Biol. Chem. 277, 32353–32359 10.1074/jbc.M111263200 [DOI] [PubMed] [Google Scholar]
- Urano Y., Hayashi I., Isoo N., Reid P. C., Shibasaki Y., Noguchi N., et al. (2005). Association of active gamma-secretase complex with lipid rafts. J. Lipid Res. 46, 904–912 10.1194/jlr.M400333-JLR200 [DOI] [PubMed] [Google Scholar]
- Utermann G., Weisgraber K. H., Weber W., Mahley R. W. (1984). Genetic polymorphism of apolipoprotein E: a variant form of apolipoprotein E2 distinguished by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. J. Lipid Res. 25, 378–382 [PubMed] [Google Scholar]
- Vanier M. T. (2010). Niemann-Pick disease type C. Orphanet. J. Rare Dis. 5:16 10.1186/1750-1172-5-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., et al. (1999). Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 10.1126/science.286.5440.735 [DOI] [PubMed] [Google Scholar]
- Vetrivel K. S., Cheng H., Kim S. H., Chen Y., Barnes N. Y., Parent A. T., et al. (2005). Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 280, 25892–25900 10.1074/jbc.M503570200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel K. S., Cheng H., Lin W., Sakurai T., Li T., Nukina N., et al. (2004). Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954 10.1074/jbc.M407986200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel K. S., Thinakaran G. (2010). Membrane rafts in Alzheimer's disease beta-amyloid production. Biochim. Biophys. Acta 1801, 860–867 10.1016/j.bbalip.2010.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J. M., Tsivkovskii R., Lutsenko S. (2002). Metallochaperone Atox1 transfers copper to the NH2-terminal domain of the Wilson's disease protein and regulates its catalytic activity. J. Biol. Chem. 277, 27953–27959 10.1074/jbc.M203845200 [DOI] [PubMed] [Google Scholar]
- Wang M. L., Motamed M., Infante R. E., Abi-Mosleh L., Kwon H. J., Brown M. S., et al. (2010). Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 12, 166–173 10.1016/j.cmet.2010.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. P., Wang X. H., Bao Y. C., Zhou J. N. (2003). Apolipoprotein E genotypes in Chinese patients with Wilson's disease. QJM 96, 541–542 10.1093/qjmed/hcg093 [DOI] [PubMed] [Google Scholar]
- Weisgraber K. H., Innerarity T. L., Mahley R. W. (1982). Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J. Biol. Chem. 257, 2518–2521 [PubMed] [Google Scholar]
- Weiss K. C., Linder M. C. (1985). Copper transport in rats involving a new plasma protein. Am. J. Physiol. 249, E77–E88 [DOI] [PubMed] [Google Scholar]
- White A. R., Bush A. I., Beyreuther K., Masters C. L., Cappai R. (1999a). Exacerbation of copper toxicity in primary neuronal cultures depleted of cellular glutathione. J. Neurochem. 72, 2092–2098 10.1046/j.1471-4159.1999.0722092.x [DOI] [PubMed] [Google Scholar]
- White A. R., Multhaup G., Maher F., Bellingham S., Camakaris J., Zheng H., et al. (1999b). The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 19, 9170–9179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. R., Reyes R., Mercer J. F., Camakaris J., Zheng H., Bush A. I., et al. (1999c). Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 842, 439–444 10.1016/S0006-8993(99)01861-2 [DOI] [PubMed] [Google Scholar]
- White A. R., Cappai R. (2003). Neurotoxicity from glutathione depletion is dependent on extracellular trace copper. J. Neurosci. Res. 71, 889–897 10.1002/jnr.10537 [DOI] [PubMed] [Google Scholar]
- White A. R., Du T., Laughton K. M., Volitakis I., Sharples R. A., Xilinas M. E., et al. (2006). Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J. Biol. Chem. 281, 17670–17680 10.1074/jbc.M602487200 [DOI] [PubMed] [Google Scholar]
- Whitney K. D., Watson M. A., Collins J. L., Benson W. G., Stone T. M., Numerick M. J., et al. (2002). Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol. Endocrinol. 16, 1378–1385 10.1210/me.16.6.1378 [DOI] [PubMed] [Google Scholar]
- Wilmarth P. A., Short K. K., Fiehn O., Lutsenko S., David L. L., Burkhead J. L. (2012). A systems approach implicates nuclear receptor targeting in the Atp7b(-/-) mouse model of Wilson's disease. Metallomics 4, 660–668 10.1039/c2mt20017a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth P. L., Linder M. C. (1985). Distribution of copper among components of human serum. J. Natl. Cancer Inst. 75, 277–284 [PubMed] [Google Scholar]
- Won J. S., Im Y. B., Khan M., Contreras M., Singh A. K., Singh I. (2008). Lovastatin inhibits amyloid precursor protein (APP) beta-cleavage through reduction of APP distribution in Lubrol WX extractable low density lipid rafts. J. Neurochem. 105, 1536–1549 10.1111/j.1471-4159.2008.05283.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff-Pak D. S., Agelan A., Del Valle L. (2007). A rabbit model of Alzheimer's disease: valid at neuropathological, cognitive, and therapeutic levels. J. Alzheimers Dis. 11, 371–383 [DOI] [PubMed] [Google Scholar]
- Xie C., Lund E. G., Turley S. D., Russell D. W., Dietschy J. M. (2003). Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J. Lipid Res. 44, 1780–1789 10.1194/jlr.M300164-JLR200 [DOI] [PubMed] [Google Scholar]
- Xu P. T., Gilbert J. R., Qiu H. L., Ervin J., Rothrock-Christian T. R., Hulette C., et al. (1999). Specific regional transcription of apolipoprotein E in human brain neurons. Am. J. Pathol. 154, 601–611 10.1016/S0002-9440(10)65305-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto N., Tasaki M., Shimanouchi T., Umakoshi H., Kuboi R. (2005). Oxidation of cholesterol catalyzed by amyloid beta-peptide (Abeta)-Cu complex on lipid membrane. J. Biosci. Bioeng. 100, 455–459 10.1263/jbb.100.455 [DOI] [PubMed] [Google Scholar]
- Yount N. Y., McNamara D. J., Al-Othman A. A., Lei K. Y. (1990). The effect of copper deficiency on rat hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. J. Nutr. Biochem. 1, 21–27 10.1016/0955-2863(90)90094-2 [DOI] [PubMed] [Google Scholar]
- Zambon D., Quintana M., Mata P., Alonso R., Benavent J., Cruz-Sanchez F., et al. (2010). Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am. J. Med. 123, 267–274 10.1016/j.amjmed.2009.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Appelkvist E. L., Kristensson K., Dallner G. (1996). The lipid compositions of different regions of rat brain during development and aging. Neurobiol. Aging 17, 869–875 [DOI] [PubMed] [Google Scholar]