

Abstract
Objective:
There is increasing evidence that common genetic risk factors underlie frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). Recently, mutations in the sequestosome 1 (SQSTM1) gene, which encodes p62 protein, have been reported in patients with ALS. P62 is a multifunctional adapter protein mainly involved in selective autophagy, oxidative stress response, and cell signaling pathways. The purpose of our study was to evaluate the frequency of SQSTM1 mutations in a dataset of unrelated patients with FTLD or ALS, in comparison with healthy controls and patients with Paget disease of bone (PDB).
Methods:
Promoter region and all exons of SQSTM1 were sequenced in a large group of subjects, including patients with FTLD or ALS, healthy controls, and patients with PDB. The clinical characteristics of patients with FTLD or ALS with gene mutations were examined.
Results:
We identified 6 missense mutations in the coding region of SQSTM1 in patients with either FTLD or ALS, none of which were found in healthy controls or patients with PDB. In silico analysis suggested a pathogenetic role for these mutations. Furthermore, 7 novel noncoding SQSTM1 variants were found in patients with FTLD and patients with ALS, including 4 variations in the promoter region.
Conclusions:
SQSTM1 mutations are present in patients with FTLD and patients with ALS. Additional studies are warranted in order to better investigate the role of p62 in the pathogenesis of both FTLD and ALS.
In recent years, there has been a growing body of clinical, pathologic, and genetic evidence supporting the idea that frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) belong to the same clinicopathologic spectrum of disease.1–3
FTLD and ALS are genetically heterogeneous disorders. Mutations in the CHMP2B, FUS, OPTN, PGRN, TARDBP, UBQLN2, and VCP genes and a repeat expansion in the C9orf72 gene have been reported to be associated with both diseases.4–11 Therefore, genes linked to both diseases may converge into a common pathogenetic pathway, explaining the overlap of clinical symptoms.
The sequestosome 1 (SQSTM1) gene is located on 5q35 and encodes p62, a multifunctional protein implicated in several cellular activities. There is accumulating evidence of p62 involvement in neurodegeneration. SQSTM1 knockout mice develop memory impairment associated with the accumulation of hyperphosphorylated τ and neurofibrillary tangles.12 Pathologic studies in humans have shown increased p62 immunoreactivity in several neurodegenerative disorders, such as Alzheimer disease (AD), dementia with Lewy bodies, FTLD, Parkinson disease (PD), and Huntington disease (HD).13–15 Intriguingly, pathologic studies showed that patients with FTLD or ALS carrying the C9orf72 gene expansion present abundant neuronal p62-positive inclusions.16,17
Mutations in the SQSTM1 gene result in Paget disease of bone (PDB), a common disorder characterized by increased bone turnover.18,19 Recently, SQSTM1 mutations have been identified in patients with ALS, suggesting a role for this gene in the pathogenesis of the disease.20
The aims of this study were 1) to confirm the increased frequency of SQSTM1 mutations in an Italian dataset of patients with ALS and 2) to evaluate the frequency of SQSTM1 mutations in Italian patients with FTLD.
METHODS
Participants.
A total of 170 consecutive unrelated patients with FTLD (90 men, 80 women; mean age ± SD = 68.7 ± 9.4 years) attending the Memory Clinics of the Department of Neuroscience of the Universities of Torino and Milano (Italy) were involved in the study. The diagnosis of FTLD was made according to the criteria of Neary et al.21; 138 patients fulfilled the diagnostic criteria for behavioral variant frontotemporal dementia, 6 for semantic dementia, and 19 for progressive nonfluent aphasia. During the follow-up, 7 patients with an initial diagnosis of FTLD developed motor neuron disease. Positive family history, defined as at least 1 first-degree relative having dementia, was recorded for 42 patients (37.5%). A group of 124 patients with sporadic ALS (70 men, 54 women; mean age ± SD = 62.3 ± 9.8 years), diagnosed according to the revised El Escorial criteria,22 were collected at the ALS Centre of the University of Torino. Patients with FTLD and patients with ALS, at recruitment, showed no sign or symptom of altered bone metabolism. A group of 145 healthy subjects (78 men, 67 women; mean age ± SD = 65.7 ± 7.9 years) was used as a control. Finally, in order to estimate the frequency of SQSTM1 mutations in PDB, 288 patients were recruited at the Unit of Geriatrics and Metabolic Bone Diseases, of the University of Torino (152 men, 136 women; mean age ± SD = 68.6 ± 12.8 years). At recruitment, no patient with PDB had a diagnosis of ALS or FTLD. Patients and controls were of Caucasian origin and came from the same area of Northern Italy.
Ethics.
Written informed consent was obtained from all participants, and the study was approved by the hospital ethics committees.
Genetics and sequencing analysis of the SQSTM1 gene.
Genomic DNA was isolated from peripheral blood leukocytes with the Gene Eluate Blood Genomic DNA Kit (Sigma-Aldrich, St. Louis, MO), according to the manufacturer's protocols. The SQSTM1 gene spans a 16-kb genomic segment encoding a 2,870-bp transcript. We analyzed the SQSTM1 gene by direct genomic sequencing of all 8 coding exons and 6 overlapping amplicons of the promoter region. Intronic primers covering the coding sequences were designed with at least 50 base pairs of intronic sequence 3′ and 5′ of each exon. Sequencing was done on an ABI Prism 3130 DNA sequencer with use of the BigDye 03 Terminator Sequencing Standard Kit (Applied Biosystems, Foster City, CA) and specific sequencing primers. Primers were generated with Primer3 software v0.04.0. PCR reactions were performed in a final volume of 50 μL, with use of 90 ng of genomic DNA, 0.4 unit of Taq Gold DNA polymerase (Applied Biosystems), 250 nM of each primer, 1.5 mM MgCl2, and 50 mM dNTPs. PCR conditions were as follows: an initial denaturation at 95°C for 10 minutes, followed by 35 cycles at 95°C for 1 minute, specific temperatures for each couple of primers for 40 seconds, 72°C for 1 minute, and a final elongation at 72°C for 5 minutes. The PCR products were purified for sequencing after electrophoresis on an agarose gel with a QIAquick PCR purification kit (Qiagen, Hilden, Germany). The forward primer was used for mutation screening, and all variations were confirmed by reverse sequencing. All exonic mutations were verified with use of restriction enzymes. Sequences were analyzed with Mutation Explorer v2.61 (SoftGenetics LLC, www.softgenetics.com). When a variant was identified, it was checked for the record in the dbSNP Short Genetic Variations, Exome Variant Server, and 1000 Genome Project. Patients with FTLD with mutations in the SQSTM1 gene were also screened for MAPT, PGRN, and TARDBP genes, according to previously described protocols.23,24 Furthermore, all patients with ALS were sequenced for SOD1, TARDBP, FUS, and OPTN.25 Finally, the presence of a pathologic expansion in the C9orf72 gene was excluded in all FTLD and ALS carriers of an SQSTM1 mutation, as previously described.9
Software analysis.
A multiple protein alignment was constructed with multiple alignment at the HomoloGene site (available at: http://www.ncbi.nlm.nih.gov/homologene/). The PolyPhen 2 program (http://genetics.bwh.harvard.edu/pph2/index.shtml) and SIFT program (http://sift.bii.a-star.edu.sg/) were used to predict effects on protein structure or function.
RESULTS
The complete analysis of the SQSTM1 gene was conducted on a total of 722 subjects. Several rare SQSTM1 variants were identified in the isoform 1 (NM_003900.4). These variants are summarized in table 1. Overall, 7 missense mutations (K238E, V259L, E274D, E319K, K344E, P348L, P438L) were identified in patients with FTLD or ALS. The E274D substitution is known (rs55793208) and was observed in both cases and controls (frequency: 2.9% in patients with FTLD, 8.9% in patients with ALS, and 2.8% in controls; χ2 = 0.00, p = 0.92, FTLD vs controls; χ2 = 4.74, p = 0.03, ALS vs controls). The K238E substitution (rs11548633) was observed in 1 patient with ALS and has not been reported previously to be associated with any disease. One patient with FTLD presented the known substitution E319K (rs61748794), without reported association to any disorder. The remaining 4 missense mutations—V259L, K344E, P348L, P438L—are novel. The V259L substitution in exon 6 and the K344E substitution in exon 7 were present in 2 patients with FTLD. The P348L substitution in exon 7 and the P438L substitution in exon 8 were identified in 2 patients with ALS. DNA analysis of the 145 healthy controls and 288 patients with PDB failed to detect any of the 4 novel variants.
Table 1.
SQSTM1 rare genetic variants in patients with FTLD, patients with ALS, and controlsa
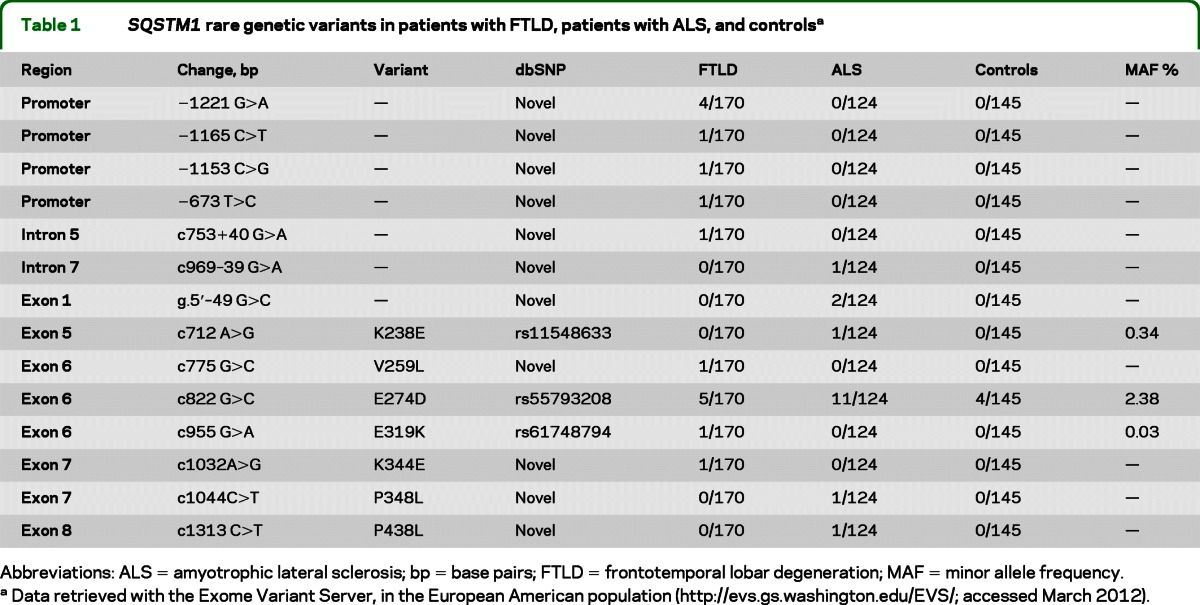
Abbreviations: ALS = amyotrophic lateral sclerosis; bp = base pairs; FTLD = frontotemporal lobar degeneration; MAF = minor allele frequency.
Data retrieved with the Exome Variant Server, in the European American population (http://evs.gs.washington.edu/EVS/; accessed March 2012).
The SQSTM1 gene codes a 440–amino acid protein (p62) with several different domains, including PB1, ZZ, TRAF6, PEST, and UBA, enabling the protein to act as a scaffold for the regulation of ubiquination.26 K238E and V259L are located in or nearby a tumor necrosis factor receptor–associated factor 6 (TRAF6) binding site, E319K does not affect any known domain, K344E is in the region that interacts with LC3, P348L is in the PEST domain, and P438L is in the C-terminal region.
In addition to the aforementioned mutations, the analysis of noncoding regions revealed 3 novel variations. In the 5′UTR region we detected g5′ − 49 G>C in 1 patient with ALS, whereas in intron 2 we detected c753 + 40 G>A variant in 1 patient with FTLD and c969 − 39 G>A variant in 1 patient with ALS. The entire region of the promoter (around 1,700 bp) was sequenced, and novel variants (−1165 C>T, −1153 C>G, −673 T>C) were identified. Furthermore, a polymorphism in the region of the transcription factor–binding protein C-ets-1 (ETS-1) was found at −1221 G>A.
Neither patients with FTLD nor patients with ALS carrying SQSTM1 missense mutations showed mutations in known ALS or FTLD genes. As expected, 17.7% of patients with PDB showed several mutations in the UBA domain (P387L, Y383X, P392L, E396X, M404V, D423X, and G425R) of SQSTM1, which is in agreement with previous reports.18,19
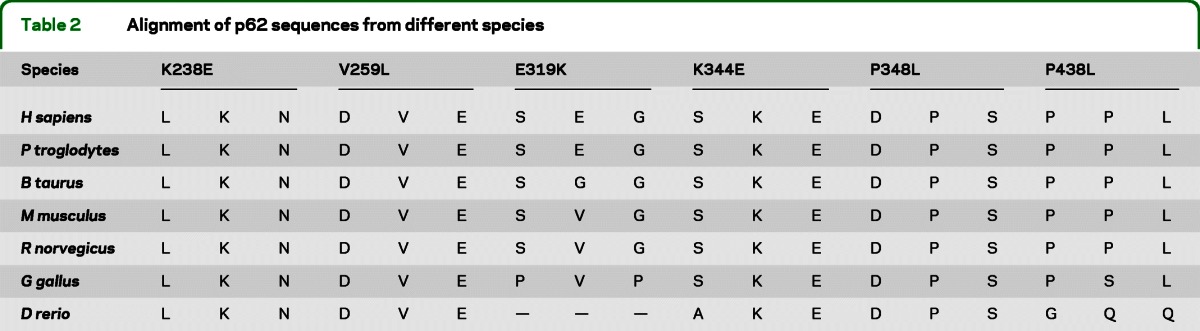
Analyses of the potential functional significance of the SQSTM1 mutations that were detected in either patients with ALS or patients with FTLD showed that 4 of the 6 mutated residues are highly conserved in evolution (K238, K344, V259, and P348), whereas E319 and P348 are only semiconserved residues (table 2). Five of the mutations were predicted to have a damaging role, by at least 1 of the 2 programs. Only E319K was predicted to be benign.
Table 2.
Alignment of p62 sequences from different species
Clinical characteristics of patients.
Table 3 shows the demographic and clinical characteristics of the patients carrying the SQSTM1 gene missense mutations. Three patients had an initial diagnosis of FTLD and 3 of ALS. All patients with FTLD carrying SQSTM1 mutation (E319K, V259L, and K344E) presented the behavioral variant of the disease, showing aggressiveness, changes of mood, and social detachment. MRI examinations showed asymmetric frontotemporal atrophy. In 1 patient, CSF phospho-τ concentration was increased, whereas total-τ and β-amyloid were normal.
Table 3.
Demographic and clinical features of patients
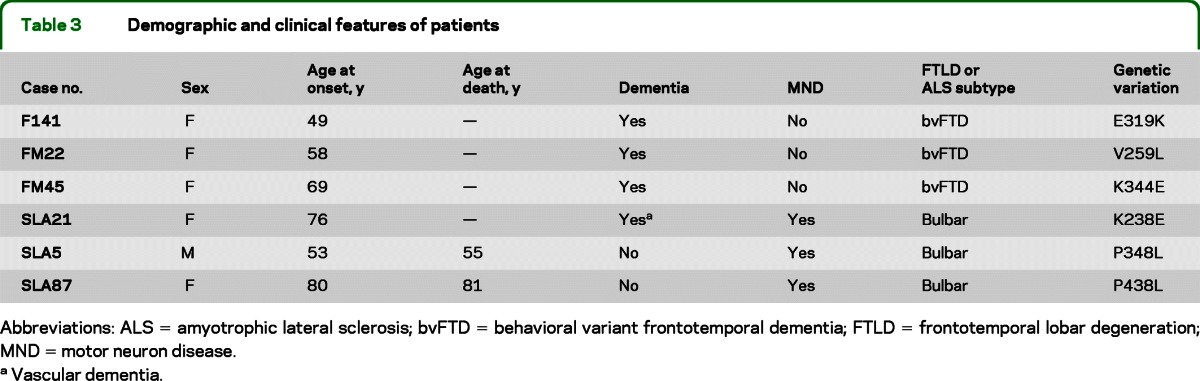
Abbreviations: ALS = amyotrophic lateral sclerosis; bvFTD = behavioral variant frontotemporal dementia; FTLD = frontotemporal lobar degeneration; MND = motor neuron disease.
Vascular dementia.
Patients with ALS with SQSTM1 mutation showed remarkable variation in age at onset. The K238E mutation was identified in a 78-year-old patient who presented with bulbar dysfunction and comorbid vascular dementia. The P348L mutation was identified in a 53-year-old patient who had a rapidly worsening clinical condition and died at age 55 years. The patient with P438L variant died at the age of 81 years, of respiratory insufficiency after an 8-month course of progressive motor neuron disease with bulbar onset.
DISCUSSION
Our study confirms the presence of SQSTM1 mutations in patients with ALS. In addition, we detected SQSTM1 mutations in patients with FTLD. As for previously reported genes, such as TARDBP and FUS, the frequency of SQSTM1 gene mutations in either FTLD or ALS is low, around 3% for our dataset. None of these mutations was present in our patients with PDB, and they have not previously been reported to occur in such patients. This is the first report describing the presence of SQSTM1 mutations in patients with FTLD, and additional studies are warranted in order to support a role for this gene in the pathogenesis of the disease.
The neurobiological bases linking SQSTM1 with neurodegenerative diseases like FTLD and ALS are unclear. P62 is a multifunctional protein containing several protein–protein interaction domains that enable the protein to exert complex physiologic actions. Furthermore, p62 forms highly stable dimers that interfere with its ability to bind ubiquitin.27 Several of the genetic variants highlighted in our study may significantly alter the protein–protein interactions or the UBA-related dimerization process, thereby promoting protein aggregation and neurodegeneration. However, the biological significance of the detected variations requires assessment in future functional studies.
In one of our patients with ALS, we found the K238E mutation in exon 5 of the SQSTM1 gene. In a recent study, a deletion at the same codon was found in 2 North American patients with ALS.20 This substitution occurs in a TRAF6 binding site, where p62 interacts with TRAF6, a critical component of the NF-κB pathway involved in regulating many aspects of cellular activity, especially in response to proinflammatory cytokines.28 Impairment of these functions may be of relevance for both FTLD and ALS pathogenesis. One of the missense mutations found in our patients with FTLD is located at codon 344 (K>E), and this could therefore interfere with the binding to LC3. P62 directly interacts with LC3 to facilitate the degradation of aggregated proteins. The surface of LC3 has a narrow channel, and p62 binds within the latter, assuming an elongated shape.29 Furthermore, mutations in p62 cause a reduced ability to bind to LC3. In experimental animals, the expression of mutants with low affinity for LC3 results in the formation of inclusions positive for p62 and ubiquitinated proteins; in particular, the interaction of LC3 with p62 was found to be severely reduced in the p62 LRS mutant 1 (L343A) and was virtually abolished in the p62 LRS mutant 2 (D337/D338/D339A).30
One of our patients with ALS has a mutation in the PEST domain (P348L) that is predicted to be damaging. There are 2 PEST sequences in p62 (regions from amino acids 266 to 294 and 345 to 377).31 The PEST domain is rich in proline, glutamate, serine, and threonine; it has been found in many short-lived proteins and acts as a signal peptide for rapid protein degradation. Finally, the P438L is located in the C-terminal tail of the p62 UBA domain. In transgenic mice lacking these residues, p62 UBA is unable to form dimers, and this may play a role in regulating the lifetime of p62 in cells32; therefore, C-terminal amino acid residues may be important for SQSTM1 functions. Mutations in the UBA domain of SQSTM1 are a common cause of PDB. Our patients with PDB show a mutation frequency of 17.7% in the UBA domain, although mutations detected in either patients with ALS or patients with FTLD were not found in our PDB cohort and were located outside of the UBA domain.
We also identified several genetic variants in the promoter region of the gene, exclusively in patients with FTLD. A number of potential binding sites for known transcription factors are present in the p62 promoter region, revealing multiple regulatory features of the p62 promoter for responding to different signals. The expression of p62 is regulated at the transcriptional level: the promoter of the gene is enriched in CpG and can be altered by oxidative stress, causing a reduction in transcription levels of the protein. A recent study showed the presence, in several neurodegenerative processes such as AD, FTLD, HD, and PD, of an oxidative process in the promoter of p62, which results in reduced expression of the protein.33 This oxidative process has been reported to be associated mainly with FTLD. Of the observed variants, the −1221 G>A mutation is of particular interest, being localized in the binding site for the transcription factor ETS-1, which is part of a family of transcription factors that share a highly conserved DNA domain. All ETS factors bind to a nucleotide sequence of the type “GGAA/T,” so alterations in the amino acid sequence of the transcription factor can lead to changes in binding specificity. The pathogenic role of these variants in the promoter region needs to be further investigated.
A large number of experimental and clinical studies provided evidence that p62 plays a major role in autophagy, an evolutionarily conserved pathway for the degradation of long-lived proteins and organelles. Autophagy dysfunction may contribute to the pathology of various neurodegenerative disorders, which manifest with abnormal protein accumulation. The autophagy pathway comprises 4 steps: initiation/nucleation, autophagosome formation, trafficking/maturation, and recycling/release. Distinct proteins act concertedly at each step to execute successful autophagic recycling. P62 helps target polyubiquitinated proteins and aggregates to the autophagy machinery, facilitated by its ability to bind LC3 proteins that are necessary for autophagosome formation.29 A recent study showed that aggregation of TDP-43, the main protein found in neurons of both patients with FTLD and patients with ALS, is significantly influenced by p62: overexpression of p62 reduces TDP-43 aggregation in an autophagy- and proteasome-dependent manner.34 Defective autophagy has been implicated in the accumulation of ubiquitinated TDP-43 inclusions in ALS, and in ALS motor neuron degeneration due to mutations in endosomal sorting complexes required for transport subunit III (ESCRTIII) and charged multivesicular body protein 2B (CHMP2B).35 It is of interest to note that 2 other genes, VCP and the recently discovered UBQLN2, mutated in families with FTLD or ALS, are involved in different steps of the autophagic process.8,11
It is well known that mutations in the same gene may be responsible for different diseases. Intriguingly, mutations in VCP encoding the multifunctional valosin-containing protein cause hereditary inclusion body myopathy associated with PDB and frontotemporal dementia, and it has been proposed that, as for SQSTM1, VCP mutations cause PDB by compromising ubiquitin binding and targeting similar cellular pathways.36 Subsequently, mutations in the VCP gene were also found in patients with ALS.8 In our study, we found that mutations in the SQSTM1 gene may be associated with FTLD, ALS, and PDB, thus supporting the idea that common molecular mechanisms may be involved in the pathogenesis of these diseases. In addition, our results suggest that patients presenting with signs and symptoms of either FTLD or ALS should be monitored for altered bone metabolism, whereas patients with PDB must be carefully evaluated for signs of dementia and motor neuron disease.
Finally, a new role for p62 in maintaining mitochondrial integrity has recently been described. A portion of p62 directly localizes within the mitochondria and supports stable electron transport by forming heterogeneous protein complexes. P62 interacts with several oxidation-prone proteins, including a few components of the electron transport chain complexes, as well as multiple chaperone molecules and redox regulatory enzymes. Accordingly, p62-deficient mitochondria exhibited compromised electron transport.37 Mutations in the Parkin gene are frequent causes of recessive PD.38 Parkin is an E3 ubiquitin ligase that recruits p62 to mitochondria, mediating the aggregation of dysfunctional mitochondria through polymerization via its PB1 domain.39 Intriguingly, a recent review highlighted the role of p62 in several neurodegenerative diseases other than PD but also in cancer, obesity, and insulin resistance, suggesting that p62 could be critical for several pathophysiologic pathways.40
We reported on extensive genetic screening of patients with FTLD or ALS, showing different previously unknown genetic variants that may be involved in the pathogenetic mechanisms of neurodegeneration. Our study enlarged the clinical spectrum of the neurodegenerative phenotype associated with SQSTM1 mutations, confirming the association with ALS and supporting the role of this protein also in FTLD pathogenesis. Whether SQSTM1 is a major gene or a modifier gene for both FTLD and ALS is not well defined. Additional clinical and experimental studies are needed in order to better elucidate the role of this gene in FTLD and ALS and to evaluate possible therapeutic targets.
Supplementary Material
Glossary
GLOSSARY
- AD
Alzheimer disease
- ALS
amyotrophic lateral sclerosis
- FTLD
frontotemporal lobar degeneration
- HD
Huntington disease
- PD
Parkinson disease
- PDB
Paget disease of bone
- SQSTM1
sequestosome 1 gene
Footnotes
Editorial, page 1526
Supplemental data at www.neurology.org
Preliminary data of the present study were presented at the annual congress of the Italian Society of Neurology (SIN) in 2010 and at the annual congress of the Italian Society for the Study of Dementias (SINDEM) in 2011.
AUTHOR CONTRIBUTIONS
Dr. Rubino conceived and supervised the project and drafted the manuscript with Dr. Rainero. Dr. Galimberti, Dr. Bruni, Dr. Scarpini, and the TODEM Group members were responsible for FTLD patient collection. Dr. Chiò and Dr. Calvo were responsible for ALS patient characterization and sample collection. Dr. Isaia was responsible for PDB patient collection. Dr. Fenoglio performed laboratory and statistical analyses. Dr. Gallone, Dr. Rogaeva, Dr.Grinberg, and Dr. St. George-Hyslop assisted in experimental design and execution as well as in data interpretation. Dr. Gentile and Dr. Pinessi edited the manuscript for intellectual content. All authors critically reviewed and approved the final manuscript.
DISCLOSURE
E. Rubino reports no disclosures. I. Rainero received speaker honoraria from Abbott, Actelion, and Novartis and has received research support from Ministero dell'Istruzione, dell'Università e della Ricerca Scientifica (MIUR) of Italy, and Regione Piemonte (Italy). A. Chiò received speaker honoraria from Biogen Idec and has received research support from Italian Ministry of Health (Ricerca Finalizzata and CCM grants), Regione Piemonte (Ricerca Finalizzata), Italian Ministry of University and Research, University of Torino, Fondazione Vialli e Mauro for ALS Research, ONLUS, Compagnia San Paolo, Federazione Italiana Giuoco Calcio, and European Commission (Health Seventh Framework Programme) under grant agreement 259867. E. Rogaeva has received research support from W. Garfield Weston Foundation, the Canadian Institutes of Health Research, and Ontario Research Fund. D. Galimberti has received research support from the Fondazione Mondino. P. Fenoglio, Y. Grinberg, and G. Isaia report no disclosures. A. Calvo has received research support from Regione Piemonte and Compagnia di San Paolo. S. Gentile reports no disclosures. A.C. Bruni received speaker honoraria from Pfizer, Novartis, and Lundbeck and has received research support from Assessorato alla Salute–Regione Calabria, Italian Ministry of Health (Ricerca Finalizzata and CCM grants), and Associazione per la Ricerca Neurogenetica Onlus. P.H. St. George-Hyslop has received research support from the Canadian Institutes of Health Research, Ontario Research Fund, the Howard Hughes Medical Institute, The Wellcome Trust, the Alzheimer Society of Ontario, the Canada Foundation for Innovation, the Ontario Mental Health Foundation, Genome Canada, and the Alzheimer Society of Canada. E. Scarpini and S. Gallone report no disclosures. L. Pinessi received speaker honoraria from Actelion, Novartis, Biogen, Pzifer, and Eisai and has received research support from Ministero dell'Istruzione, dell'Università e della Ricerca Scientifica (MIUR) of Italy, and Regione Piemonte (Italy). Go to Neurology.org for full disclosures.
REFERENCES
- 1. Mackenzie IR, Feldman HH. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol 2005; 64: 730– 739. [DOI] [PubMed] [Google Scholar]
- 2. Lillo P, Hodges JR. Frontotemporal dementia and motor neuron disease: overlapping clinic-pathological disorders. J Clin Neurosci 2009; 16: 1131– 1135. [DOI] [PubMed] [Google Scholar]
- 3. Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain 2011; 134: 2582– 2594. [DOI] [PubMed] [Google Scholar]
- 4. Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 2007; 114: 63– 70. [DOI] [PubMed] [Google Scholar]
- 5. Parkinson N, Ince PG, Smith MO, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006; 67: 1074– 1077. [DOI] [PubMed] [Google Scholar]
- 6. Schymick JC, Yang Y, Andersen PM, et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry 2007; 78: 754– 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mackenzie IRA, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010; 9: 995– 1007. [DOI] [PubMed] [Google Scholar]
- 8. Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010; 68: 857– 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeJesus-Hernandez M, Mackenzie IRA, Boev BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245– 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010; 465: 223– 226. [DOI] [PubMed] [Google Scholar]
- 11. Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011; 477: 211– 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Babu JR, Lamar Seibenhener M, Peng J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 2008; 106: 107– 120. [DOI] [PubMed] [Google Scholar]
- 13. Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer's disease: possible role in tangle formation. Neuropathol Appl Neurobiol 2002; 28: 228– 237. [DOI] [PubMed] [Google Scholar]
- 14. Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002; 160: 255– 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakaso K, Yoshimoto Y, Nakano T, et al. Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson's disease. Brain Res 2004; 1012: 42– 51. [DOI] [PubMed] [Google Scholar]
- 16. Murray E, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropath 2011; 122: 673– 690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 2011; 122: 691– 702. [DOI] [PubMed] [Google Scholar]
- 18. Laurin N, Brown JP, Morisette J, Raymond V. Recurrent mutation of the gene encoding sequestome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70: 1582– 1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gennari L, Gianfrancesco F, Di Stefano M, et al. SQSTM1 gene analysis and gene-environment interaction in Paget's disease of bone. J Bone Miner 2010; 25: 1375– 1384. [DOI] [PubMed] [Google Scholar]
- 20. Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011; 68: 1440– 1446. [DOI] [PubMed] [Google Scholar]
- 21. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546– 1554. [DOI] [PubMed] [Google Scholar]
- 22. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motoneuron Disord 2000; 1: 293– 299. [DOI] [PubMed] [Google Scholar]
- 23. Dumanchin C, Camuzat A, Campion D, et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet 1998; 7: 1825– 1829. [DOI] [PubMed] [Google Scholar]
- 24. Cortini F, Fenoglio C, Guidi I, et al. Novel exon 1 progranulin gene variant in Alzheimer's disease. Eur J Neurol 2008; 15: 1111– 1117. [DOI] [PubMed] [Google Scholar]
- 25. Tsai CP, Soong BW, Lin KP, Tu PH, Lin JL, Lee YC. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol Aging 2011; 32: 553. [DOI] [PubMed] [Google Scholar]
- 26. Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci 2007; 32: 95– 100. [DOI] [PubMed] [Google Scholar]
- 27. Long J, Garner TP, Pandya MJ, et al. Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF-κB signaling. J Mol Biol 2010; 396: 178– 194. [DOI] [PubMed] [Google Scholar]
- 28. Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 2004; 14: 289– 301. [DOI] [PubMed] [Google Scholar]
- 29. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131– 24145. [DOI] [PubMed] [Google Scholar]
- 30. Ichimura Y, Kumanomidou T Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283: 22847– 22857. [DOI] [PubMed] [Google Scholar]
- 31. Geetha T, Wooten MW. Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett 2002; 512: 19– 24. [DOI] [PubMed] [Google Scholar]
- 32. Isogai S, Morimoto D, Arita K, et al. Crystal structure of the UBA domain of p62 and its interaction with ubiquitin. J Biol Chem 2011; 286: 31864– 31874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Du Y, Wooten MC, Wooten MW. Oxidative damage to the promoter region of SQSTM/p62 is common to neurodegenerative disease. Neurobiol Dis 2009; 35: 302– 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem 2011; 116: 248– 259. [DOI] [PubMed] [Google Scholar]
- 35. Chen S, Zhang X, Song L, Le W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol 2012; 22: 110– 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul Disord 2009; 19: 308– 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee M, Shin J. Triage of oxidation-prone proteins by Sqstm1/p62 within the mitochondria. Biochem Biophys Res Commun 2011; 413: 122– 127. [DOI] [PubMed] [Google Scholar]
- 38. Lücking CB, Dürr A, Bonifati V, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000; 342: 1560– 1567. [DOI] [PubMed] [Google Scholar]
- 39. Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on Huntington-induced cell death. J Cell Biol 2005; 171: 603– 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geetha T, Vishwaprakash N, Sycheva M, Babu JR. Sequestosome 1/p62: across diseases. Biomarkers 2012; 17: 99– 103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.