

Abstract
Transforming Growth Factor-β (TGF-β) and Epidermal Growth Factor (EGF) signaling pathways are both independently implicated as key regulators in tumor formation and progression. Here, we report that the tumor-associated overexpression of epidermal growth factor receptor (EGFR) desensitizes TGF-β signaling and its cytostatic regulation through specific and persistent Stat3 activation and Smad7 induction in vivo. In human tumor cell lines, reduction of TGF-β-mediated Smad2 phosphorylation, nuclear translocation and Smad3 target gene activation were observed when EGFR was overexpressed, but not in cells that expressed EGFR at normal levels. We identified Stat3, which is activated specifically and persistently by overexpressed EGFR, as a key signaling molecule responsible for the reduced TGF-β sensitivity. Stable knockdown of Stat3 using small hairpin RNA(shRNA) in Head and Neck (HN5) and Epidermoid (A431) tumor cell lines resulted in reduced growth compared with control shRNA-transfected cells when grown as subcutaneous tumor xenografts. Furthermore, xenografts with Stat3 knockdown displayed increased Smad3 transcriptional activity, increased Smad2 phosphorylation and decreased Smad7 expression compared with control xenografts in vivo. Consistently, Smad7 mRNA and protein expression was also significantly reduced when EGFR activity was blocked by a specific tyrosine kinase inhibitor, AG1478, or in Stat3 knockdown tumors. Similarly, Smad7 knockdown also resulted in enhanced Smad3 transcriptional activity in vivo. Importantly, there was no uptake of subcutaneous HN5 xenografts with Smad7 knockdown. Taken together, we demonstrate here that targeting Stat3 or Smad7 for knockdown results in resensitization of TGF-β's cytostatic regulation in vivo. Overall, these results establish EGFR/Stat3/Smad7/TGF-β signaling axis driving tumor growth, which can be targeted therapeutically.
Keywords: EGFR, TGF-β Stat3, Smad7, cancer
Introduction
Growth factor and cytokine signaling networks control many aspects of cell behavior such as proliferation, survival, migration, invasive capabilities, transformation and differentiation. In normal cells, these complex signaling pathways are tightly regulated. Alterations of these signals are often found to cause, directly or indirectly, tumor formation. Transforming Growth Factor-β (TGF-β) and Epidermal Growth Factor (EGF) signaling pathways are both independently implicated as key regulators in tumor formation, and as such are potential therapeutic targets.1, 2, 3, 4
TGF-β regulates a wide range of cellular processes including cell proliferation, differentiation, migration, organization and death.3 It is widely accepted that TGF-β can act as a tumor suppressor, given the frequent occurrence of tumor formation when TGF-β or its downstream signaling is disrupted by gene targeting in mice or the identification of loss-of-function mutations within several TGF-β signaling components in many types of human cancers.5, 6, 7 Biological responses to TGF-β are mediated mainly by the type I (TβRI) and II (TβRII) transmembrane cell surface receptors,4, 8 which contain cytoplasmic domains with serine/threonine kinase activity. The activated ligand–receptor complex then binds and phosphorylates, through TβRI, the intracellular signaling molecules Smad2 and Smad3.4, 8, 9 Once phosphorylated, these regulatory Smads (R-Smad) form complexes with Smad4 (also called DPC4 for deleted in pancreatic carcinoma locus 4) and translocate into the nucleus. In the nucleus, they associate with transcription factors to form transcriptionally active DNA complexes.4, 8, 9
TGF-β signaling can be negatively regulated by multiple proteins located both extracellularly and intracellularly,4 including Smad7, which directly competes with Smad2/3 for binding to TβRI. Perturbation of TGF-β signaling by Smad710 as a transgene leads to various forms of tumor formation in mice. However, it remains to be answered whether the TGF-β signaling pathway is so fundamental in maintaining homeostasis that not only its direct disruptions but also other oncogenic signals acting through the impairment of TGF-β signaling leads to tumor formation.
Epidermal growth factor receptor (EGFR) signaling is elicited by ligand binding, initiating the activation of many downstream signaling cascades including the following: Ras-Raf-Erk1/2, Phospholipase Cγ, the PTEN-regulated phosphatidylinositol 3-kinase (PI3-K)-Akt-mTOR, Src and Signal transducer and activator of transcription (Stat) family members.11 In turn, these signaling intermediates trigger gene transcription of numerous proteins involved in regulating a variety of cellular functions.12 Not surprisingly, genetic alterations leading to EGFR overexpression or mutation are frequently observed in almost all epithelial cancer types.13, 14, 15 However, which downstream molecules are specifically activated by tumor-associated and overexpressed EGFR and are responsible for EGFR's tumorigenic function have yet to be clearly identified.
Stat3 is a member of the Stat family of cytoplasmic transcription factors that are activated by many cytokine and growth factor receptors including the EGFR.16, 17 Phosphorylated Stat3 transmits its signal from the EGFR to the nucleus, where it initiates transcription of multiple genes known to promote cancer development.7, 16, 18 Indeed, Stat3 activation has been detected at a high frequency in many types of tumors, and its activity is required for the growth of head-and-neck cancer and multiple-myeloma cells.19, 20 We have recently discovered21 that persistent activation of Stat3 causes 100% occurrence of tumor formation in a gastric mouse model. Importantly, this study identified that only persistent, not transient, activation of Stat3 desensitizes TGF-β signaling via increased Stat3-mediated Smad7 expression, releasing cells from its cytostatic regulation, thereby allowing tumor formation.
Here we establish a novel EGFR–Stat3–Smad7–TGF-β signaling molecular axis where tumor-associated overexpression of EGFR in epithelial cells results in sustained hyperactivation of Stat3, which induces Smad7 expression, compromising TGF-β's cytostatic regulation of the epithelium and consequent tumor formation and growth.
Results
Overexpression of EGFR inhibits TGF-β signaling
Previously, we found that aberrant IL-6/gp130 signaling causes the desensitization of TGF-β signaling in the gastric epithelium.21 To test the effect of EGFR signaling on TGF-β signaling, seven human cell lines with varying EGFR expression levels were used (Figure 1a). Of those, A431, HN5, 293T-EGFR and U87MG-EGFR overexpress EGFR, whereas the 293T, A549 and U87MG do not. The Smad3 luciferase reporter construct pCAGA12-luc was transiently transfected into those cell lines to quantitatively determine TGF-β signaling sensitivity. Stimulation with TGF-β-activated pCAGA12-luc activity was seen in all seven cell lines used (Figure 1b). Interestingly, this increased pCAGA12-luc activity was significantly reduced in all four EGFR-overexpressing cells, but not in the low levels of EGFR-expressing cells lines when cells were cotreated with EGF (Figure 1b), suggesting that EGF inhibited pCAGA12-luc activity in cells with high levels of EGFR expression. Furthermore, EGF reduced the TGF-β-mediated phospho-Smad2 levels in the EGFR-overexpressing cell lines A431, HN5 (Figure 1c) and 293T-EGFR cells (Supplementary Figure 1A), without affecting phospho-Smad2 levels in normal EGFR-expressing cells (293T and A549; Supplementary Figure 1A). Consequently, EGF treatment caused a marked decrease of Smad2 nuclear localization by TGF-β in A431 cells (Figure 1d). In addition, EGF reduced the expression of TGF-β-induced p21WAF1, an inhibitor of cyclin-dependent kinases and mediator of growth arrest (Supplementary Figure 1B).
Figure 1.

Overexpression of EGFR desensitizes the TGF-β pathway. (a) Cell lines as indicated were lysed and examined for EGFR and Actin protein expression or (b) transfected with the Smad3 reporter construct pCAGA12-luc and allowed to adhere overnight. Cells were then treated with 2 ng/ml of TGF-β in the presence (□) or absence (▪) of EGF (20 ng/ml) for a further 24 h, and then lysed and assessed for luciferase activity. Data are expressed as relative Smad3 luciferase activity (fold change) by standardizing the luciferase activity of unstimulated cells to 1, and accordingly normalizing all other raw values. (c) Cells were treated with EGF (20 ng/ml) overnight, then stimulated with TGF-β (0, 0.2 and 2 ng/ml) for 30 min and lysed and examined for phospho-Smad2 and total Smad2 expression. (d) A431 cells were stimulated with or without EGF (20 ng/ml) overnight, then treated with or without TGF-β (0.2 ng/ml) for 15 min. Cells were then fixed in formaldehyde, permeablized in 0.2% Triton-X-100 and stained with anti-Smad2 antibody. Localization of Smad2 was visualized with Alexa488-conjugated secondary antibody using confocal microscopy. (e) A431 (i) and HN5 (ii) cells were transfected with pCAGA12-luc, pretreated with (▪) or without (□) AG1478 for 4 h, and then stimulated with TGF-β (2 ng/ml) and/or EGF (20 ng/ml) for a further 24 h. Cells were then lysed and assessed for luciferase activity and expressed as outlined in Figure 1b. (f) HN5 cells were treated with or without TGF-β in the presence (▪) or absence (□) of AG1478 for 48 h. Cells were then incubated with 0.2 μCi of [3H]-thymidine/well for 4 h, lysed with 0.5 ℳ NaOH, harvested and then measured for incorporated [3H]-thymidine.
To confirm that EGFR activation was responsible for the observed desensitization of the TGF-β signaling, AG1478, a specific inhibitor of EGFR22 (Supplementary Figure 1C), was used. EGF-mediated reduction of the TGF-β reporter activity was reversed when A431 and HN5 cells were cotreated with AG1478 (Figure 1e), confirming that activation of overexpressed EGFR mediates the desensitization of the TGF-β signaling. Furthermore, blockade of EGFR activity in HN5 cells by AG1478 resensitized these cells to the growth-inhibitory effects of TGF-β, producing a reduction in [3H]-thymidine incorporation >50% (Figure 1f).
EGF-mediated inhibition of TGF-β signaling is not dependent on PI3-K and MEK activity
The two most documented signaling pathways activated upon EGFR phosphorylation are the Ras-MAPKs (MEKs) and the PI3-K/Akt pathways. Both pathways have been implicated in modulating Smad activation.23, 24, 25 To examine whether these pathways were involved in the desensitization of TGF-β signaling by overexpressed EGFR, we used pharmacological inhibitors to block either MEK (U0126) or PI3-K (LY294002) activity without affecting phospho-EGFR levels (Figures 2a and b; Supplementary Figures 2A and B). Unlike the EGFR inhibitor AG1478, neither U0126 nor LY294002 resensitized the TGF-β reporter activity in HN5 and 293T-EGFR cells (Figure 2c and Supplementary Figure 2C), suggesting that the inhibition of the TGF-β pathway mediated by the overexpression of EGFR is not dependent on MEK and PI3K signaling.
Figure 2.

EGF-mediated desensitization of the TGF-β pathway is MEK and PI3K independent. HN5 cells were treated with (a) U0126 (0, 2 and 10 μM) or (b) LY294002 (0, 2 and 10 μM) for 4 h, then stimulate with or without EGF (20 ng/ml) for 10 min. Cells were then lysed and examined for phosphorylated and total expression of EGFR, Akt and Erk1/2. (c) HN5 cells were transfected with pCAGA12-luc, pre-treated with (▪) or without (□) (i). U0126 and (ii). LY294002 for 4 h, then stimulated with TGF-β (2 ng/ml) and/or EGF (20 ng/ml) for a further 24 h. Cells were then lysed and assessed for luciferase activity and expressed as outlined in Figure 1b.
EGFR overexpression induces specifically sustained Stat3 phosphorylation and transcriptional activity
As we had previously shown that sustained Stat3 activation results in the desensitization of TGF-β signaling in the IL-6/gp130 signaling system,21 we next set out to determine whether EGFR activated Stat3 in several human cell lines. Although EGF stimulation resulted in increased phospho-EGFR, phospho-Erk1/2 and phospho-Akt levels in all five cell lines used, phosphorylation of Stat3 was only observed in cell lines overexpressing EGFR (A431, HN5 and 293T-EGFR) (Figure 3a). Furthermore, this EGFR-overexpression-specific Stat3 activation was sustained for at least 8 h post EGF stimulation (Figure 3b). In fact, the EGFR-mediated Stat3 phosphorylation observed correlated with an increase in Stat3 transcriptional activation as measured by the luciferase activity using the pAPRE-luc reporter construct in EGFR-overexpressing cells (Supplementary Figures 3A and B). TGF-β stimulation did not affect EGF-mediated Stat3 activity (Supplementary Figure 3C). There was minimal Stat3 reporter activation (<twofold) in the cells expressing normal or low levels of EGFR (293T, A549 and U87MG; Supplementary Figure 3B). Taken together, these results demonstrate that overexpression of EGFR mediates specifically sustained Stat3 phosphorylation and transcriptional activity.
Figure 3.

Overexpression of the EGFR leads to sustained Stat3 phosphorylation and desensitized TGF-β signaling. Cells were stimulated with or without EGF (20 ng/ml) for (a) 10 min or (b) 0.2, 1, 2, 4 and 8 h and then lysed and examined for phosphorylated and total expression of EGFR, Stat3, Akt and Erk1/2. (c) At 24 h following transient transfection with pCAGA12-luc, A431 (i) and HN5 (ii) cells were retransfected with control (□) or Stat3 (▪) siRNA, seeded and allowed to adhere for 24 h. Cells were then stimulated with TGF-β (2 ng/ml)±EGF (20 ng/ml) as indicated above for a further 24 h. Cells were then lysed and assessed for luciferase activity and expressed as relative luciferase activity (fold change). (d) Following transient transfection with control (□) or Stat3 (▪) siRNA, HN5 cells were seeded and allowed to adhere overnight. Cells were then treated with or without TGF-β for 48 h and measured for incorporated [3H]-thymidine as outlined in Figure 1f.
Our results so far demonstrate an inverse correlation between the specific activation of Stat3 and the TGF-β signaling sensitivity specifically in cells overexpressing EGFR. To verify conclusively that Stat3 mediates desensitization of the TGF-β signaling, we knocked down Stat3 protein expression by small interfering RNA (siRNA) (Supplementary Figure 4A). The activity of the TGF-β reporter pCAGA12-luc was increased when the endogenous Stat3 levels were knocked down in both A431 and HN5 cells (Figure 3c), indicating that Stat3 is indeed required for the overexpressed EGFR-mediated desensitization of the TGF-β signaling. Importantly, Stat3 knockdown restored TGF-β-mediated growth suppression of HN5 cells (Figure 3d).
Stable knockdown of Stat3 in HN5 and A431 cells results in reduced tumor growth
To confirm our in vitro results and to examine the role of Stat3 in an in vivo setting, we generated HN5 and A431 cells with stable knockdown of Stat3 expression using Stat3-specific small hairpin RNA(shRNA). HN5 and A431 cells with reduced Stat3 expression (designated S1, S2 and S3 for HN5 and A-S1 and A-S2 for A431 cells) expressed similar levels of EGFR compared with control cells (Figure 4a and Supplementary Figure 4B). As expected, S1, S2 and S3 cells also displayed reduced EGF-mediated Stat3 transcriptional activity (Supplementary Figure 4C) and phosphorylated Stat3 compared with the control HN5 cell line, although no effect was seen on phosphorylated EGFR or phospho-Erk1/2 levels (Supplementary Figure 4D). Most importantly, the tumor growth of these clones was significantly reduced when grown as xenografts in BALB/cnu/nu mice, indicating a key role of Stat3 in mediating tumorigenicity of HN5 and A431 cells (Figure 4b and Supplementary Figure 4E). Two tumors from each HN5 subclone were removed with subsequent western blot analysis of these tumors, showing that the knockdown of Stat3 had remained intact in vivo throughout the course of the experiment (Supplementary Figure 4F). As with transient knockdown of Stat3, stable knockdown of Stat3 increased the TGF-β-mediated activity of pCAGA12-luc and circumvented the inhibitory effects of EGF stimulation (Figure 4c). Furthermore, pCAGA12-luc activity was also increased by approximately 10-fold in HN5 cells with stable Stat3 knockdown compared with control cells in vivo (Figure 4d), whereas phospho-Smad2 expression was detected in approximately 4% of HN5 cells with Stat3 knockdown versus 1% of HN5 control cells in vivo (Figure 4e). These data confirm that Stat3 is required for the overexpressed EGFR-mediated desensitization of the TGF-β signaling.
Figure 4.

Stable Stat3 knockdown desensitizes the TGF-β signaling pathway in vitro and in vivo. (a) HN5-control and Stat3 shRNA clones (S1, S2, S3) were lysed and examined for EGFR, Stat3 and Actin protein expression or (b) inoculated subcutaneously into both flanks of BALB/cnu−/nu− female mice and measured for tumor volume (n=10/ group) as indicated in Materials and methods; HN5-control (▪) or Stat3 shRNA (open symbols). (c) HN5-control, S1, S2 and S3 cells were transfected with pCAGA12-luc and allowed to adhere overnight. Cells were then treated with 2 ng/ml of TGF-β in the presence (□) or absence (▪) of EGF (20 ng/ml) for a further 24 h, and then either lysed and assessed for luciferase activity as outlined in Figure 1b or (d) inoculatedsubcutaneously into both flanks of BALB/cnu−/nu− female mice and analyzed for Smad3 reporter activity using the IVIS lumina Animal Imager as outlined in Materials and methods 24 h post inoculation. Cells were infected with the adenoviral smad3 reporter construct (Ad-pCAGA-luc) overnight, 24 h before inoculation into mice. Analysis of total photon emissions (Smad3 transcriptional activity) was performed using the IVIS lumina image analysis software and was presented relative to control tumors±s.d. (n=4). (e) HN5-control and Stat3 shRNA xenografts were removed and fixed in formaldehyde. Sections were stained for phospho-Smad2 expression and visualized with Alexa546-conjugated secondary antibody using confocal microscopy. Percentage positive staining was determined by counting approximately 3000 cells per group.
Smad7-dependent desensitization of TGF-β signaling by EGFR
As IL-6/gp130 signaling sustained Stat3 activation and induced the expression of the negative regulator of TGF-β signaling, Smad7,21 we next examined whether the EGFR-Stat3-mediated desensitization of TGF-β signaling is also dependent on Smad7. Indeed, blockade of EGFR activation by AG1478 significantly reduced Smad7 gene promoter activity in A431 and HN5 cells (Figure 5a), whereas EGF stimulation enhanced Smad7 promoter activity (Supplementary Figure 5A). AG1478 treatment also reduced Smad7 mRNA and protein levels in HN5 cells (Figures 5b and c). In addition, EGF inhibited TGF-β-mediated TGFβRI/Smad2 association presumably through Smad7's well-recognized role of competing for TGFβRI binding (Supplementary Figure 5B). Furthermore, both Smad7 promoter activity and Smad7 protein expression were reduced when the Stat3 expression levels were knocked down transiently by siRNA (Figure 6a and b). Comparison of HN5 control and stable Stat3 knockdown xenografts also revealed reduced Smad7 mRNA expression (Figure 6c) and protein expression in vivo (Figure 6d). These data demonstrate that upregulation of Smad7 protein levels in EGFR-overexpressing tumor cells is through Stat3-mediated Smad7 promoter activation. To determine whether EGFR-Stat3-mediated desensitization of TGF-β signaling is due to increased Smad7 expression, we use Smad7 siRNA to knock down its expression. Indeed, in HN5 tumor cells, Smad7 knockdown largely restored the TGF-β reporter activity when EGFR is activated by EGF treatment (Figure 6e). It is noteworthy that the reporter activity in the absence of EGF treatment is also vastly increased by Smad7 knockdown, confirming the effect of increased basal Smad7 expression in EGFR-overexpressing cells. Importantly, Smad7 knockdown, similar to Stat3 knockdown, restored HN5 tumor cell growth inhibitory sensitivity to TGF-β (Figure 6f). Finally, and similar to stable knockdown of Stat3, stable Smad7 knockdown by shRNA in HN5 cells resulted in increased pCAGA12-luc activity by approximately 10-fold compared with control cells in vivo (Figure 6g). Taken together, this signaling pathway cross talk mediated by EGFR–Stat3–Smad7–TGF-β operates specifically in tumor-associated EGFR-overexpressing cells.
Figure 5.
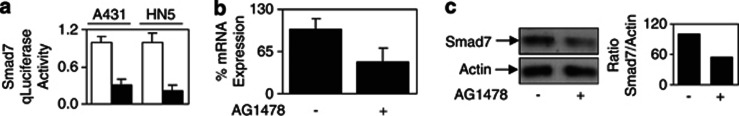
EGF-mediated desensitization of the TGF-β signaling pathway is Smad7 dependent. (a) A431 and HN5 cells were transfected with the Smad7 reporter construct pSmad7-luc and allowed to adhere overnight. Cells were then treated with (▪) or without (□) AG1478 for a further 24 h, lysed and assessed for luciferase activity and expressed as outlined in Figure 1b. HN5 cells were treated with or without AG1478 overnight and then analyzed for Smad7 (b) mRNA and (c) protein expression.
Figure 6.

Stat3 knockdown desensitizes the TGF-β signaling pathway by reducing Smad7 expression. (a) At 24 h following transient transfection with pSMAD712-luc, A431 and HN5 cells were re-transfected with control (□) or Stat3 (▪) siRNA, seeded and allowed to adhere for 24 h. Cells were then lysed and assessed for luciferase activity and expressed as outlined in Figure 1b. (b) HN5 cells were transiently transfected with control or Smad7 siRNA. After 48 h, cells were lysed and examined for Smad7 and Actin expression. (c) HN5-control or Stat3 shRNA clones (S1, S2, S3) were lysed and analyzed for Stat3 (□) and Smad7 (▪) mRNA expression. (d) HN5-control and Stat3 shRNA xenografts from Figure 4e were stained for Smad7 expression and visualized with Alexa488-conjugated secondary antibody using confocal microscopy. Percentage positive staining was determined by counting approximately 4000 cells per group. (e) At 24 h following transient transfection with pCAGA12-luc, HN5 cells were re-transfected with control (□) or Smad7 (▪) siRNA, seeded and allowed to adhere for 24 h. Cells were then stimulated with TGF-β (2 ng/ml)±EGF (20 ng/ml) as indicated above for a further 24 h. Cells were then lysed and assessed for luciferase activity. Data are expressed as relative luciferase activity (fold change) as described in Figure 3c. (f) Following transient transfection with control (□) or Smad7 (▪) siRNA, HN5 cells were seeded and allowed to adhere overnight. Cells were then measured for incorporated [3H]-thymidine as outlined in Figure 1f. (g) HN5-control or Smad7 shRNA cells were infected with the adenoviral smad3 reporter construct (Ad-pCAGA-luc) and then inoculated subcutaneously into both flanks of BALB/cnu−/nu− female mice. Smad3 reporter activity was analyzed as outlined in Figure 4d (n=4).
Discussion
Ever since its discovery as one of the first receptor tyrosine kinases, EGFR and its signaling have been studied extensively. Many downstream pathways have been identified, but little is known about the difference between the normal and tumorigenic signals transduced from EGFR. It was presumed that the two main downstream pathways, Ras/MAPK and PI3-K/Akt, were responsible for EGFR-driven tumor growth. However, it has been difficult to detect the different mode of their activation between normal and tumor cells. At best, <10% elevation in MAPK activation by a constitutively active EGFR (Δ2-7EGFR)26 was proposed to explain its tumorigenicity. Yet these downstream signals have been used to measure the efficacy of EGFR inhibition in cancer treatment.27, 28, 29 The identification of Stat3 here as a molecule specifically and persistently activated by the overexpressed and tumor-associated EGFR but not by EGFR expressed at normal levels reveals a critical signaling difference by EGFR between normal and tumor cells.
In both the normal and tumor cells, MAPK (Erk1/2) and Akt activation by EGFR are rapid and transient, regardless of the levels of EGFR expression. In contrast, low levels of EGFR do not activate Stat3, whereas only high levels of EGFR are capable of Stat3 activation. It appears that a threshold of approximately 106 EGFR/cell is required for EGF-mediated Stat3 activity based on the cell lines tested here. Surprisingly, Stat3 activation by high levels of EGFR is slower than the Erk1/2 or Akt activation (Figure 3). Furthermore, the high level of EGFR-mediated Stat3 activation is persistent, unlike the transient nature of Erk1/2 and Akt activation. This is in direct contrast to cytokines such as IL-6 and IL-11, which mediated Stat3 activation rapidly and transiently.21, 30 Our previous work demonstrated, in both animal models and human tissues, that persistent Stat3 activation leads to stomach epithelial hyperproliferation. It has become clear that Stat3 is an oncogene, and its activation is prevalent in many human cancers;16, 31, 32 however, unlike many other oncogenes, there is no genetic evidence of gain-of-function mutation. Our observation that the tumor-associated overexpressed EGFR activates Stat3 persistently identifies EGFR as a physiologically relevant upstream molecular cause of Stat3 activation.
The tumorigenic consequence of the persistent Stat3 activation is at least partially mediated through the desensitization of TGF-β signaling via its negative regulator Smad7 in stomach epithelium.21 Indeed, EGFR-mediated Stat3 activation also results in the desensitization of TGF-β signaling in many tumor cell lines with EGFR overexpression. More importantly, in the head-and-neck tumor cell line with high levels of EGFR, HN5, there is a loss of cytostatic/growth-inhibitory regulation by TGF-β (Figure 1), which can be reversed by blocking either EGFR activation or reducing Stat3 or Smad7 expression (Figures 1, 3 and 6). Restoration of TGF-β signaling as evident by enhanced cytosolic and nuclear phospho-Smad2 can also be achieved by stable knockdown of Stat3 or Smad7 expression in vivo (Figures 4 and 6).33 Thus, the loss of TGF-β cytostatic regulation on normal cell growth may represent a key molecular event during tumorigenesis driven by overexpression of EGFR through the EGFR–Stat3–Smad7–TGF-β signaling axis.
Loss of TGF-β sensitivity and thereby loss of cytostatic regulation may represent a key molecular event in tumor progression. Desensitization to TGF-β-mediated growth regulation can occur through the generation of loss-of-function mutations in either TGF-β receptors or downstream signaling molecules.34 However, the overall occurrences of such mutations in human tumors are not frequent,34 suggesting that there may exist alternatives other than direct deletion or mutation of TGF-β signaling components. Indeed, some early work showed that Ras inhibited TGF-β signaling through MAPK's modification of Smads,24, 27, 35 whereas IFN-γ inhibits TGF-β signaling through Stat1-mediated Smad7 expression.36 We have not seen any desensitization of TGF-β signaling mediated by MAPKs (Erk1/2) in cells expressing high levels of EGFR (Figure 2). At least in the context of the stomach epithelium, Stat1 did not cause desensitization of TGF-β signaling.21 We identify Stat3 as a key mediator of loss of TGF-β cytostatic regulation through signaling pathway cross talk in vivo both here and previously.21 In early tumor development where tumor growth and expansion are dominant, TGF-β acts as a tumor suppressor, whereas it promotes invasion in late stages of tumor progression. Importantly, TGF-β signaling may be necessary for the late tumor invasion, best demonstrated in colon tumor development where deletions or mutations in the TGF-β signaling pathway gave rise to a better prognosis compared with patients with colon cancers with intact TGF-β signaling.37, 38 Desensitization of TGF-β signaling through cross talk such as EGFR-Stat3-Smad7-TGF-β demonstrated here, instead of the deletion or mutation of TGF-β signaling, may enable the cells to bypass TGF-β's tumor-suppressive effect in early tumor development, although the pathway can be resensitized by different molecular means to promote tumor invasion at late stages.
Establishing the loss of cytostatic regulation by TGF-β by tumor-associated EGFR overexpression through the EGFR-Stat3-Smad7-TGF-β axis has direct implications in EGFR signaling-targeted cancer therapy. First, it provides some clear molecular targets for treating EGFR-driven tumors, namely, Stat3 and Smad7. Several research groups have developed therapeutics that target Stat3 with some success,20, 39, 40 such as administration of antisense oligonucleotides targeting Stat3-expressing hematological tumors in mice39 and a synthesized triterpenoid, CDDO-Imidazolide, to inhibit Stat3 phosphorylation in human myeloma and lung cancer cells.34, 41 A similar strategy may be used to target Smad7 expression in tumors. A combination of Stat3 and/or Smad7 with EGFR targeting may provide a much more effective treatment. Second, to determine the efficacy of any EGFR-targeted treatment, in addition to its effects on Ras/MAPK and PI3-K/Akt pathways, it may be necessary to measure the effects on Stat3 activation, Smad7 expression and TGF-β-signaling sensitivity. Given the availability of the sensitive luciferase reporters for Stat3, Smad7 and TGF-β signaling, it is not unreasonable to envisage them being used to directly monitor the real effects of any EGFR-targeting therapy on its more specifically tumorigenic downstream signaling. More importantly, they can be used in vivo in real time when coupled with live imaging techniques to optimize treatment regimen, especially regarding dosages and schedules.
Materials and Methods
Antibodies and reagents
Rabbit polyclonal antibodies directed against Erk1, TGFβRI, p21WAF1 and Stat3 and the polyclonal goat Akt antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The phospho-Erk1/2, phospho-Stat3 and Phospho-Akt rabbit polyclonal antibodies were from Cell Signaling Technology (Danvers, MA, USA), whereas the mouse phospho-tyrosine monoclonal antibody (4G10) was from Upstate Biotechnology (Lake Placid, NY, USA). The anti-mouse Actin and Smad2 antibodies were purchased from Sigma (St Louis, MO, USA) and BD transduction Laboratories (Rockville, MD, USA), respectively. The Anti-rabbit phospho-Smad2 antibody was kindly provided by Professor P Ten Dijke (Leiden University Medical Center, The Netherlands). The anti-rabbit Smad7 antibody was from Imgenex (Imgenex Corporation, San Diego, CA, USA). The anti-rabbit Alexa488-conjugated and Alexa546-conjugated secondary antibodies and the [3H]-thymidine were from Invitrogen (Mulgrave, VIC, Australia). The mouse anti-EGF receptor antibody was provided by the Melbourne Centre for Clinical Sciences (Ludwig Institute for Cancer Research). AG1478 and Recombinant mouse EGF were kindly provided by the Melbourne Tumor Biology Branch, Ludwig Institute for Cancer Research, and recombinant human TGF-β1 was purchased from R & D Systems (Minneapolis, MN, USA). The MEK inhibitor U0126 and the PI3K inhibitor LY294002 were purchased from Calbiochem (Merck KGaA, Darmstadt, Germany). Human Stat3 and Smad7 siRNAs were from Santa Cruz, whereas the fluorescein-labeled control siRNA was from Qiagen (Qiagen, SA Biosciences, Frederick, MD, USA).
Cells and cell culture
The epidermoid carcinoma cell line A431, the head-and-neck carcinoma cell line HN5, the human embryonic kidney cell line HEK-293T (293T) and the EGFR-Flag tagged stably transfected HEK-293T-EGFR (293T-EGFR) cell line, the lung carcinoma cell line A549, the glioblastoma cell line U87MG and EGFR stable transfected U87MG-EGFR have all been previously described.42, 43, 44, 45, 46 The HN5-Stat3 shRNA clones (designated S1, S2 and S3) and A431-Stat3 shRNA clones (designated A-S1 and A-S2) were generated by transfecting cells with the SureSilencing Stat3-shRNA construct (Qiagen) using FuGENE HD transfection reagent (Roche, Basel, Switzerland) following the manufacturer's instructions, and selected for using Geneticin (Roche). Similarly, the HN5-Smad7 shRNA clones were generated by transfecting cells with the SureSilencing Smad7-shRNA construct (Qiagen) as above. Control HN5 cells were generated by stably transfecting a negative-control shRNA (Qiagen). All cells were maintained in Dulbecco's Modified Eagle's Medium containing 10% fetal bovine serum (FBS) (DKSH, Hallam, VIC, Australia), 2 mℳ glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Cells were incubated in a humidified atmosphere of 90% air and 10% CO2 at 37 °C.
Western blot and immunoprecipitation analysis
Cells were lysed in a lysis buffer (50 mℳ Tris (pH 7.4), 150 mℳ NaCl, 1% Triton-X-100, 50 mℳ NaF, 2 mℳ MgCl2, 1 mℳ Na3VO4 and protease inhibitor cocktail (Roche)) and clarified by centrifugation (13 000g for 15 min at 4 °C). Tumor xenografts were snap-frozen and lysed in RIPA buffer (50 mℳ Tris (pH 7.4), 5 mℳ EDTA, 150 mℳ NaCl, 0.5% Na Deoxycholate, 0.5% SDS, 10 mℳ NaF and protease inhibitor cocktail (Roche)) and also clarified by centrifugation. For Immunoprecipitation experiments, anti-TGFβRI antibody and protein-A-Sepharose beads (Amersham Biosciences Corporation, Piscataway, NJ, USA) were added to clarified proteins for 4 h at 4 °C. Immune complexes were washed three times in ice-cold PBS. Proteins were then separated by SDS–PAGE (Invitrogen), blotted onto nitrocellulose and probed with the indicated primary antibodies. The signal was visualized using the ECL chemoluminescence detection kit (GE Healthcare, Rydelmere, NSW, Australia) following incubation with appropriate secondary antibodies.
Luciferase assays
The firefly luciferase constructs pAPRE-luc,47 pSmad7-luc48 and pCAGA12-luc49 have all been previously described. Cells were transiently transfected with the construct using the FuGENE HD transfection kit (Roche). After a 24-h transfection period, cells were washed with PBS and cultured with TGF-β, EGF, AG1478, U0126 and/or LY294002 at the concentrations indicated for a further 24 h. Cells were then lysed and assessed for luciferase activity using the Luciferase Reporter Assay Kit (Promega Corporation, Madison, WI, USA) following the manufacturer's instructions. To assess for the effects of Stat3 and Smad7 knockdown on Smad3 and Smad7 promoter activity, cells were transiently transfected with Stat3, Smad7 or fluorescein-labeled control siRNA using the HiPerFect transfection reagent (Qiagen) as per the manufacturer's instructions 24 h after transfection with p(CAGA)12-luc and pSmad7-luc constructs. The creation of the Adenoviral Smad3 reporter (Ad-pCAGA-luc) has been described previously.50
[3H]-thymidine incorporation assays
Cells were plated in 96-well plates in DMEM-10% FBS and allowed to adhere overnight. Quadruplicate wells were treated with TGF-β and/or AG1478 at the concentrations indicated for 48 h and then incubated with 0.2 μCi of [3H]-thymidine/well for an additional 4 h. Cells were lysed with 0.5 ℳ NaOH, harvested using a Filtermate Harvester (Packard Instrument Co., Meriden, CT, USA) and the incorporated [3H]-thymidine measured with a Microplate Scintillation Counter (Packard Instrument Co.). To assess for the effects of Stat3 and Smad7 knockdown on [3H]-thymidine incorporation, cells were transiently transfected with Stat3, Smad7 siRNA or fluorescein-labeled control siRNA using the HiPerFect transfection reagent in solution and seeded in 96-well plates 24 h before TGF-β treatment.
Immunofluorescence staining and confocal microscopy
A431 cells were seeded onto coverslips in six-well plates in DMEM-10% FBS and allowed to adhere overnight. Cells were stimulated with or without EGF in serum-free media overnight, followed by treatment with TGF-β for 15 min. Cells were then washed twice in PBS, fixed in formaldehyde and permeabilized with PBS containing 0.2% Triton-X-100. Following blocking in PBS-Tween20 containing 5% BSA, cells were stained with anti-Smad2 antibody. Tumor xenografts were excised from mice at the end of the experiment and processed in 10% formalin/PBS before being embedded in paraffin. Tumor sections (4 μm) were then dewaxed, fixed, permeablized and blocked as described above and stained with either pSmad2 or Smad7 antibody. Visualization was achieved with either Alexa546- or Alexa488-conjugated secondary antibody using confocal microscopy as described.49
Tumor growth and bioluminescence Imaging in vivo
A431 cells and all derivatives (3 × 106) or HN5 cell and all derivatives (1 × 107) were inoculated subcutaneously into both flanks of 4- to 6-week-old BALB/cnu/nu female mice (Animal Research Center, WA, Australia). Tumor volume in mm3 was determined using the formula (length × width2)/2, where length was the longest axis and width the measurement at right angles to the length.51 This research project was approved by the Animal Ethics Committee of the Ludwig Institute for Cancer Research and Department of Surgery, University of Melbourne at the Royal Melbourne Hospital. For bioluminescence imaging, cells were infected with an Adenoviral Smad3 reporter construct (Ad-pCAGA12-Luc) 24 h before subcutaneous inoculation into BALB/cnu/nu female mice. Mice were then injected intraperitonealy with 150mg/kg 𝒟-luciferin (Xenogen Corp., Alameda, CA, USA) in PBS, anesthetized with isofluorane and placed under the IVIS camera (Xenogen Corp). The bioluminescence images were recorded between 10 and 20 min after each 𝒟-luciferin injection, and bioluminescence intensity was quantified as the sum of detected photons per second within the region of interest using the LivingImage software (Xenogen Corp).
RNA extraction and qRT–PCR
RNA from cells or xenograft lysates were extracted using TRIzol reagent (Invitrogen), and converted to cDNA using SuperScript III First-Strand Synthesis SuperMix as per the manufacturer's instructions (Invitrogen). RT-PCR was performed using an ABI 7700 Real-Time PCR system (Applied Biosystems, Life Technologies, Mulgrave, Victoria, Australia). The following primers were used: Stat3, 5′-GAGGACTGAGCATCGAGCA-3′ (forward) and 5′-CATGTGATCTGACACCCTGAA-3′ (reverse); Smad7, 5′-CTGCAACCCCCATCACCTTA-3′ (forward) and 5′-CCCTGTTTCAGCGGAGGAA-3′ (reverse); and GAPDH, 5′-TGCACCACCAACTGCTTAGC-3′ (forward) and 5′-GGCATGGACTGTGGTCATGAG-3′ (reverse). Triplicate reactions with SYBR Green (Invitrogen) were performed using a two-step amplification program of initial denaturation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 20 s and 60 °C for 30 s. A melting-curve analysis step was carried out at the end of the amplification, consisting of denaturation at 95 °C for 1 min and re-annealing at 55 °C for 1 min. Each change in gene expression is expressed as percentage compared with the control and normalized to GAPDH Data acquisition, and analysis was performed using the ABI 7700 Real-Time PCR system software.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council (NHMRC) to H-JZ. RBL is a recipient of the Winter and Glover Fellowship for Cancer Research from the University of Melbourne.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Supplementary Material
References
- Ettinger DS. Clinical implications of EGFR expression in the development and progression of solid tumors: focus on non-small cell lung cancer. Oncologist. 2006;11:358–373. doi: 10.1634/theoncologist.11-4-358. [DOI] [PubMed] [Google Scholar]
- Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Burgess AW. Regulation of transforming growth factor-beta signaling. Mol Cell Biol Res Commun. 2001;4:321–330. doi: 10.1006/mcbr.2001.0301. [DOI] [PubMed] [Google Scholar]
- Das P, Maduzia LL, Padgett RW. Genetic approaches to TGFbeta signaling pathways. Cytokine Growth Factor Rev. 1999;10:179–186. doi: 10.1016/s1359-6101(99)00014-3. [DOI] [PubMed] [Google Scholar]
- Luwor RB, Kaye AH, Zhu HJ. Transforming growth factor-beta (TGF-beta) and brain tumours. J Clin Neurosci. 2008;15:845–855. doi: 10.1016/j.jocn.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329–340. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- Wrana JL. Regulation of Smad activity. Cell. 2000;100:189–192. doi: 10.1016/s0092-8674(00)81556-1. [DOI] [PubMed] [Google Scholar]
- Kuang C, Xiao Y, Liu X, Stringfield TM, Zhang S, Wang Z, et al. In vivo disruption of TGF-beta signaling by Smad7 leads to premalignant ductal lesions in the pancreas. Proc Natl Acad Sci USA. 2006;103:1858–1863. doi: 10.1073/pnas.0508977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors. 2008;26:263–274. doi: 10.1080/08977190802312844. [DOI] [PubMed] [Google Scholar]
- Grandal MV, Madshus IH. Epidermal growth factor receptor and cancer: control of oncogenic signalling by endocytosis. J Cell Mol Med. 2008;12:1527–1534. doi: 10.1111/j.1582-4934.2008.00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JE. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth in vitro. J Clin Invest. 1998;102:1385–1392. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins BJ, Grail D, Nheu T, Najdovska M, Wang B, Waring P, et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat Med. 2005;11:845–852. doi: 10.1038/nm1282. [DOI] [PubMed] [Google Scholar]
- Nagane M, Narita Y, Mishima K, Levitzki A, Burgess AW, Cavenee WK, et al. Human glioblastoma xenografts overexpressing a tumor-specific mutant epidermal growth factor receptor sensitized to cisplatin by the AG1478 tyrosine kinase inhibitor. J Neurosurg. 2001;95:472–479. doi: 10.3171/jns.2001.95.3.0472. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- Montgomery RB, Moscatello DK, Wong AJ, Cooper JA, Stahl WL. Differential modulation of mitogen-activated protein (MAP) kinase/extracellular signal-related kinase kinase and MAP kinase activities by a mutant epidermal growth factor receptor. J Biol Chem. 1995;270:30562–30566. doi: 10.1074/jbc.270.51.30562. [DOI] [PubMed] [Google Scholar]
- Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, et al. Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res. 2001;61:6500–6510. [PubMed] [Google Scholar]
- Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res. 2003;9:2316–2326. [PubMed] [Google Scholar]
- Normanno N, De Luca A, Maiello MR, Campiglio M, Napolitano M, Mancino M, et al. The MEK/MAPK pathway is involved in the resistance of breast cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol. 2006;207:420–427. doi: 10.1002/jcp.20588. [DOI] [PubMed] [Google Scholar]
- Tenney R, Stansfield K, Pekala PH. Interleukin 11 signaling in 3T3-L1 adipocytes. J Cell Physiol. 2005;202:160–166. doi: 10.1002/jcp.20100. [DOI] [PubMed] [Google Scholar]
- Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol. 2005;2:315–324. doi: 10.1038/ncponc0195. [DOI] [PubMed] [Google Scholar]
- Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004;23:8017–8023. doi: 10.1038/sj.onc.1208159. [DOI] [PubMed] [Google Scholar]
- Itman C, Small C, Griswold M, Nagaraja AK, Matzuk MM, Brown CW, et al. Developmentally regulated SMAD2 and SMAD3 utilization directs activin signaling outcomes. Dev Dyn. 2009;238:1688–1700. doi: 10.1002/dvdy.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Howe PH, Dobrowolski SF, Reddy KB, Stacey DW. Release from G1 growth arrest by transforming growth factor beta 1 requires cellular ras activity. J Biol Chem. 1993;268:21448–21452. [PubMed] [Google Scholar]
- Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- Iacopetta BJ, Soong R, House AK, Hamelin R. Gastric carcinomas with microsatellite instability: clinical features and mutations to the TGF-beta type II receptor, IGFII receptor, and BAX genes. J Pathol. 1999;187:428–432. doi: 10.1002/(SICI)1096-9896(199903)187:4<428::AID-PATH264>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, et al. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11:623–629. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- Liby K, Voong N, Williams CR, Risingsong R, Royce DB, Honda T, et al. The synthetic triterpenoid CDDO-Imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin Cancer Res. 2006;12 (14 Part 1:4288–4293. doi: 10.1158/1078-0432.CCR-06-0215. [DOI] [PubMed] [Google Scholar]
- Place AE, Suh N, Williams CR, Risingsong R, Honda T, Honda Y, et al. The novel synthetic triterpenoid, CDDO-imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin Cancer Res. 2003;9:2798–2806. [PubMed] [Google Scholar]
- Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- Luwor RB, Johns TG, Murone C, Huang HJ, Cavenee WK, Ritter G, et al. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 2001;61:5355–5361. [PubMed] [Google Scholar]
- Luwor RB, Zhu HJ, Walker F, Vitali AA, Perera RM, Burgess AW, et al. The tumor-specific de2-7 epidermal growth factor receptor (EGFR) promotes cells survival and heterodimerizes with the wild-type EGFR. Oncogene. 2004;23:6095–6104. doi: 10.1038/sj.onc.1207870. [DOI] [PubMed] [Google Scholar]
- Wild R, Fager K, Flefleh C, Kan D, Inigo I, Castaneda S, et al. Cetuximab preclinical antitumor activity (monotherapy and combination based) is not predicted by relative total or activated epidermal growth factor receptor tumor expression levels. Mol Cancer Ther. 2006;5:104–113. doi: 10.1158/1535-7163.MCT-05-0259. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Iaria J, Orchard S, Walker F, Burgess AW. Epidermal growth factor receptor: association of extracellular domain negatively regulates intracellular kinase activation in the absence of ligand. Growth Factors. 2003;21:15–30. doi: 10.1080/0897719031000096424. [DOI] [PubMed] [Google Scholar]
- Wang Y, Robledo O, Kinzie E, Blanchard F, Richards C, Miyajima A, et al. Receptor subunit-specific action of oncostatin M in hepatic cells and its modulation by leukemia inhibitory factor. J Biol Chem. 2000;275:25273–25285. doi: 10.1074/jbc.M002296200. [DOI] [PubMed] [Google Scholar]
- Brodin G, Ahgren A, ten Dijke P, Heldin CH, Heuchel R. Efficient TGF-beta induction of the Smad7 gene requires cooperation between AP-1, Sp1, and Smad proteins on the mouse Smad7 promoter. J Biol Chem. 2000;275:29023–29030. doi: 10.1074/jbc.M002815200. [DOI] [PubMed] [Google Scholar]
- Stenvers KL, Tursky ML, Harder KW, Kountouri N, Amatayakul-Chantler S, Grail D, et al. Heart and liver defects and reduced transforming growth factor beta2 sensitivity in transforming growth factor beta type III receptor-deficient embryos. Mol Cell Biol. 2003;23:4371–4385. doi: 10.1128/MCB.23.12.4371-4385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luwor RB, Wang B, Nheu TV, Iaria J, Tsantikos E, Hibbs ML, et al. New reagents for improved in vitro and in vivo examination of TGF-beta signalling. Growth Factors. 29:211–218. doi: 10.3109/08977194.2011.615311. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Hobbs K, Clark GM. Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res. 1985;45:584–590. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.